Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention

 ,
,
Abstract
1. Introduction
2. Risk Factors of UC-CRC
2.1. Disease-Related Factors
2.2. Patient-Related Factors
2.3. Pathology-Related Factors
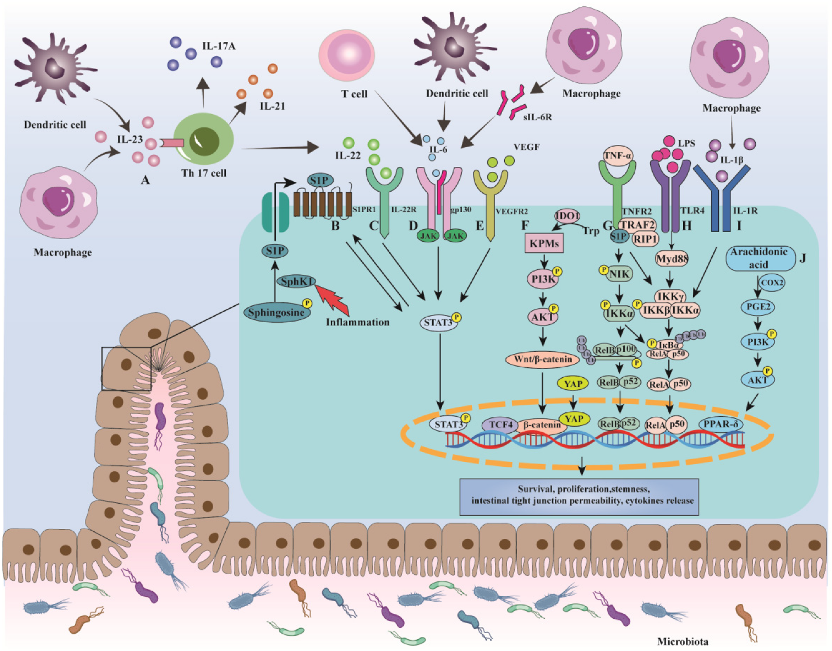
3. Mechanisms of Colorectal Cancer in Ulcerative Colitis
3.1. NF-κB
3.2. IL-6/STAT3
3.3. IL-23/Th17
3.4. COX-2/PGE2
3.5. Wnt/β-Catenin
3.6. UC-CRC and Gut Microbiota
4. Surveillance for CRC in UC
4.1. Surveillance Strategies for CRC in UC
4.2. Comparison between Surveillance Techniques

{kind=link}
| Study | Year | IBD Type | Type of Endoscopy | Number of Patients | Patients with Dysplasia, n (%) | Number of Dysplasia | Number of CRC Patients | p Value | Withdrawal Time (Minutes) |
|---|---|---|---|---|---|---|---|---|---|
| Subramanian et al. [130] | 2008–2010 | IBD | SD-WLE HD-WLE | 160 (UC101/CD59) 209 (UC147/CD62) | 8 (5.00) 24 (11.48) | 11 (HGD1) 32 (HGD2) | 2 5 | 0.03 | - - |
| Marion et al. [131] | 2005–2011 | IBD | SD-WLE CE | 68 (UC55/CD13) | 11 (16.18) 27 (39.71) | 11 27 | - - | 0.001 | - - |
| Yang et al. [134] | 2013–2015 | UC | HD-WLE HD-CE | 108 102 | 6 (5.56) 4 (3.92) | - - | - - | 0.749 | 17.6 16.5 |
| Alexandersson et al. [133] | 2011–2016 | IBD | HD-WLE HD-CE | 153 (UC90/CD62/Indeterminate colitis1) 152 (UC96/CD54/Indeterminate colitis2) | 7 (4.58) 17 (11.18) | 7 (LGD5/HGD2) 24 (LGD21/HGD3) | 0 0 | 0.032 | 17 ± 8 24 ± 11 |
| Bisschops et al. [136] | 2008–2013 | UC | CE NBI | 66 65 | 14 (21.21) 14 (21.54) | 30 (LGD30) 20 (LGD19/HGD1) | 0 1 | 0.964 | 27.0 18.5 |
| Iacucci et al. [137] | 2014–2016 | IBD | HD-WLE CE i-SCAN | 90 (UC42/CD44/ Indeterminate colitis4) 90 (UC43/CD47/ Indeterminate colitis0) 90 (UC44/CD45/ Indeterminate colitis1) | 23 (25.56) 22 (24.44) 14 (15.56) | 22 (LGD22) 15 (LGD15) 11 (LGD11) | 0 1 0 | 0.84 | 15.4 16.2 15.3 |
| Vleugels et al. [139] | 2013–2017 | UC | CE AFI | 105 105 | 20 (19.05) 13 (12.38) | 37 (36LGD/1HGD) 14 (14LGD) | 1 0 | 0.18 | 25.1 18.0 |
| Kiesslich et al. [142] | - | UC | SD-WLE CLE | 73 80 | 4 (5.48) 11 (13.75) | 4 (3LGD/1HGD) 19 (12LGD/7HGD) | 0 0 | 0.097 | - - |
| Leong et al. [149] | 2014–2015 | IBD | FVC FUSE | 27 (UC15/CD12) 25 (UC14/CD11) | 2 6 | 7 19 | 0 0 | 0.018 | 12.0 15.8 |
4.3. Comparison of Random Biopsies (Rb) vs. Targeted Biopsies (Tb)
5. Chemoprevention of CRC in UC
5.1. 5-ASA
5.2. Thiopurines
5.3. Ursodeoxycholic Acid
5.4. Biologics
| Drugs | Study Design | Area | Study Population | Primary Outcome | OR/RR/HR | 95% CI | p Value | Effect | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 5-ASA | Case–control study | French | IBD | CRC | 0.59 | 0.37–0.94 | 0.026 | Protective | [156] |
| Case–control study | Finland | IBD | CRC | 0.17 | 0.03–1.01 | 0.051 | Protective | [157] | |
| Case–control study | UK | UC | CRC | 0.60 | 0.38–0.96 | - | Protective | [158] | |
| Cohort study | China | IBD | CRC | 1.22 | 0.60–2.48 | 0.593 | No effect | [162] | |
| Case–control study | - | IBD | CRN | 0.27 | 0.08–0.88 | - | Protective | [160] | |
| Case–control study | Asian | UC | CRC | 0.25 | 0.06–1.02 | 0.037 | Protective | [159] | |
| Meta-analysis | - | IBD | CRN | 0.57 | 0.45–0.71 | - | Protective | [161] | |
| Thiopurine | Cohort study | Spanish | UC | CRN | 0.21 | 0.06–0.74 | 0.015 | Protective | [167] |
| Case–control study | French | IBD | CRC | 0.76 | 0.43–1.34 | 0.347 | No effect | [156] | |
| Case–control study | Netherlands | IBD | CRC | 0.30 | 0.16–0.56 | <0.001 | Protective | [178] | |
| Case–control study | - | IBD | CRN | 0.18 | 0.05–0.70 | - | Protective | [160] | |
| Case–control study | USA | UC | CRN | 1.06 | 0.59–1.93 | No effect | [166] | ||
| Case–control study | Finland | IBD | CRC | 0.09 | 0.02–0.33 | 0.0003 | Protective | [157] | |
| Meta-analysis | - | UC | CRN | 0.67 | 0.45–0.98 | 0.006 | Protective | [168] | |
| Meta-analysis | - | IBD | CRN | 0.71 | 0.54–0.94 | 0.017 | Protective | [184] | |
| Ursodeoxycholic acid | Cohort study | USA | UC | CRC | 0.59 | 0.26–1.36 | - | No effect | [185] |
| Case–control study | USA | UC | CRC | 0.26 | 0.06–0.92 | 0.034 | Protective | [173] | |
| Meta-analysis | - | IBD | aCRN | 0.35 | 0.17–0.73 | - | Protective | [172] | |
| Anti-TNFα agents | Cohort study | USA | UC | CRC | 0.78 | 0.73–0.83 | <0.0001 | Protective | [14] |
| Case–control study | Netherlands | IBD | CRC | 0.09 | 0.01–0.68 | <0.02 | Protective | [178] | |
| Cohort study | French | UC | CRC | 0.41 | 0.20–0.86 | - | Protective | [179] |
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rubin, D.; Ananthakrishnan, A.; Siegel, C.; Sauer, B.; Long, M. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.; Hart, A.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wanders, L.; Dekker, E.; Pullens, B.; Bassett, P.; Travis, S.; East, J. Cancer risk after resection of polypoid dysplasia in patients with longstanding ulcerative colitis: A meta-analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.; Guadagni, I. From inflammation to colitis-associated colorectal cancer in inflammatory bowel disease: Pathogenesis and impact of current therapies. Dig. Liver Dis. 2021, 53, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Konishi, T.; Kishimoto, J.; Kotake, K.; Muto, T.; Sugihara, K. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: A nationwide Japanese study. Inflamm. Bowel Dis. 2011, 17, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Leowardi, C.; Schneider, M.; Hinz, U.; Harnoss, J.; Tarantino, I.; Lasitschka, F.; Ulrich, A.; Büchler, M.; Kadmon, M. Prognosis of Ulcerative Colitis-Associated Colorectal Carcinoma Compared to Sporadic Colorectal Carcinoma: A Matched Pair Analysis. Ann. Surg. Oncol. 2016, 23, 870–876. [Google Scholar] [CrossRef]
- Rogler, G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014, 345, 235–241. [Google Scholar] [CrossRef]
- Wan, M.; Wang, Y.; Zeng, Z.; Deng, B.; Zhu, B.; Cao, T.; Li, Y.; Xiao, J.; Han, Q.; Wu, Q. Colorectal cancer (CRC) as a multifactorial disease and its causal correlations with multiple signaling pathways. Biosci. Rep. 2020, 40, BSR20200265. [Google Scholar] [CrossRef]
