Morphogenetic and Imaging Characteristics in Giant Cell Glioblastoma

 ,
,  ,
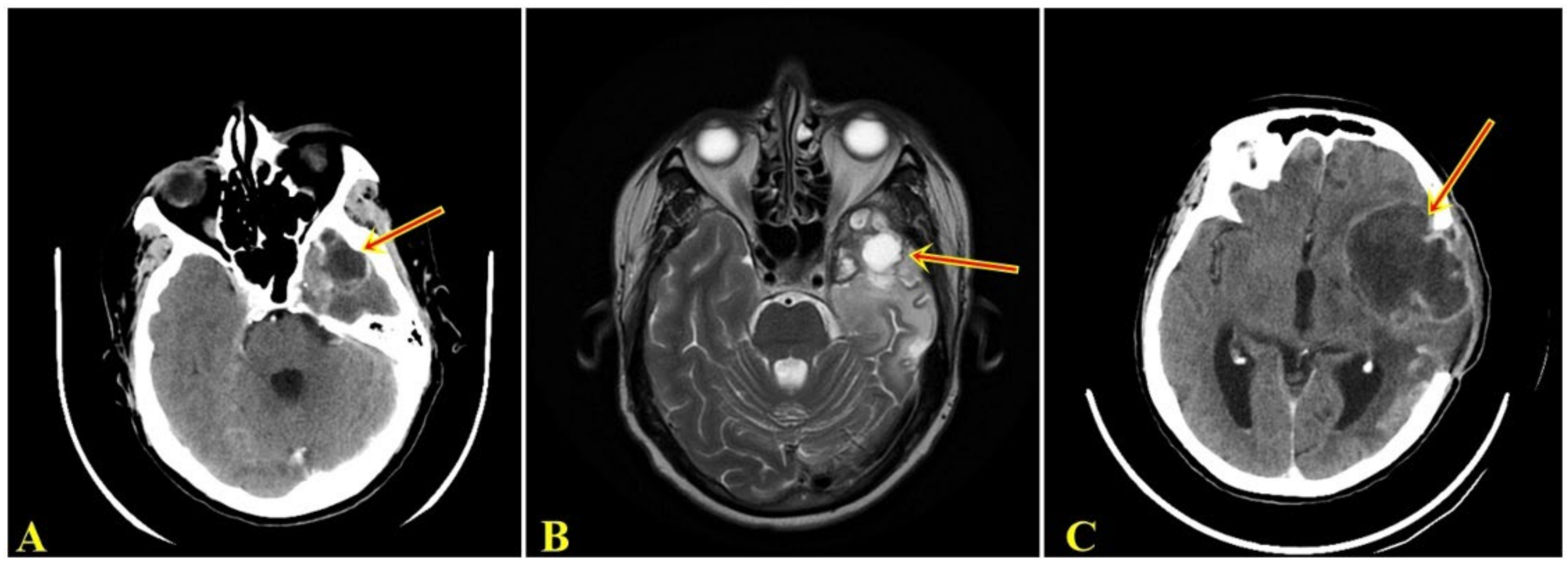
, 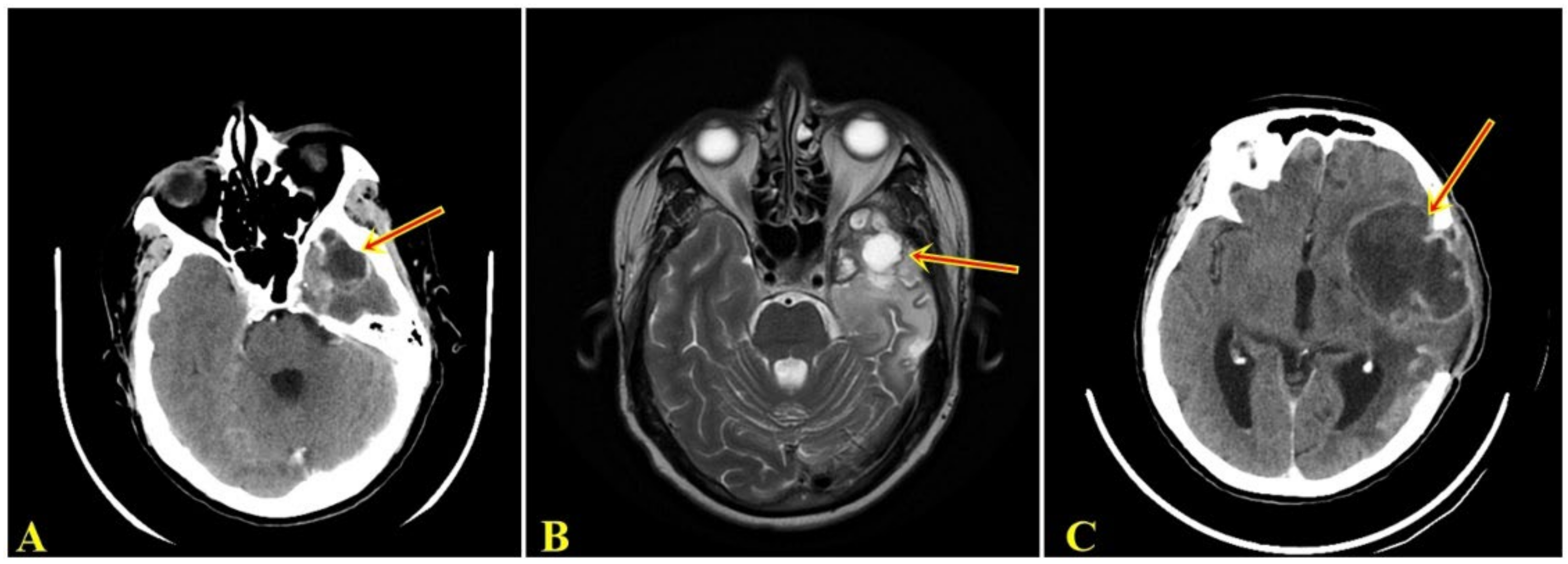{kind=link}
{kind=link}
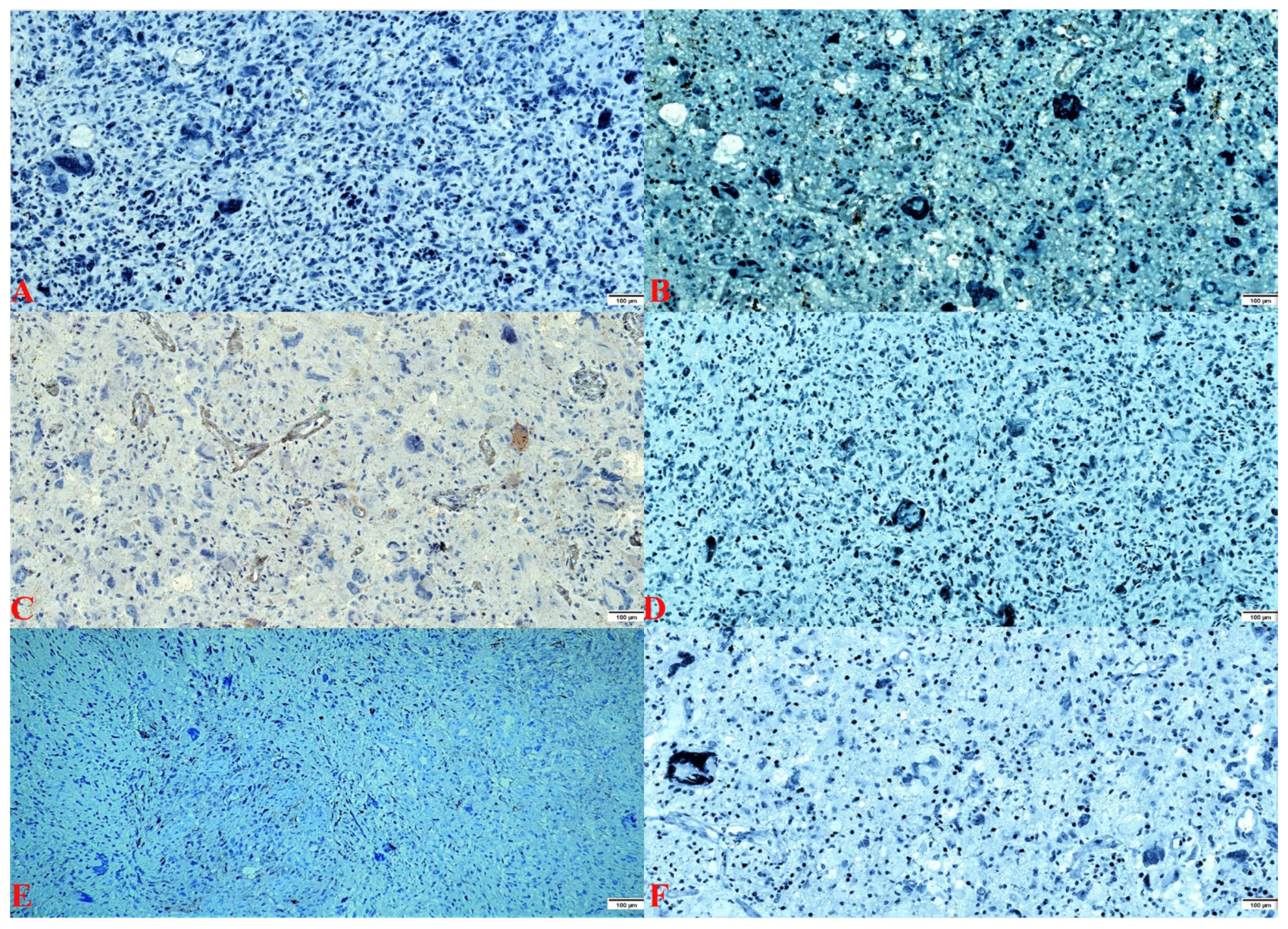{kind=link}
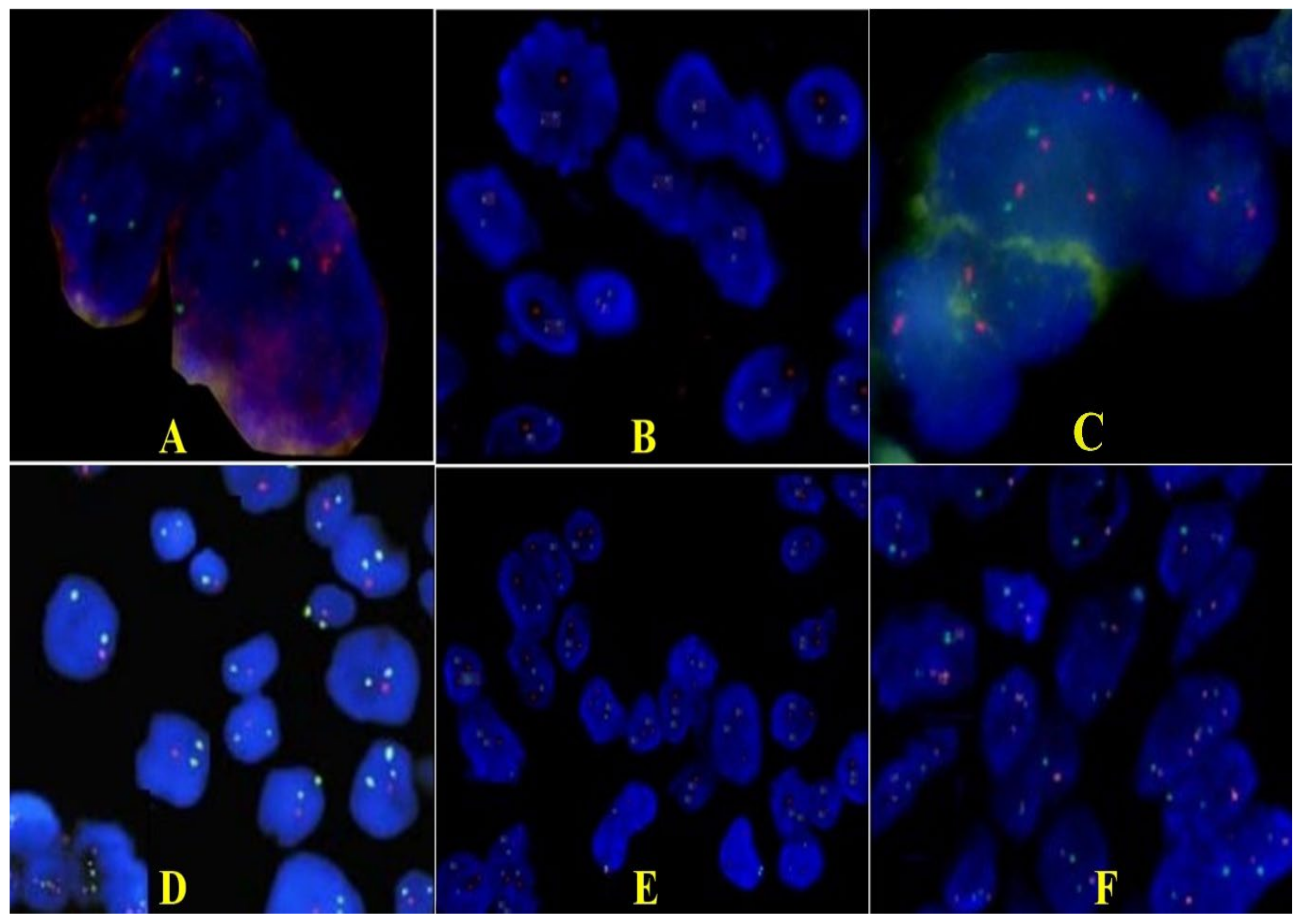{kind=link}
Abstract
:1. Introduction
2. Case
2.1. Clinical Summary
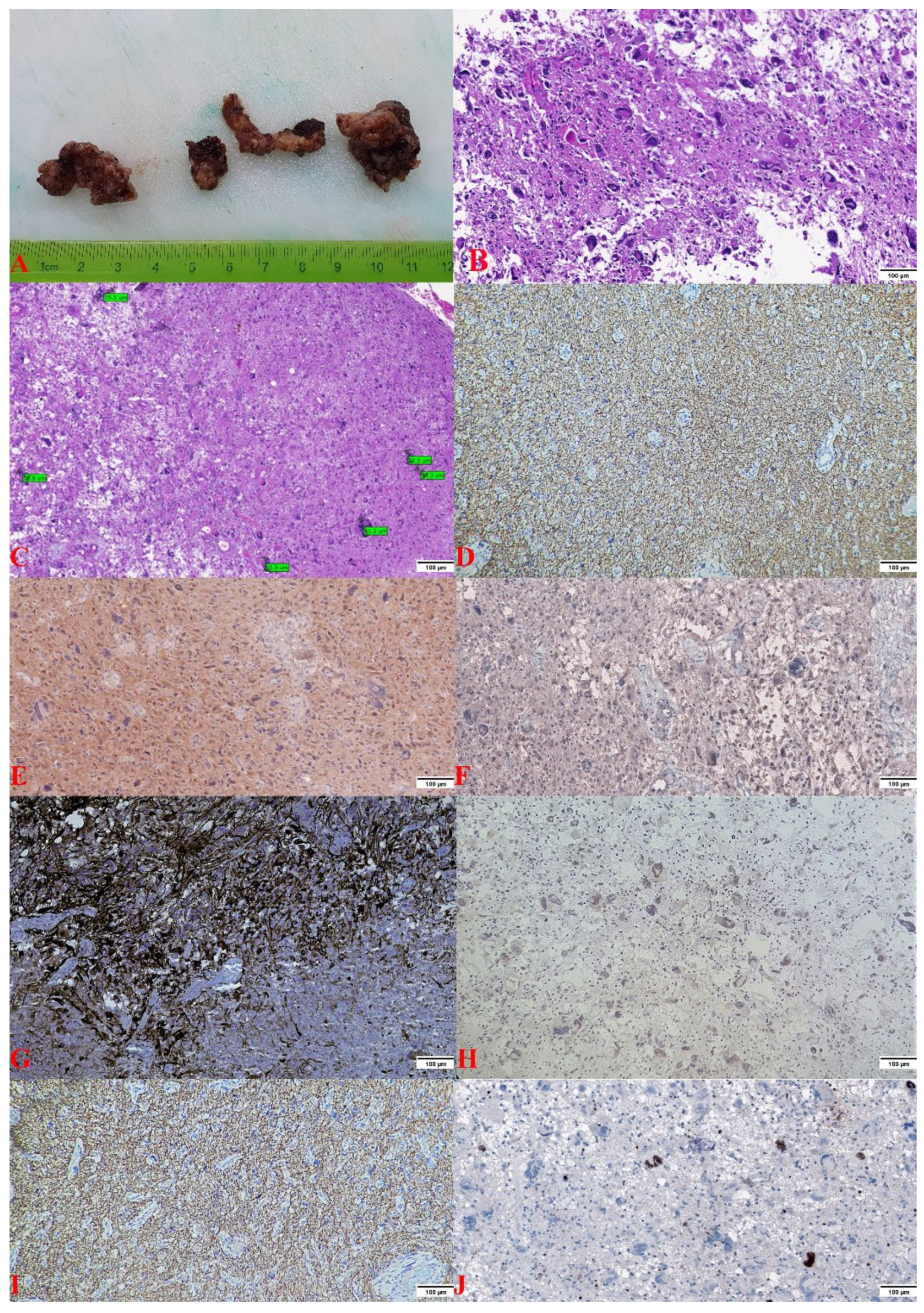
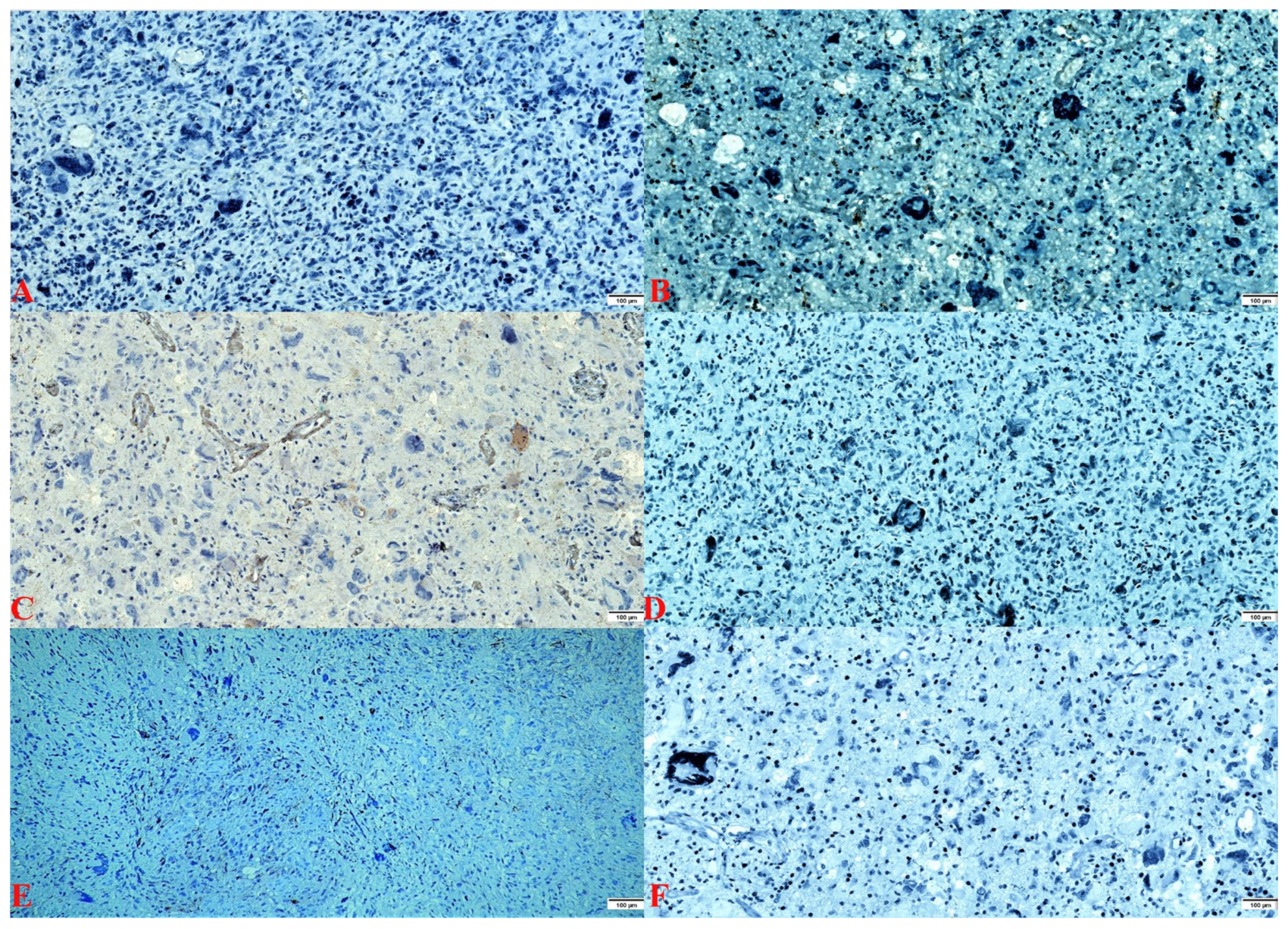
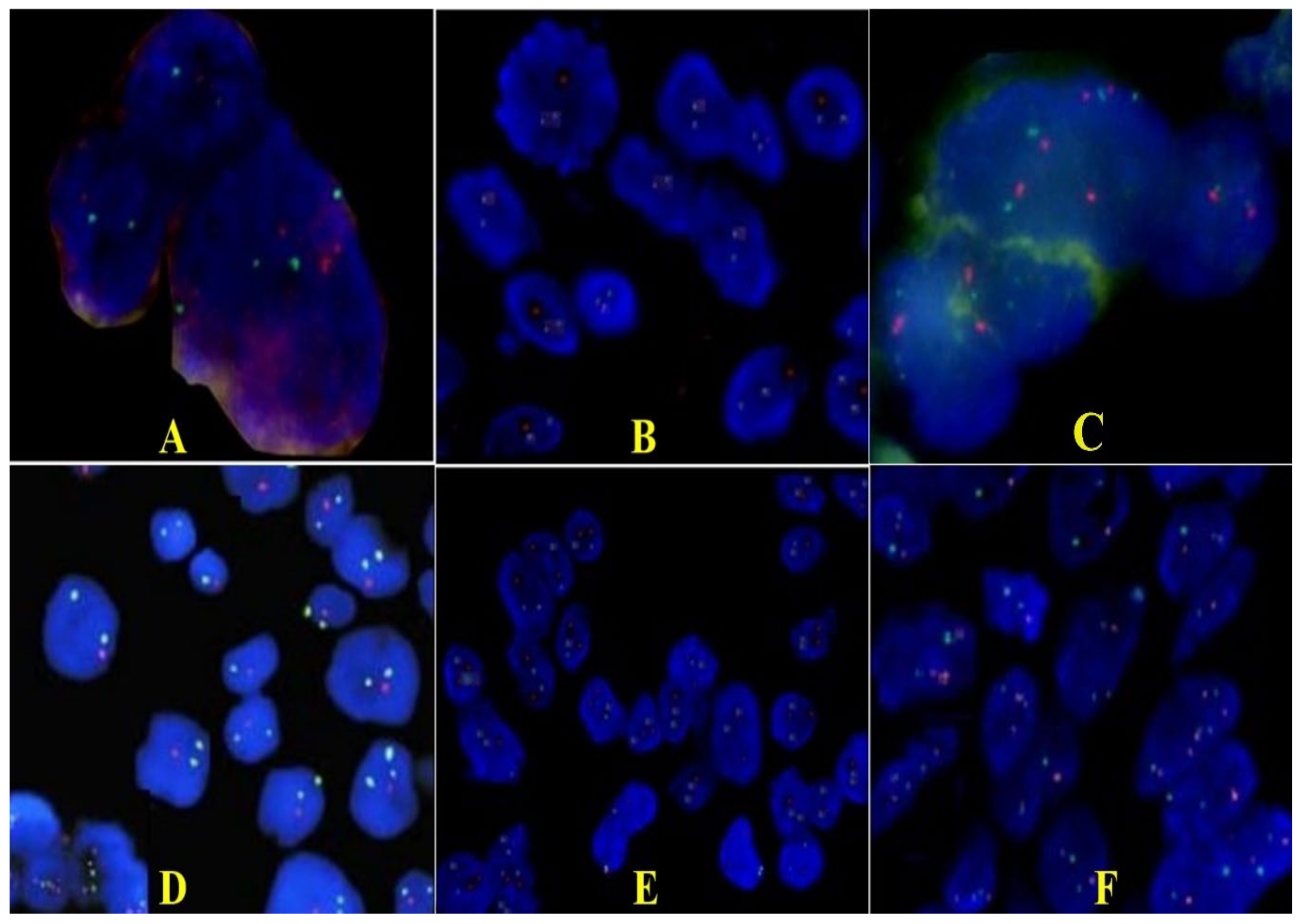
2.2. Pathology Findings
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Figarella-Branger, D.; Perry, A.; Reifenberger, G.; von Deimling, A. WHO Classification of Tumors of the Central Nervous System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016; pp. 46–47. [Google Scholar]
