Real-World Clinical Outcomes after Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Data Source and Extraction
2.3. Study Population
2.4. Treatment Lines
2.5. Genomic Profiling
2.6. Patient Outcomes
2.7. Statistical Analysis
3. Results
3.1. Patients
3.2. First-Line Treatments
3.3. ctDNA Testing Results
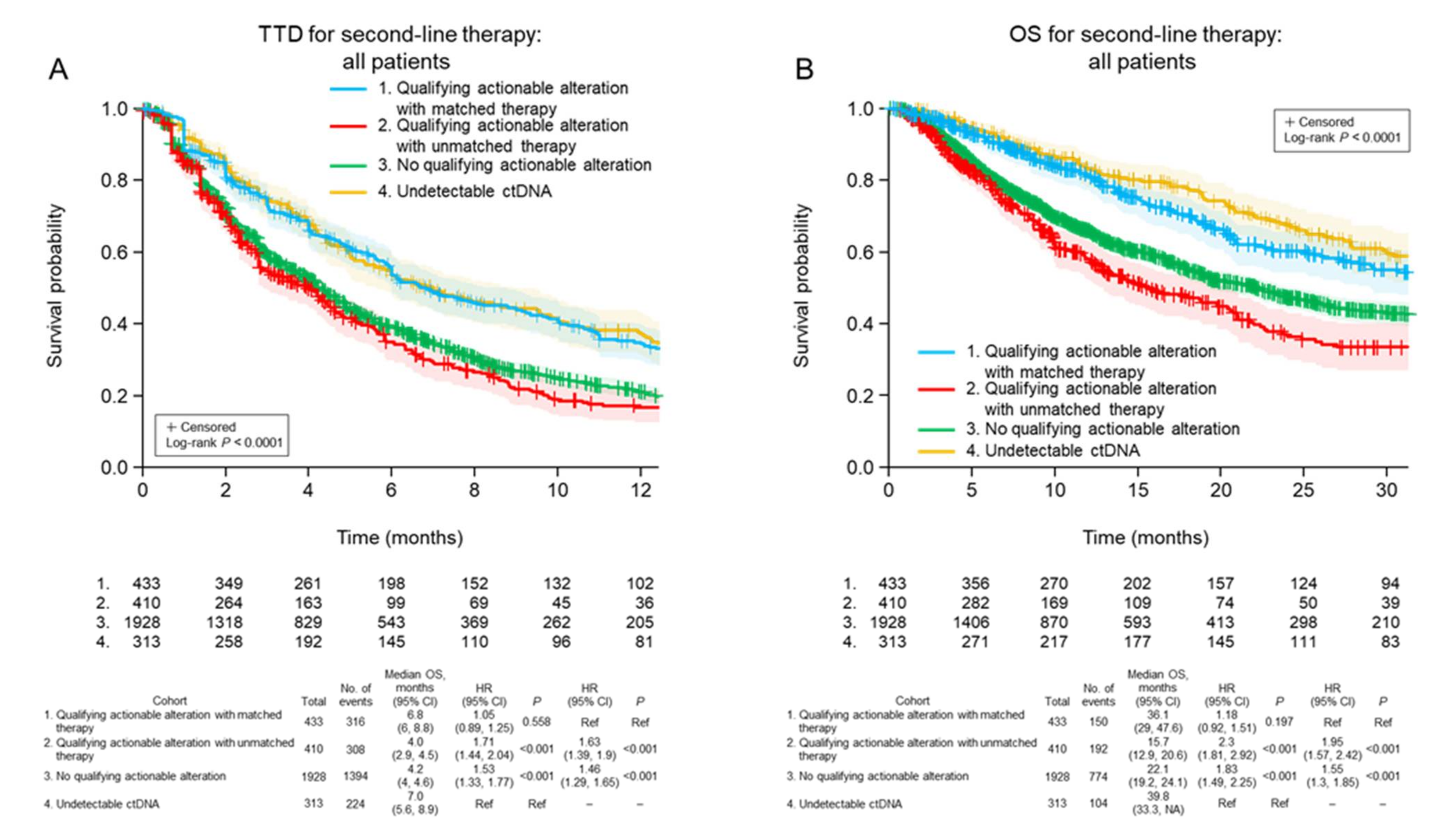
3.4. Treatment Decisions after ctDNA Testing
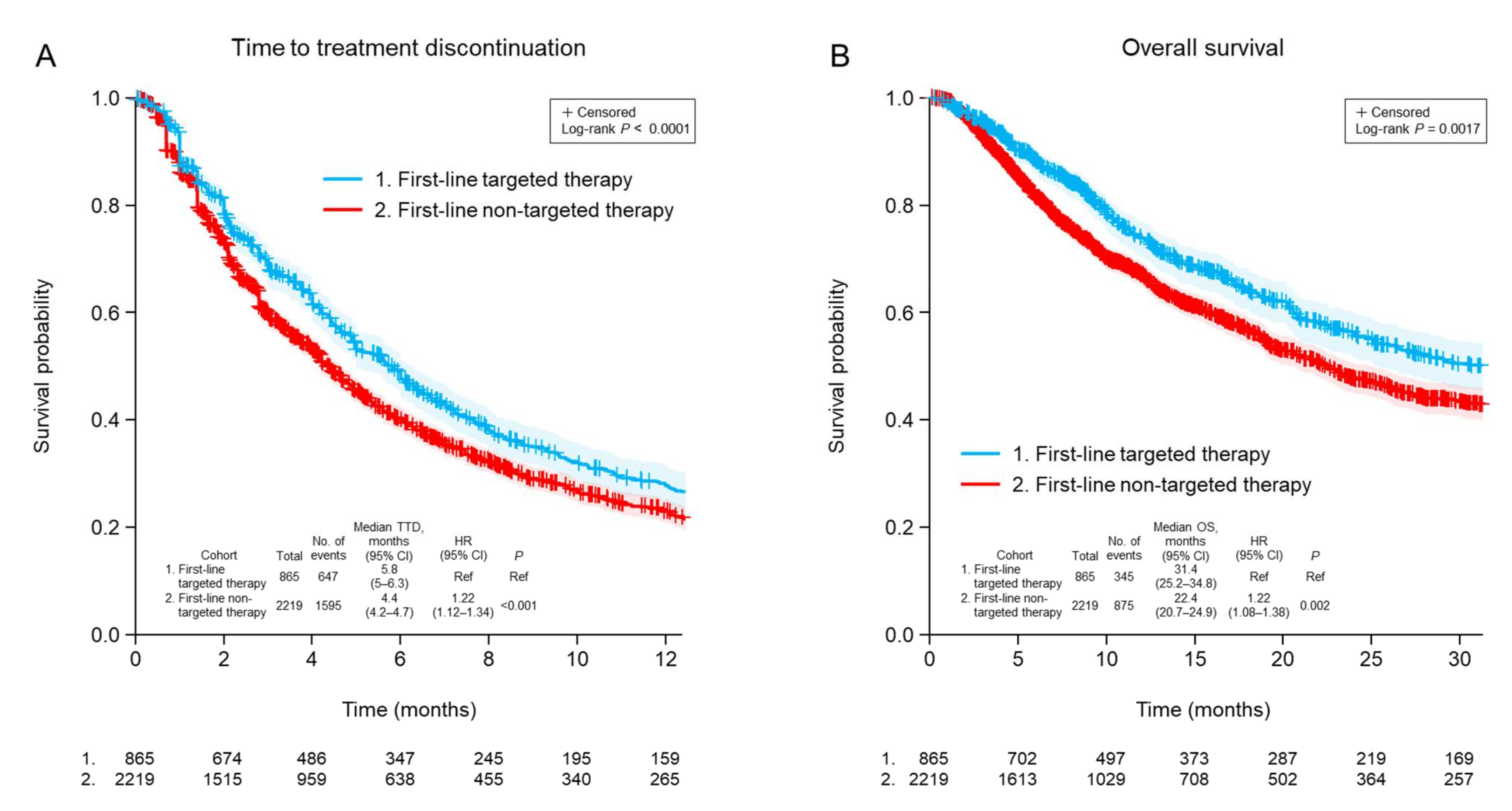
3.5. Clinical Outcomes
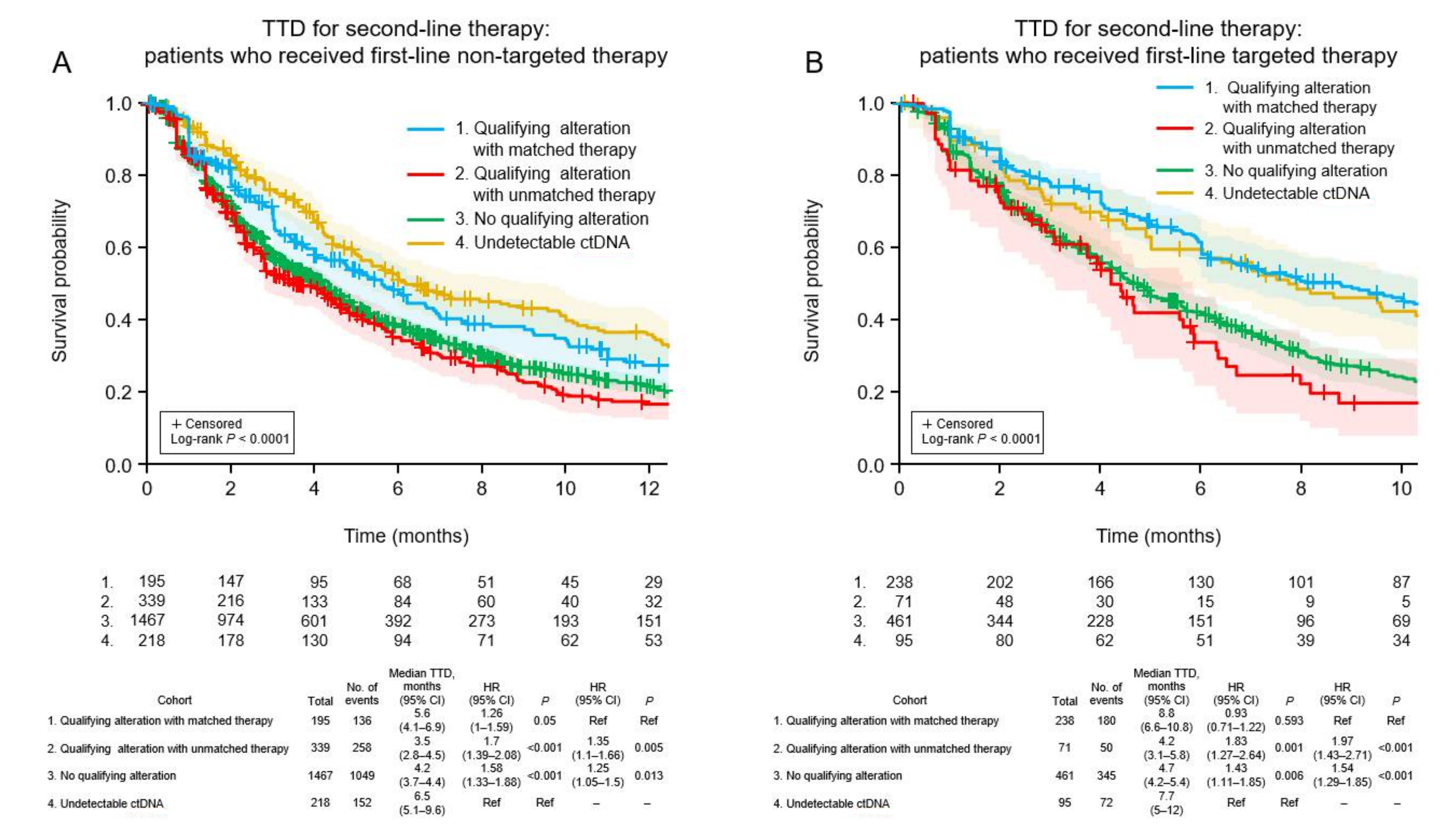
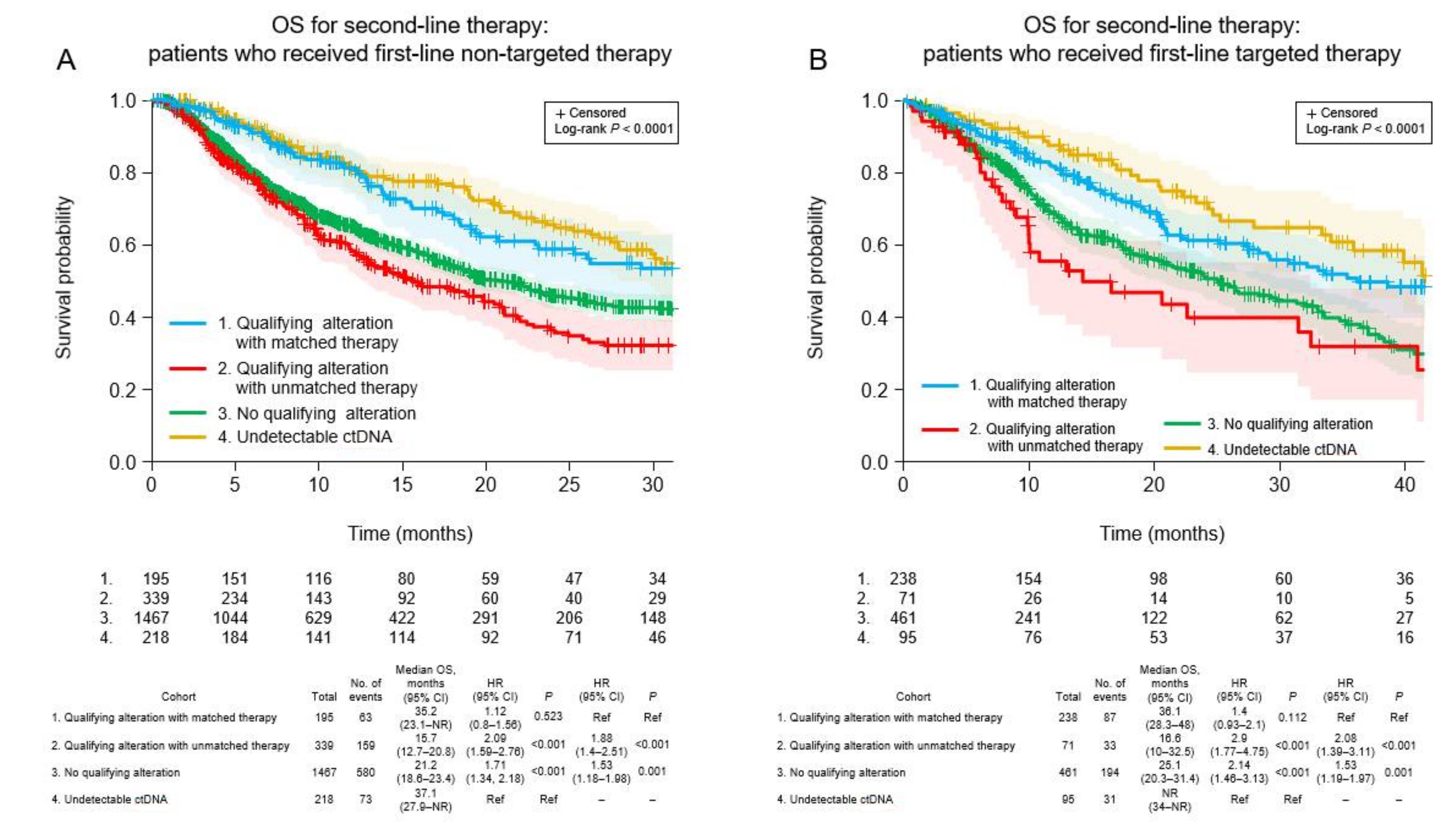
3.6. Clinical Outcomes of Patients Who Received Non-Targeted First-Line Therapy
3.7. Clinical Outcomes of Patients Who Received Targeted First-Line Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanna, N.H.; Robinson, A.G.; Temin, S.; Baker, S., Jr.; Brahmer, J.R.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D.; et al. Therapy for Stage IV Non–Small-Cell Lung Cancer With Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. J. Clin. Oncol. 2021, 39, 1040–1091. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Thorac. Oncol. 2018, 13, 323–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-small Cell Lung Cancer Version 1.2022—7 December 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 6 April 2022).