- Murthy, S.; Feuerstein, J.; Nguyen, G.; Velayos, F. AGA Clinical Practice Update on Endoscopic Surveillance and Management of Colorectal Dysplasia in Inflammatory Bowel Diseases: Expert Review. Gastroenterology 2021, 161, 1043–1051.e4. [Google Scholar] [CrossRef]
- Wijnands, A.; de Jong, M.; Lutgens, M.; Hoentjen, F.; Elias, S.; Oldenburg, B. Prognostic Factors for Advanced Colorectal Neoplasia in Inflammatory Bowel Disease: Systematic Review and Meta-analysis. Gastroenterology 2021, 160, 1584–1598. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.; Cagan, A.; Cai, T.; Gainer, V.; Shaw, S.; Churchill, S.; Karlson, E.; Murphy, S.; Kohane, I.; Liao, K. Colonoscopy is associated with a reduced risk for colon cancer and mortality in patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 2015, 13, 322–329.e1. [Google Scholar] [CrossRef] [PubMed]
- Bezzio, C.; Festa, S.; Saibeni, S.; Papi, C. Chemoprevention of colorectal cancer in ulcerative colitis: Digging deep in current evidence. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Alkhayyat, M.; Abureesh, M.; Gill, A.; Khoudari, G.; Abou Saleh, M.; Mansoor, E.; Regueiro, M. Lower Rates of Colorectal Cancer in Patients with Inflammatory Bowel Disease Using Anti-TNF Therapy. Inflamm. Bowel Dis. 2021, 27, 1052–1060. [Google Scholar] [CrossRef]
- Flores, B.; O’Connor, A.; Moss, A. Impact of mucosal inflammation on risk of colorectal neoplasia in patients with ulcerative colitis: A systematic review and meta-analysis. Gastrointest. Endosc. 2017, 86, 1006–1011.e8. [Google Scholar] [CrossRef]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef]
- Beaugerie, L.; Svrcek, M.; Seksik, P.; Bouvier, A.; Simon, T.; Allez, M.; Brixi, H.; Gornet, J.; Altwegg, R.; Beau, P.; et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology 2013, 145, 166–175.e8. [Google Scholar] [CrossRef]
- Roda, G.; Narula, N.; Pinotti, R.; Skamnelos, A.; Katsanos, K.; Ungaro, R.; Burisch, J.; Torres, J.; Colombel, J. Systematic review with meta-analysis: Proximal disease extension in limited ulcerative colitis. Aliment. Pharmacol. Ther. 2017, 45, 1481–1492. [Google Scholar] [CrossRef]
- Selinger, C.; Andrews, J.; Titman, A.; Norton, I.; Jones, D.; McDonald, C.; Barr, G.; Selby, W.; Leong, R. Long-term follow-up reveals low incidence of colorectal cancer, but frequent need for resection, among Australian patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2014, 12, 644–650. [Google Scholar] [CrossRef]
- Choi, C.; Rutter, M.; Askari, A.; Lee, G.; Warusavitarne, J.; Moorghen, M.; Thomas-Gibson, S.; Saunders, B.; Graham, T.; Hart, A. Forty-Year Analysis of Colonoscopic Surveillance Program for Neoplasia in Ulcerative Colitis: An Updated Overview. Am. J. Gastroenterol. 2015, 110, 1022–1034. [Google Scholar] [CrossRef]
- Bopanna, S.; Ananthakrishnan, A.; Kedia, S.; Yajnik, V.; Ahuja, V. Risk of colorectal cancer in Asian patients with ulcerative colitis: A systematic review and meta-analysis. Lancet. Gastroenterol. Hepatol. 2017, 2, 269–276. [Google Scholar] [CrossRef]
- Lindström, L.; Boberg, K.; Wikman, O.; Friis-Liby, I.; Hultcrantz, R.; Prytz, H.; Sandberg-Gertzén, H.; Sangfelt, P.; Rydning, A.; Folvik, G.; et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Aliment. Pharmacol. Ther. 2012, 35, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Castagliuolo, I.; Castoro, C.; Pozza, A.; Scarpa, M.; Kotsafti, A.; Angriman, I. Inflammatory colonic carcinogenesis: A review on pathogenesis and immunosurveillance mechanisms in ulcerative colitis. World J. Gastroenterol. 2014, 20, 6774–6785. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Ten Hove, J.; Castaneda, D.; Palmela, C.; Mooiweer, E.; Colombel, J.; Harpaz, N.; Ullman, T.; van Bodegraven, A.; Jansen, J.; et al. High Risk of Advanced Colorectal Neoplasia in Patients with Primary Sclerosing Cholangitis Associated with Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 1106–1113.e3. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Cancer surveillance in longstanding ulcerative colitis: Endoscopic appearances help predict cancer risk. Gut 2004, 53, 1813–1816. [Google Scholar] [CrossRef]
- Velayos, F.; Loftus, E.; Jess, T.; Harmsen, W.; Bida, J.; Zinsmeister, A.; Tremaine, W.; Sandborn, W. Predictive and protective factors associated with colorectal cancer in ulcerative colitis: A case-control study. Gastroenterology 2006, 130, 1941–1949. [Google Scholar] [CrossRef]
- Mahmoud, R.; Shah, S.; Ten Hove, J.; Torres, J.; Mooiweer, E.; Castaneda, D.; Glass, J.; Elman, J.; Kumar, A.; Axelrad, J.; et al. No Association between Pseudopolyps and Colorectal Neoplasia in Patients with Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 1333–1344.e3. [Google Scholar] [CrossRef]
- de Jong, M.; Gillis, V.; Derikx, L.; Hoentjen, F. No Increased Risk of Colorectal Neoplasia in Patients with Inflammatory Bowel Disease and Postinflammatory Polyps. Inflamm. Bowel Dis. 2020, 26, 1383–1389. [Google Scholar] [CrossRef]
- Axelrad, J.; Faye, A.; Slaughter, J.; Harpaz, N.; Itzkowitz, S.; Shah, S. Colorectal Strictures in Patients with Inflammatory Bowel Disease Do not Independently Predict Colorectal Neoplasia. Inflamm. Bowel Dis. 2022, 28, 855–861. [Google Scholar] [CrossRef]
- Choi, J.; Kim, D.; Shin, S.; Park, E.; Kang, J. Effect of Ulcerative Colitis on Incidence of Colorectal Cancer: Results from the Nationwide Population-Based Cohort Study (2003–2013). J. Cancer 2016, 7, 681–686. [Google Scholar] [CrossRef][Green Version]
- Söderlund, S.; Granath, F.; Broström, O.; Karlén, P.; Löfberg, R.; Ekbom, A.; Askling, J. Inflammatory bowel disease confers a lower risk of colorectal cancer to females than to males. Gastroenterology 2010, 138, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Samadder, N.; Valentine, J.; Guthery, S.; Singh, H.; Bernstein, C.; Leighton, J.; Wan, Y.; Wong, J.; Boucher, K.; Pappas, L.; et al. Family History Associates with Increased Risk of Colorectal Cancer in Patients with Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2019, 17, 1807–1813.e1. [Google Scholar] [CrossRef] [PubMed]