- WHO Classification of Tumors Editorial Board. Central Nervous System Tumors. WHO Classification of Tumors, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 39, 46–47, 53. [Google Scholar]
- Kozak, K.R.; Moody, J.S. Giant cell glioblastoma: A glioblastoma subtype with distinct epidemiology and superior prognosis. Neuro-Oncology 2009, 11, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naydenov, E.; Bussarsky, V.; Nachev, S.; Hadjidekova, S.; Toncheva, D. Long-Term Survival of a Patient with Giant Cell Glioblastoma: Case Report and Review of the Literature. Case Rep. Oncol. 2009, 2, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.V.; Kaiser, A.E.; Liuzzi, F.J. Characterization and Histological Examination of a Rare Giant Cell Glioblastoma. Cureus 2020, 12, e9237. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Homagain, S.; Raut, A.; Sedhain, G.; Bhatta, S.; Shrivastav, S. Giant cell glioblastoma in 6-year-old kid: Report of an unusual case. Clin. Case Rep. 2020, 8, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.C.; Wu, A.; Xiang, M.; Azad, T.D.; Soltys, S.G.; Li, G.; Pollom, E.L. Prognostic Factors and Treatment Patterns in the Management of Giant Cell Glioblastoma. World Neurosurg. 2019, 128, e217–e224. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Harle, L. Difficult diagnosis of brainstem glioblastoma multiforme in a woman: A case report and review of the literature. J. Med. Case Rep. 2009, 3, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thong, P.M.; Minh Duc, N. Malignant Ependymoblastoma Mimicking a Benign Pilocytic Astrocytoma. Neurol. Int. 2020, 12, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Belsuzarri, T.A.B.; Araujo, J.F.; Catanoce, A.P.; Neves, M.W.F.; Sola, R.A.S.; Navarro, J.N.; Brito, L.G.; Silva, N.R.; Pontelli, L.O.C.; Mattos, L.G.A.; et