- de Campos Silva, E.; Baima, J.; de Barros, J.; Tanni, S.; Schreck, T.; Saad-Hossne, R.; Sassaki, L. Risk factors for ulcerative colitis-associated colorectal cancer: A retrospective cohort study. Medicine 2020, 99, e21686. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Simonsen, J.; Jørgensen, K.; Pedersen, B.; Nielsen, N.; Frisch, M. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology 2012, 143, 375–381.e1. [Google Scholar] [CrossRef]
- Annese, V.; Beaugerie, L.; Egan, L.; Biancone, L.; Bolling, C.; Brandts, C.; Dierickx, D.; Dummer, R.; Fiorino, G.; Gornet, J.; et al. European Evidence-based Consensus: Inflammatory Bowel Disease and Malignancies. J. Crohn’s Colitis 2015, 9, 945–965. [Google Scholar] [CrossRef]
- Fumery, M.; Dulai, P.; Gupta, S.; Prokop, L.; Ramamoorthy, S.; Sandborn, W.; Singh, S. Incidence, Risk Factors, and Outcomes of Colorectal Cancer in Patients with Ulcerative Colitis with Low-Grade Dysplasia: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2017, 15, 665–674.e5. [Google Scholar] [CrossRef]
- Lamb, C.; Kennedy, N.; Raine, T.; Hendy, P.; Smith, P.; Limdi, J.; Hayee, B.; Lomer, M.; Parkes, G.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef]
- Gupta, R.; Harpaz, N.; Itzkowitz, S.; Hossain, S.; Matula, S.; Kornbluth, A.; Bodian, C.; Ullman, T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: A cohort study. Gastroenterology 2007, 133, 1099–1105. [Google Scholar] [CrossRef]
- Choi, C.; Al Bakir, I.; Ding, N.; Lee, G.; Askari, A.; Warusavitarne, J.; Moorghen, M.; Humphries, A.; Ignjatovic-Wilson, A.; Thomas-Gibson, S.; et al. Cumulative burden of inflammation predicts colorectal neoplasia risk in ulcerative colitis: A large single-centre study. Gut 2019, 68, 414–422. [Google Scholar] [CrossRef]
- Yvellez, O.; Rai, V.; Sossenheimer, P.; Hart, J.; Turner, J.; Weber, C.; El Jurdi, K.; Rubin, D. Cumulative Histologic Inflammation Predicts Colorectal Neoplasia in Ulcerative Colitis: A Validation Study. Inflamm. Bowel Dis. 2021, 27, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Gracio, D.; Teixeira, J.P.; Magro, F. Oxidative Stress and DNA Damage: Implications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.J.; Neurath, M.F. Mechanisms of Immune Signaling in Colitis-Associated Cancer. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Irrazabal, T.; Thakur, B.K.; Kang, M.; Malaise, Y.; Streutker, C.; Wong, E.O.Y.; Copeland, J.; Gryfe, R.; Guttman, D.S.; Navarre, W.W.; et al. Limiting oxidative DNA damage reduces microbe-induced colitis-associated colorectal cancer. Nat. Commun. 2020, 11, 1802. [Google Scholar] [CrossRef]
- Ullman, T.; Itzkowitz, S. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Hussain, S.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.; Shields, P.; Ham, A.; Swenberg, J.; Marrogi, A.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar]
- Luo, C.; Zhang, H. The Role of Proinflammatory Pathways in the Pathogenesis of Colitis-Associated Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 5126048. [Google Scholar] [CrossRef]
- Hirano, T.; Hirayama, D.; Wagatsuma, K.; Yamakawa, T.; Yokoyama, Y.; Nakase, H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 3062. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Suzuki, M.; Nagaishi, T.; Yamazaki, M.; Onizawa, M.; Watabe, T.; Sakamaki, Y.; Ichinose, S.; Totsuka, M.; Oshima, S.; Okamoto, R.; et al. Myosin light chain kinase expression induced via tumor necrosis factor receptor 2 signaling in the epithelial cells regulates the development of colitis-associated carcinogenesis. PLoS ONE 2014, 9, e88369. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Guo, S.; Ye, D.; Rawat, M.; Ma, T. TNF-α Modulation of Intestinal Tight Junction Permeability Is Mediated by NIK/IKK-α Axis Activation of the Canonical NF-κB Pathway. Am. J. Pathol. 2016, 186, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Harikumar, K.; Hait, N.; Allegood, J.; Strub, G.; Kim, E.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Yuza, K.; Nagahashi, M.; Shimada, Y.; Nakano, M.; Tajima, Y.; Kameyama, H.; Nakajima, M.; Takabe, K.; Wakai, T. Upregulation of phosphorylated sphingosine kinase 1 expression in colitis-associated cancer. J. Surg. Res. 2018, 231, 323–330. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Ye, D.; Dokladny, K.; Ma, T. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J. Immunol. 2008, 180, 5653–5661. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Ye, D.; Said, H.; Ma, T. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am. J. Pathol. 2010, 177, 2310–2322. [Google Scholar] [CrossRef]
- Yin, Q.; Pi, X.; Jiang, Y.; Ren, G.; Liu, Z.; Liu, H.; Wang, M.; Sun, W.; Li, S.; Gao, Z.; et al. An immuno-blocking agent targeting IL-1β and IL-17A reduces the lesion of DSS-induced ulcerative colitis in mice. Inflammation 2021, 44, 1724–1736. [Google Scholar] [CrossRef]
- Rafa, H.; Benkhelifa, S.; AitYounes, S.; Saoula, H.; Belhadef, S.; Belkhelfa, M.; Boukercha, A.; Toumi, R.; Soufli, I.; Morales, O.; et al. All-Trans Retinoic Acid Modulates TLR4/NF-kappaB Signaling Pathway Targeting TNF-alpha and Nitric Oxide Synthase 2 Expression in Colonic Mucosa during Ulcerative Colitis and Colitis Associated Cancer. Mediat. Inflamm. 2017, 2017, 7353252. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef]
- Fisher, R.; Bellamkonda, K.; Alex Molina, L.; Xiang, S.; Liska, D.; Sarvestani, S.; Chakrabarti, S.; Berg, A.; Jorgensen, M.; Hatala, D.; et al. Disrupting Inflammation-Associated CXCL8-CXCR1 Signaling Inhibits Tumorigenicity Initiated by Sporadic- and Colitis-Colon Cancer Stem Cells. Neoplasia 2019, 21, 269–281. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Saadatdoust, Z.; Pandurangan, A.; Ananda Sadagopan, S.; Mohd Esa, N.; Ismail, A.; Mustafa, M. Dietary cocoa inhibits colitis associated cancer: A crucial involvement of the IL-6/STAT3 pathway. J. Nutr. Biochem. 2015, 26, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.; von Burstin, V.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Matsumoto, S.; Hara, T.; Mitsuyama, K.; Yamamoto, M.; Tsuruta, O.; Sata, M.; Scheller, J.; Rose-John, S.; Kado, S.; Takada, T. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J. Immunol. 2010, 184, 1543–1551. [Google Scholar] [CrossRef]
- Schreiber, S.; Aden, K.; Bernardes, J.; Conrad, C.; Tran, F.; Höper, H.; Volk, V.; Mishra, N.; Blase, J.; Nikolaus, S.; et al. Therapeutic Interleukin-6 Trans-signaling Inhibition by Olamkicept (sgp130Fc) in Patients with Active Inflammatory Bowel Disease. Gastroenterology 2021, 160, 2354–2366.e11. [Google Scholar] [CrossRef]
- Waldner, M.J.; Wirtz, S.; Jefremow, A.; Warntjen, M.; Neufert, C.; Atreya, R.; Becker, C.; Weigmann, B.; Vieth, M.; Rose-John, S.; et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J. Exp. Med. 2010, 207, 2855–2868. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.; Harikumar, K.; Yamada, A.; Huang, W.; Hait, N.; Allegood, J.; Price, M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Bao, Y.; Guo, Y.; Zhang, C.; Fan, F.; Yang, W. Sphingosine Kinase 1 and Sphingosine-1-Phosphate Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 2109. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Wang, J.; Goren, I.; Yang, B.; Lin, S.; Li, J.; Elias, M.; Fiocchi, C.; Rieder, F. Review article: The sphingosine 1 phosphate/sphingosine 1 phosphate receptor axis—A unique therapeutic target in inflammatory bowel disease. Aliment. Pharm. 2022, 55, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity 2018, 48, 45–58.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, X.; Ma, Y.; Hua, S. IL-23 and dendritic cells: What are the roles of their mutual attachment in immune response and immunotherapy? Cytokine 2019, 120, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Zepp, J.A.; Zhao, J.; Liu, C.; Bulek, K.; Wu, L.; Chen, X.; Hao, Y.; Wang, Z.; Wang, X.; Ouyang, W.; et al. IL-17A-Induced PLET1 Expression Contributes to Tissue Repair and Colon Tumorigenesis. J. Immunol. 2017, 199, 3849–3857. [Google Scholar] [CrossRef]
- Jiang, R.; Wang, H.; Deng, L.; Hou, J.; Shi, R.; Yao, M.; Gao, Y.; Yao, A.; Wang, X.; Yu, L.; et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 2013, 13, 59. [Google Scholar] [CrossRef]
- Yu, L.; Wang, H.; Yang, S.; Yuan, Z.; Xu, F.; Sun, C.; Shi, R. Expression of interleukin-22/STAT3 signaling pathway in ulcerative colitis and related carcinogenesis. World J. Gastroenterol. 2013, 19, 2638–2649. [Google Scholar] [CrossRef]
- Dumoutier, L.; de Meester, C.; Tavernier, J.; Renauld, J. New activation modus of STAT3: A tyrosine-less region of the interleukin-22 receptor recruits STAT3 by interacting with its coiled-coil domain. J. Biol. Chem. 2009, 284, 26377–26384. [Google Scholar] [CrossRef]
- Hyun, Y.; Han, D.; Lee, A.; Eun, C.; Youn, J.; Kim, H. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef]
- Qi, H.; Yang, H.; Xu, G.; Ren, J.; Hua, W.; Shi, Y.; Torsvik, M.; Florholmen, J.; Cui, G. Therapeutic efficacy of IL-17A antibody injection in preventing the development of colitis associated carcinogenesis in mice. Immunobiology 2015, 220, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Zenewicz, L.; Huber, F.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Gao, X.; Lin, Y.; Yue, C.; Wang, Y.; Quan, F.; Zhang, Z.; Liu, X.; Lu, Y.; Zhan, Y.; et al. Thymopentin ameliorates dextran sulfate sodium-induced colitis by triggering the production of IL-22 in both innate and adaptive lymphocytes. Theranostics 2019, 9, 7490–7505. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Zhang, X.; Liu, J.; Fu, Z.; Shi, X.; Li, Q.; Liu, J.; Zhu, Q. Elevated expression of interleukin-21 and its correlation to T-cell subpopulation in patients with ulcerative colitis. Cent. -Eur. J. Immunol. 2015, 40, 331–336. [Google Scholar] [CrossRef]
- Araki, A.; Jin, L.; Nara, H.; Takeda, Y.; Nemoto, N.; Gazi, M.; Asao, H. IL-21 Enhances the Development of Colitis-Associated Colon Cancer: Possible Involvement of Activation-Induced Cytidine Deaminase Expression. J. Immunol. 2019, 202, 3326–3333. [Google Scholar] [CrossRef]
- Chandramouli, A.; Onyeagucha, B.; Mercado-Pimentel, M.; Stankova, L.; Shahin, N.; LaFleur, B.; Heimark, R.; Bhattacharyya, A.; Nelson, M. MicroRNA-101 (miR-101) post-transcriptionally regulates the expression of EP4 receptor in colon cancers. Cancer Biol. Ther. 2012, 13, 175–183. [Google Scholar] [CrossRef]
- Sheibanie, A.; Yen, J.; Khayrullina, T.; Emig, F.; Zhang, M.; Tuma, R.; Ganea, D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J. Immunol. 2007, 178, 8138–8147. [Google Scholar] [CrossRef]
- Niho, N.; Mutoh, M.; Kitamura, T.; Takahashi, M.; Sato, H.; Yamamoto, H.; Maruyama, T.; Ohuchida, S.; Sugimura, T.; Wakabayashi, K. Suppression of azoxymethane-induced colon cancer development in rats by a prostaglandin E receptor EP1-selective antagonist. Cancer Sci. 2005, 96, 260–264. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Shi, Q.; Katkuri, S.; Walhi, W.; Desvergne, B.; Das, S.; Dey, S.; DuBois, R. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell 2004, 6, 285–295. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Ning, W.; Guo, L.; Sun, X.; Dey, S.; Chaturvedi, R.; Wilson, K.; DuBois, R. Peroxisome proliferator-activated receptor δ promotes colonic inflammation and tumor growth. Proc. Natl. Acad. Sci. USA 2014, 111, 7084–7089. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Santos, L.; Dias, R.; Quadros, C.; Bezerra, D. Emerging agents that target signaling pathways to eradicate colorectal cancer stem cells. Cancer Commun. 2021, 41, 1275–1313. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Peng, L.; Li, Z.; Tan, G.; Liang, E.; Chen, S.; Zhao, X.; Zhi, F. YAP triggers the Wnt/beta-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death Dis. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ding, Y.; Xu, C.; Hao, M.; Li, H.; Ding, L. Cldn-7 deficiency promotes experimental colitis and associated carcinogenesis by regulating intestinal epithelial integrity. Oncoimmunology 2021, 10, 1923910. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kawai, T.; Akira, S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016246. [Google Scholar] [CrossRef]