al. Giant cells glioblastoma: Case report and pathological analysis from this uncommon subtype of glioma. Rare Tumors 2015, 7, 5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.E.; Ohta, T.; Nonoguchi, N.; Satomi, K.; Capper, D.; Pierscianek, D.; Sure, U.; Vital, A.; Paulus, W.; Mittelbronn, M.; et al. Genetic Alterations in Gliosarcoma and Giant Cell Glioblastoma. Brain Pathol. 2016, 26, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Valle-Folgueral, J.M.; Mascarenhas, L.; Costa, J.A.; Viera, F.; Soare-Fernandes, J.; Beleza, P.; Alegria, C. Giant cell glioblastoma: Review of the literature and illustrated case. Neurocirugia 2008, 19, 343–349. [Google Scholar] [CrossRef]
- Ogawa, K.; Kurose, A.; Kamataki, A.; Asano, K.; Katayama, K.; Kurotaki, H. Giant cell glioblastoma is a distinctive subtype of glioma characterized by vulnerability to DNA damage. Brain Tumor Pathol. 2020, 37, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantero, D.; Mollejo, M.; Sepúlveda, J.M.; D’Haene, N.; Gutiérrez-Guamán, M.J.; Rodríguez de Lope, Á.; Fiaño, C.; Castresana, J.S.; Lebrun, L.; Rey, J.A.; et al. TP53, ATRX alterations, and low tumor mutation load feature IDH-wildtype giant cell glioblastoma despite exceptional ultra-mutated tumors. Neurooncol. Adv. 2020, 2, vdz059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogazianou, A.P.; Chan, R.; Bäcklund, L.M.; Pearson, D.M.; Liu, L.; Langford, C.F.; Gregory, S.G.; Collins, V.P.; Ichimura, K. Distinct patterns of 1p and 19q alterations identify subtypes of human gliomas that have different prognoses. Neuro-Oncology 2010, 12, 