- Peri, F.; Piazza, M.; Calabrese, V.; Damore, G.; Cighetti, R. Exploring the LPS/TLR4 signal pathway with small molecules. Biochem. Soc. Trans. 2010, 38, 1390–1395. [Google Scholar] [CrossRef]
- Song, X.; Gao, H.; Lin, Y.; Yao, Y.; Zhu, S.; Wang, J.; Liu, Y.; Yao, X.; Meng, G.; Shen, N.; et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 2014, 40, 140–152. [Google Scholar] [CrossRef]
- Pastille, E.; Faßnacht, T.; Adamczyk, A.; Ngo Thi Phuong, N.; Buer, J.; Westendorf, A. Inhibition of TLR4 Signaling Impedes Tumor Growth in Colitis-Associated Colon Cancer. Front. Immunol. 2021, 12, 669747. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007, 317, 124–127. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Iyadorai, T.; Mariappan, V.; Vellasamy, K.M.; Wanyiri, J.W.; Roslani, A.C.; Lee, G.K.; Sears, C.; Vadivelu, J. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS ONE 2020, 15, e0228217. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Dechelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Zhu, W.; Miyata, N.; Winter, M.G.; Arenales, A.; Hughes, E.R.; Spiga, L.; Kim, J.; Sifuentes-Dominguez, L.; Starokadomskyy, P.; Gopal, P.; et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J. Exp. Med. 2019, 216, 2378–2393. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Aldeguer, X.; Sabat-Mir, M.; Serra-Pages, M.; Duncan, S.H.; Flint, H.J.; Garcia-Gil, L.J.; Martinez-Medina, M. Evaluation of bacterial biomarkers to aid in challenging inflammatory bowel diseases diagnostics and subtype classification. World J. Gastrointest. Pathophysiol. 2020, 11, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hong, X.L.; Sun, T.T.; Huang, X.W.; Wang, J.L.; Xiong, H. Fusobacterium nucleatum exacerbates colitis by damaging epithelial barriers and inducing aberrant inflammation. J. Dig. Dis. 2020, 21, 385–398. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Kostic, A.; Chun, E.; Robertson, L.; Glickman, J.; Gallini, C.; Michaud, M.; Clancy, T.; Chung, D.; Lochhead, P.; Hold, G.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Ye, X.; Wang, R.; Bhattacharya, R.; Boulbes, D.; Fan, F.; Xia, L.; Adoni, H.; Ajami, N.; Wong, M.; Smith, D.; et al. Fusobacterium Nucleatum Subspecies Influences Proinflammatory Cytokine Expression and Monocyte Activation in Human Colorectal Tumors. Cancer Prev. Res. 2017, 10, 398–409. [Google Scholar] [CrossRef]
- Brennan, C.; Clay, S.; Lavoie, S.; Bae, S.; Lang, J.; Fonseca-Pereira, D.; Rosinski, K.; Ou, N.; Glickman, J.; Garrett, W. Fusobacterium nucleatum drives a pro-inflammatory intestinal microenvironment through metabolite receptor-dependent modulation of IL-17 expression. Gut Microbes 2021, 13, 1987780. [Google Scholar] [CrossRef]
- Yu, M.; Kim, H.; Park, H. Fusobacterium nucleatum Accelerates the Progression of Colitis-Associated Colorectal Cancer by Promoting EMT. Cancers 2020, 12, 2728. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.; Destefano Shields, C.; Wu, S.; Huso, D.; Wu, X.; Murray-Stewart, T.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.; Sears, C.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Thiele Orberg, E.; Geis, A.; Chan, J.; Fu, K.; DeStefano Shields, C.; Dejea, C.; Fathi, P.; Chen, J.; Finard, B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, Z.; Yan, Y.; Ji, L.; He, J.; Xuan, B.; Shen, C.; Ma, Y.; Jiang, S.; Ma, D.; et al. Enterotoxigenic Bacteroidesfragilis Promotes Intestinal Inflammation and Malignancy by Inhibiting Exosome-Packaged miR-149-3p. Gastroenterology 2021, 161, 1552–1566.e12. [Google Scholar] [CrossRef]
- Kostic, A.; Xavier, R.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Richard, M.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.; Pierluigi Di Simone, M.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2018, 9, 131–142. [Google Scholar] [CrossRef]
- Oh, N.; Lee, J.; Kim, Y.; Kim, S.; Lee, J. Lactobacillus gasseriCancer-protective effect of a synbiotic combination between 505 and a leaf extract on colitis-associated colorectal cancer. Gut Microbes 2020, 12, 1785803. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, K.; Wu, W.; Lv, L.; Bian, X.; Yang, L.; Wang, Q.; Li, Y.; Ye, J.; Fang, D.; et al. Administration of Bifidobacterium bifidum CGMCC 15068 modulates gut microbiota and metabolome in azoxymethane (AOM)/dextran sulphate sodium (DSS)-induced colitis-associated colon cancer (CAC) in mice. Appl. Microbiol. Biotechnol. 2020, 104, 5915–5928. [Google Scholar] [CrossRef]
- Bye, W.; Ma, C.; Nguyen, T.; Parker, C.; Jairath, V.; East, J. Strategies for Detecting Colorectal Cancer in Patients with Inflammatory Bowel Disease: A Cochrane Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2018, 113, 1801–1809. [Google Scholar] [CrossRef]
- Farraye, F.; Odze, R.; Eaden, J.; Itzkowitz, S. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology 2010, 138, 746–774.e4. [Google Scholar] [CrossRef]
- Mowat, C.; Cole, A.; Windsor, A.; Ahmad, T.; Arnott, I.; Driscoll, R.; Mitton, S.; Orchard, T.; Rutter, M.; Younge, L.; et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011, 60, 571–607. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.; Shah, S. Diagnosis and management of inflammatory bowel disease-associated neoplasia: Considerations in the modern era. Ther. Adv. Gastroenterol. 2020, 13, 1756284820920779. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Dickman, P.; Karlén, P.; Broström, O.; Lapidus, A.; Löfberg, R.; Ekbom, A. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology 2001, 120, 1356–1362. [Google Scholar] [CrossRef]
- Krugliak Cleveland, N.; Rubin, D.; Hart, J.; Weber, C.; Meckel, K.; Tran, A.; Aelvoet, A.; Pan, I.; Gonsalves, A.; Gaetano, J.; et al. Patients with Ulcerative Colitis and Primary Sclerosing Cholangitis Frequently Have Subclinical Inflammation in the Proximal Colon. Clin. Gastroenterol. Hepatol. 2018, 16, 68–74. [Google Scholar] [CrossRef]