664–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.B.; Blair, H.; Ballman, K.V.; Giannini, C.; Arusell, R.M.; Law, M.; Flynn, H.; Passe, S.; Felten, S.; Brown, P.D.; et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006, 66, 9852–9861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marker, D.F.; Pearce, T.M. Homozygous deletion of CDKN2A by fluorescence in situ hybridization is prognostic in grade 4, but not grade 2 or 3, IDH-mutant astrocytomas. Acta Neuropathol. Commun. 2020, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- Etcheverry, A.; Aubry, M.; de Tayrac, M.; Vauleon, E.; Boniface, R.; Guenot, F.; Saikali, S.; Hamlat, A.; Riffaud, L.; Menei, P.; et al. DNA methylation in glioblastoma: Impact on gene expression and clinical outcome. BMC Genom. 2010, 11, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Fletcher, M.; Gu, Z.; Wang, Q.; Costa, B.; Bertoni, A.; Man, K.H.; Schlotter, M.; Felsberg, J.; Mangei, J.; et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat. Commun. 2020, 11, 6434. [Google Scholar] [CrossRef] [PubMed]
- Abdelkareem, R.M.; Elnashar, A.T.; Fadle, K.N.; Muhammad, E.M.S. Immunohistochemical expression of Nestin as Cancer Stem Cell Marker in gliomas. J. Neurosci. Neurol. Disord. 2019, 3, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Rutkowski, M.J.; Safaee, M.; Sun, M.Z.; Sayegh, E.T.; Bloch, O.; Tihan, T.; Parsa, A.T. Survival outcomes of giant cell glioblastoma: Institutional experience in the management of 20 patients. J. Clin. Neurosci. 2014, 21, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orasanu, C.I.; Aschie, M.; Deacu, M.; Mocanu, L.; Voda, R.I.; Topliceanu, T.S.; Cozaru, G.C. Morphogenetic and Imaging Characteristics in Giant Cell Glioblastoma. Curr. Oncol. 2022, 29, 5316-5323. https://doi.org/10.3390/curroncol29080422
Orasanu CI, Aschie M, Deacu M, Mocanu L, Voda RI, Topliceanu TS, Cozaru GC. Morphogenetic and Imaging Characteristics in Giant Cell Glioblastoma. Current Oncology. 2022; 29(8):5316-5323. https://doi.org/10.3390/curroncol29080422
Chicago/Turabian StyleOrasanu, Cristian Ionut, Mariana Aschie, Mariana Deacu, Liliana Mocanu, Raluca Ioana Voda, Theodor Sebastian Topliceanu, and Georgeta Camelia Cozaru. 2022. "Morphogenetic and Imaging Characteristics in Giant Cell Glioblastoma" Current Oncology 29, no. 8: 5316-5323. https://doi.org/10.3390/curroncol29080422
APA StyleOrasanu, C. I., Aschie, M., Deacu, M., Mocanu, L., Voda, R. I., Topliceanu, T. S., & Cozaru, G. C. (2022). Morphogenetic and Imaging Characteristics in Giant Cell Glioblastoma. Current Oncology, 29(8), 5316-5323. https://doi.org/10.3390/curroncol29080422