- Sørensen, J.; Nielsen, O.; Andersson, M.; Ainsworth, M.; Ytting, H.; Bélard, E.; Jess, T. Inflammatory bowel disease with primary sclerosing cholangitis: A Danish population-based cohort study 1977–2011. Liver Int. 2018, 38, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Ballester, M.P.; Mesonero, F.; Florez-Diez, P.; Gomez, C.; Fuentes-Valenzuela, E.; Martin, N.; Senosiain, C.; Vela, M.; Fernandez-Clotet, A.; Perez, P.; et al. Adherence to endoscopic surveillance for advanced lesions and colorectal cancer in inflammatory bowel disease: An AEG and GETECCU collaborative cohort study. Aliment. Pharm. 2022, 55, 1402–1413. [Google Scholar] [CrossRef]
- Muthukrishnan, M.; Arnold, L.; James, A. Patients’ self-reported barriers to colon cancer screening in federally qualified health center settings. Prev. Med. Rep. 2019, 15, 100896. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, J.; Shah, S.; Shaffer, S.; Bernstein, C.; Castaneda, D.; Palmela, C.; Mooiweer, E.; Elman, J.; Kumar, A.; Glass, J.; et al. Consecutive negative findings on colonoscopy during surveillance predict a low risk of advanced neoplasia in patients with inflammatory bowel disease with long-standing colitis: Results of a 15-year multicentre, multinational cohort study. Gut 2019, 68, 615–622. [Google Scholar] [CrossRef]
- Laine, L.; Kaltenbach, T.; Barkun, A.; McQuaid, K.R.; Subramanian, V.; Soetikno, R.; Panel, S.G.D. SCENIC international consensus statement on surveillance and management of dysplasia in inflammatory bowel disease. Gastroenterology 2015, 148, 639–651.e28. [Google Scholar] [CrossRef]
- Subramanian, V.; Ramappa, V.; Telakis, E.; Mannath, J.; Jawhari, A.U.; Hawkey, C.J.; Ragunath, K. Comparison of high definition with standard white light endoscopy for detection of dysplastic lesions during surveillance colonoscopy in patients with colonic inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 350–355. [Google Scholar] [CrossRef]
- Marion, J.F.; Waye, J.D.; Israel, Y.; Present, D.H.; Suprun, M.; Bodian, C.; Harpaz, N.; Chapman, M.; Itzkowitz, S.; Abreu, M.T.; et al. Chromoendoscopy is More Effective Than Standard Colonoscopy in Detecting Dysplasia during Long-term Surveillance of Patients with Colitis. Clin. Gastroenterol. Hepatol. 2016, 14, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, J.; Rakowsky, S.; Sattler, L.; Yadav, A.; Foromera, J.; Grossberg, L.; Cheifetz, A. Meta-analysis of dye-based chromoendoscopy compared with standard- and high-definition white-light endoscopy in patients with inflammatory bowel disease at increased risk of colon cancer. Gastrointest. Endosc. 2019, 90, 186–195.e1. [Google Scholar] [CrossRef] [PubMed]
- Alexandersson, B.; Hamad, Y.; Andreasson, A.; Rubio, C.A.; Ando, Y.; Tanaka, K.; Ichiya, T.; Rezaie, R.; Schmidt, P.T. High-Definition Chromoendoscopy Superior to High-Definition White-Light Endoscopy in Surveillance of Inflammatory Bowel Diseases in a Randomized Trial. Clin. Gastroenterol. Hepatol. 2020, 18, 2101–2107. [Google Scholar] [CrossRef]
- Yang, D.H.; Park, S.J.; Kim, H.S.; Park, Y.S.; Park, D.I.; Lee, K.M.; Jung, S.A.; Choi, C.H.; Koo, J.S.; Cheon, J.H.; et al. High-Definition Chromoendoscopy Versus High-Definition White Light Colonoscopy for Neoplasia Surveillance in Ulcerative Colitis: A Randomized Controlled Trial. Am. J. Gastroenterol. 2019, 114, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- El-Dallal, M.; Chen, Y.; Lin, Q.; Rakowsky, S.; Sattler, L.; Foromera, J.; Grossberg, L.; Cheifetz, A.; Feuerstein, J. Meta-analysis of Virtual-based Chromoendoscopy Compared with Dye-spraying Chromoendoscopy Standard and High-definition White Light Endoscopy in Patients with Inflammatory Bowel Disease at Increased Risk of Colon Cancer. Inflamm. Bowel Dis. 2020, 26, 1319–1329. [Google Scholar] [CrossRef]
- Bisschops, R.; Bessissow, T.; Joseph, J.; Baert, F.; Ferrante, M.; Ballet, V.; Willekens, H.; Demedts, I.; Geboes, K.; De Hertogh, G.; et al. Chromoendoscopy versus narrow band imaging in UC: A prospective randomised controlled trial. Gut 2018, 67, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Iacucci, M.; Kaplan, G.G.; Panaccione, R.; Akinola, O.; Lethebe, B.C.; Lowerison, M.; Leung, Y.; Novak, K.L.; Seow, C.H.; Urbanski, S.; et al. A Randomized Trial Comparing High Definition Colonoscopy Alone with High Definition Dye Spraying and Electronic Virtual Chromoendoscopy for Detection of Colonic Neoplastic Lesions during IBD Surveillance Colonoscopy. Am. J. Gastroenterol. 2018, 113, 225–234. [Google Scholar] [CrossRef]
- Gulati, S.; Dubois, P.; Carter, B.; Cornelius, V.; Martyn, M.; Emmanuel, A.; Haji, A.; Hayee, B. A Randomized Crossover Trial of Conventional vs. Virtual Chromoendoscopy for Colitis Surveillance: Dysplasia Detection, Feasibility, and Patient Acceptability (CONVINCE). Inflamm. Bowel Dis. 2019, 25, 1096–1106. [Google Scholar] [CrossRef]
- Vleugels, J.L.A.; Rutter, M.D.; Ragunath, K.; Rees, C.J.; Ponsioen, C.Y.; Lahiff, C.; Ket, S.N.; Wanders, L.K.; Samuel, S.; Butt, F.; et al. Chromoendoscopy versus autofluorescence imaging for neoplasia detection in patients with longstanding ulcerative colitis (FIND-UC): An international, multicentre, randomised controlled trial. Lancet Gastroenterol. Hepatol. 2018, 3, 305–316. [Google Scholar] [CrossRef]
- Vleugels, J.L.A.; Rutter, M.D.; Ragunath, K.; Rees, C.J.; Ponsioen, C.Y.; Lahiff, C.; Ket, S.N.; Wanders, L.K.; Samuel, S.; Butt, F.; et al. Diagnostic Accuracy of Endoscopic Trimodal Imaging and Chromoendoscopy for Lesion Characterization in Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 1438–1447. [Google Scholar] [CrossRef]
- Bojarski, C.; Waldner, M.; Rath, T.; Schürmann, S.; Neurath, M.; Atreya, R.; Siegmund, B. Innovative Diagnostic Endoscopy in Inflammatory Bowel Diseases: From High-Definition to Molecular Endoscopy. Front. Med. 2021, 8, 655404. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, R.; Goetz, M.; Lammersdorf, K.; Schneider, C.; Burg, J.; Stolte, M.; Vieth, M.; Nafe, B.; Galle, P.; Neurath, M. Chromoscopy-guided endomicroscopy increases the diagnostic yield of intraepithelial neoplasia in ulcerative colitis. Gastroenterology 2007, 132, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Hurlstone, D.; Kiesslich, R.; Thomson, M.; Atkinson, R.; Cross, S. Confocal chromoscopic endomicroscopy is superior to chromoscopy alone for the detection and characterisation of intraepithelial neoplasia in chronic ulcerative colitis. Gut 2008, 57, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Lord, R.; Burr, N.; Mohammed, N.; Subramanian, V. Colonic lesion characterization in inflammatory bowel disease: A systematic review and meta-analysis. World J. Gastroenterol. 2018, 24, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Hundorfean, G.; Pereira, S.; Karstensen, J.; Vilmann, P.; Saftoiu, A. Modern Endoscopic Imaging in Diagnosis and Surveillance of Inflammatory Bowel Disease Patients. Gastroenterol. Res. Pract. 2018, 2018, 5738068. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.; Malek, N.; Kiesslich, R. Microscopic imaging in endoscopy: Endomicroscopy and endocytoscopy. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 11–18. [Google Scholar] [CrossRef]
- Kudo, S.E.; Wakamura, K.; Ikehara, N.; Mori, Y.; Inoue, H.; Hamatani, S. Diagnosis of colorectal lesions with a novel endocytoscopic classification—A pilot study. Endoscopy 2011, 43, 869–875. [Google Scholar] [CrossRef]
- Kudo, S.E.; Maeda, Y.; Ogata, N.; Misawa, M.; Ogawa, Y.; Takishima, K.; Ishiyama, M.; Mochizuki, K.; Minegishi, Y.; Ogura, Y.; et al. Combined endocytoscopy with pit pattern diagnosis in ulcerative colitis-associated neoplasia: Pilot study. Dig. Endosc. 2022, 34, 133–143. [Google Scholar] [CrossRef]
- Leong, R.W.; Ooi, M.; Corte, C.; Yau, Y.; Kermeen, M.; Katelaris, P.H.; McDonald, C.; Ngu, M. Full-Spectrum Endoscopy Improves Surveillance for Dysplasia in Patients with Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 1337–1344.e3. [Google Scholar] [CrossRef]
- Watanabe, T.; Ajioka, Y.; Mitsuyama, K.; Watanabe, K.; Hanai, H.; Nakase, H.; Kunisaki, R.; Matsuda, K.; Iwakiri, R.; Hida, N.; et al. Comparison of Targeted vs. Random Biopsies for Surveillance of Ulcerative Colitis-Associated Colorectal Cancer. Gastroenterology 2016, 151, 1122–1130. [Google Scholar] [CrossRef]
- Gasia, M.; Ghosh, S.; Panaccione, R.; Ferraz, J.; Kaplan, G.; Leung, Y.; Novak, K.; Seow, C.; Iacucci, M. Targeted Biopsies Identify Larger Proportions of Patients with Colonic Neoplasia Undergoing High-Definition Colonoscopy, Dye Chromoendoscopy, or Electronic Virtual Chromoendoscopy. Clin. Gastroenterol. Hepatol. 2016, 14, 704–712.e4. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Kochhar, G.; Venkatesh, P.; Bennett, A.; Rizk, M.; Shen, B.; Kiran, R. Random biopsies during surveillance colonoscopy increase dysplasia detection in patients with primary sclerosing cholangitis and ulcerative colitis. J. Crohn’s Colitis 2013, 7, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Moussata, D.; Allez, M.; Cazals-Hatem, D.; Treton, X.; Laharie, D.; Reimund, J.M.; Bertheau, P.; Bourreille, A.; Lavergne-Slove, A.; Brixi, H.; et al. Are random biopsies still useful for the detection of neoplasia in patients with IBD undergoing surveillance colonoscopy with chromoendoscopy? Gut 2018, 67, 616–624. [Google Scholar] [CrossRef]
- Stolfi, C.; De Simone, V.; Pallone, F.; Monteleone, G. Mechanisms of action of non-steroidal anti-inflammatory drugs (NSAIDs) and mesalazine in the chemoprevention of colorectal cancer. Int. J. Mol. Sci. 2013, 14, 17972–17985. [Google Scholar] [CrossRef] [PubMed]
- Le Berre, C.; Roda, G.; Nedeljkovic Protic, M.; Danese, S.; Peyrin-Biroulet, L. Modern use of 5-aminosalicylic acid compounds for ulcerative colitis. Expert Opin. Biol. Ther. 2020, 20, 363–378. [Google Scholar] [CrossRef]
- Carrat, F.; Seksik, P.; Colombel, J.F.; Peyrin-Biroulet, L.; Beaugerie, L.; Group, C.S. The effects of aminosalicylates or thiopurines on the risk of colorectal cancer in inflammatory bowel disease. Aliment. Pharm. 2017, 45, 533–541. [Google Scholar] [CrossRef]
- Nieminen, U.; Jussila, A.; Nordling, S.; Mustonen, H.; Färkkilä, M. Inflammation and disease duration have a cumulative effect on the risk of dysplasia and carcinoma in IBD: A case-control observational study based on registry data. Int. J. Cancer 2014, 134, 189–196. [Google Scholar] [CrossRef]
- van Staa, T.; Card, T.; Logan, R.; Leufkens, H. 5-Aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: A large epidemiological study. Gut 2005, 54, 1573–1578. [Google Scholar] [CrossRef]
- Soon, A.Y.; Neo, S.; Thia, K.T.; Li, H.; Ling, K.-L.; Kong, S.C.; Ooi, C.-J. Risk of Colorectal Cancer and Dysplasia in Asian Ulcerative Colitis Patients. Gastroenterology 2011, 140, S430–S431. [Google Scholar] [CrossRef]
- DOP079. Predictors of first neoplastic colonic lesion in patients with inflammatory bowel disease undergoing colonoscopic surveillance. J. Crohn’s Colitis 2016, 10, S80. [CrossRef][Green Version]
- Bonovas, S.; Fiorino, G.; Lytras, T.; Nikolopoulos, G.; Peyrin-Biroulet, L.; Danese, S. Systematic review with meta-analysis: Use of 5-aminosalicylates and risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharm. 2017, 45, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.; So, J.; Tang, W.; Yip, T.; Leung, W.; Li, M.; Lo, F.; Ng, K.; Sze, S.; Leung, C.; et al. Cancer risk and chemoprevention in Chinese inflammatory bowel disease patients: A population-based cohort study. Scand. J. Gastroenterol. 2020, 55, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E. A practical perspective on ulcerative colitis: Patients’ needs from aminosalicylate therapies. Inflamm. Bowel Dis. 2006, 12, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Lenti, M.; Selinger, C. Medication non-adherence in adult patients affected by inflammatory bowel disease: A critical review and update of the determining factors, consequences and possible interventions. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 215–226. [Google Scholar] [CrossRef]
- Fraser, A.; Orchard, T.; Robinson, E.; Jewell, D. Long-term risk of malignancy after treatment of inflammatory bowel disease with azathioprine. Aliment. Pharmacol. Ther. 2002, 16, 1225–1232. [Google Scholar] [CrossRef]
- Matula, S.; Croog, V.; Itzkowitz, S.; Harpaz, N.; Bodian, C.; Hossain, S.; Ullman, T. Chemoprevention of colorectal neoplasia in ulcerative colitis: The effect of 6-mercaptopurine. Clin. Gastroenterol. Hepatol. 2005, 3, 1015–1021. [Google Scholar] [CrossRef]
- Gordillo, J.; Cabré, E.; Garcia-Planella, E.; Ricart, E.; Ber-Nieto, Y.; Márquez, L.; Rodríguez-Moranta, F.; Ponferrada, Á.; Vera, I.; Gisbert, J.; et al. Thiopurine Therapy Reduces the Incidence of Colorectal Neoplasia in Patients with Ulcerative Colitis. Data from the ENEIDA Registry. J. Crohn’s Colitis 2015, 9, 1063–1070. [Google Scholar] [CrossRef]
- Lu, M.; Qiu, X.; Mao, X.; Li, X.; Zhang, H. Systematic review with meta-analysis: Thiopurines decrease the risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2018, 47, 318–331. [Google Scholar] [CrossRef]
- Pasternak, B.; Svanstrom, H.; Schmiegelow, K.; Jess, T.; Hviid, A. Use of azathioprine and the risk of cancer in inflammatory bowel disease. Am. J. Epidemiol. 2013, 177, 1296–1305. [Google Scholar] [CrossRef]
- Nguyen, T.; Vacek, P.; O’Neill, P.; Colletti, R.; Finette, B. Mutagenicity and potential carcinogenicity of thiopurine treatment in patients with inflammatory bowel disease. Cancer Res. 2009, 69, 7004–7012. [Google Scholar] [CrossRef]
- Smith, M.; Irving, P.; Marinaki, A.; Sanderson, J. Review article: Malignancy on thiopurine treatment with special reference to inflammatory bowel disease. Aliment. Pharmacol. Ther. 2010, 32, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khanna, S.; Pardi, D.; Loftus, E.; Talwalkar, J. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: A systematic review and meta-analysis. Inflamm. Bowel Dis. 2013, 19, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Pardi, D.S.; Loftus, E.V., Jr.; Kremers, W.K.; Keach, J.; Lindor, K.D. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003, 124, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Chen, Z.; Xie, L.; Wang, W. Clinical effects of ursodeoxycholic acid on patients with ulcerative colitis may improve via the regulation of IL-23-IL-17 axis and the changes of the proportion of intestinal microflora. Saudi J. Gastroenterol. 2021, 27, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Cerda, S.; Wali, R.; von Lintig, F.; Tretiakova, M.; Joseph, L.; Stoiber, D.; Cohen, G.; Nimmagadda, K.; Hart, J.; et al. Ursodeoxycholic acid inhibits Ras mutations, wild-type Ras activation, and cyclooxygenase-2 expression in colon cancer. Cancer Res. 2003, 63, 3517–3523. [Google Scholar] [CrossRef]
- Raine, T.; Bonovas, S.; Burisch, J.; Kucharzik, T.; Adamina, M.; Annese, V.; Bachmann, O.; Bettenworth, D.; Chaparro, M.; Czuber-Dochan, W.; et al. ECCO Guidelines on Therapeutics in Ulcerative Colitis: Medical Treatment. J. Crohn’s Colitis 2022, 16, 2–17. [Google Scholar] [CrossRef]
- Onizawa, M.; Nagaishi, T.; Kanai, T.; Nagano, K.; Oshima, S.; Nemoto, Y.; Yoshioka, A.; Totsuka, T.; Okamoto, R.; Nakamura, T.; et al. Signaling pathway via TNF-alpha/NF-kappaB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am. J. Physiol. Gastrointest Liver Physiol. 2009, 296, G850–G859. [Google Scholar] [CrossRef]
- Baars, J.E.; Looman, C.W.; Steyerberg, E.W.; Beukers, R.; Tan, A.C.; Weusten, B.L.; Kuipers, E.J.; van der Woude, C.J. The risk of inflammatory bowel disease-related colorectal carcinoma is limited: Results from a nationwide nested case-control study. Am. J. Gastroenterol. 2011, 106, 319–328. [Google Scholar] [CrossRef]
- Charkaoui, M.; Hajage, D.; Tubach, F.; Beaugerie, L.; Kirchgesner, J. Impact of Anti-tumour Necrosis Factor Agents on the Risk of Colorectal Cancer in Patients with Ulcerative Colitis: Nationwide French Cohort Study. J. Crohn’s Colitis 2022, 16, 893–899. [Google Scholar] [CrossRef]
- Lima, C.; Queiroz, N.; Sobrado, C.; Silva, G.; Nahas, S. Critical Analysis of Anti-Tnf Use in the Era of New Biological Agents in Inflammatory Bowel Disease. Arq. Gastroenterol. 2020, 57, 323–332. [Google Scholar] [CrossRef]
- Sands, B.; Sandborn, W.; Panaccione, R.; O’Brien, C.; Zhang, H.; Johanns, J.; Adedokun, O.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.; Baert, F.; Danese, S.; Krznarić, Ž.; Kobayashi, T.; Yao, X.; Chen, J.; Rosario, M.; Bhatia, S.; Kisfalvi, K.; et al. Efficacy and Safety of Vedolizumab Subcutaneous Formulation in a Randomized Trial of Patients with Ulcerative Colitis. Gastroenterology 2020, 158, 562–572.e12. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Quirk, D.; Wang, W.; Nduaka, C.I.; Mukherjee, A.; Su, C.; Sands, B.E. Efficacy and Safety of Extended Induction with Tofacitinib for the Treatment of Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2022, 20, 1821–1830.e1823. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhu, L.; Guo, Z.; Li, Y.; Zhu, W.; Li, N.; Li, J. Use of thiopurines and risk of colorectal neoplasia in patients with inflammatory bowel diseases: A meta-analysis. PLoS ONE 2013, 8, e81487. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rybicki, L.; Lashner, B. The impact of ursodeoxycholic acid on cancer, dysplasia and mortality in ulcerative colitis patients with primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2005, 22, 783–788. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zhao, T.; Wu, D.; Li, J.; Wang, M.; Sun, Y.; Hou, S. Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention. Curr. Oncol. 2022, 29, 6091-6114. https://doi.org/10.3390/curroncol29090479
Li W, Zhao T, Wu D, Li J, Wang M, Sun Y, Hou S. Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention. Current Oncology. 2022; 29(9):6091-6114. https://doi.org/10.3390/curroncol29090479
Chicago/Turabian StyleLi, Wenqian, Tiantian Zhao, Dacheng Wu, Jiajia Li, Mei Wang, Yunyun Sun, and Sicong Hou. 2022. "Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention" Current Oncology 29, no. 9: 6091-6114. https://doi.org/10.3390/curroncol29090479
APA StyleLi, W., Zhao, T., Wu, D., Li, J., Wang, M., Sun, Y., & Hou, S. (2022). Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention. Current Oncology, 29(9), 6091-6114. https://doi.org/10.3390/curroncol29090479