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237, Correction in Ann. Oncol. 2019, 2030, 2863–2070. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [Green Version]
- Palmero, R.; Taus, A.; Viteri, S.; Majem, M.; Carcereny, E.; Garde-Noguera, J.; Felip, E.; Nadal, E.; Malfettone, A.; Sampayo, M.; et al. Biomarker Discovery and Outcomes for Comprehensive Cell-Free Circulating Tumor DNA Versus Standard-of-Care Tissue Testing in Advanced Non–Small-Cell Lung Cancer. JCO Precis. Oncol. 2021, 5, 93–102. [Google Scholar] [CrossRef]
- Park, S.; Olsen, S.; Ku, B.M.; Lee, M.S.; Jung, H.A.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.L.; et al. High concordance of actionable genomic alterations identified between circulating tumor DNA–based and tissue-based next-generation sequencing testing in advanced non–small cell lung cancer: The Korean Lung Liquid Versus Invasive Biopsy Program. Cancer 2021, 127, 3019–3028. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non–small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- U.S Food and Drug Agency; Foundation Medicine Inc. FoundationOne Liquid CDx (F1 Liquid CDx). Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p190032 (accessed on 6 April 2022).
- U.S. Food and Drug Agency; Guardant Health, Inc. Guardant360 CDx. Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p200010 (accessed on 6 April 2022).
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Madison, R.; Schrock, A.B.; Castellanos, E.; Gregg, J.P.; Snider, J.; Ali, S.M.; Miller, V.A.; Singal, G.; Alexander, B.M.; Venstrom, J.M.; et al. Retrospective analysis of real-world data to determine clinical outcomes of patients with advanced non-small cell lung cancer following cell-free circulating tumor DNA genomic profiling. Lung Cancer 2020, 148, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; Delmonte, A.; Hsia, T.C.; Laskin, J.; Kim, S.W.; He, Y.; Tsai, C.M.; et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non–small cell lung cancer. Cancer 2020, 126, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Wang, W.; Song, Z.; Zhang, Y. A Comparison of ddPCR and ARMS for detecting EGFR T790M status in ctDNA from advanced NSCLC patients with acquired EGFR-TKI resistance. Cancer Med. 2017, 6, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Takahama, T.; Azuma, K.; Shimokawa, M.; Takeda, M.; Ishii, H.; Kato, T.; Saito, H.; Daga, H.; Tsuboguchi, Y.; Okamoto, I.; et al. Plasma screening for the T790M mutation of EGFR and phase 2 study of osimertinib efficacy in plasma T790M–positive non–small cell lung cancer: West Japan Oncology Group 8815L/LPS study. Cancer 2020, 126, 1940–1948. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Gutierrez, M.E.; Choi, K.; Lanman, R.B.; Licitra, E.J.; Skrzypczak, S.M.; Pe Benito, R.; Wu, T.; Arunajadai, S.; Kaur, S.; Harper, H.; et al. Genomic Profiling of Advanced Non–small Cell Lung Cancer in Community Settings: Gaps and Opportunities. Clin. Lung Cancer 2017, 18, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Sakai, K.; Terashima, M.; Kaneda, H.; Hayashi, H.; Tanaka, K.; Okamoto, K.; Takahama, T.; Yoshida, T.; Iwasa, T.; et al. Clinical application of amplicon-based next-generation sequencing to therapeutic decision making in lung cancer. Ann. Oncol. 2015, 26, 2477–2482. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, S.; Lee, D.H.; Jang, S.J.; Chun, S.M.; Kim, S.W. Real-world utility of next-generation sequencing for targeted gene analysis and its application to treatment in lung adenocarcinoma. Cancer Med. 2021, 10, 3197–3204. [Google Scholar] [CrossRef]
- Hardtstock, F.; Myers, D.; Li, T.; Cizova, D.; Maywald, U.; Wilke, T.; Griesinger, F. Real-world treatment and survival of patients with advanced non–small cell lung Cancer: A German retrospective data analysis. BMC Cancer 2020, 20, 260. [Google Scholar] [CrossRef]
- Lee, Y.; Park, S.; Kim, W.S.; Lee, J.C.; Jang, S.J.; Choi, J.; Choi, C.M. Correlation between progression-free survival, tumor burden, and circulating tumor DNA in the initial diagnosis of advanced-stage EGFR-mutated non-small cell lung cancer. Thorac. Cancer 2018, 9, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation–Positive Advanced Non–small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.; Ku, B.M.; Park, S.; Jung, H.A.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. Longitudinal monitoring by next generation sequencing of plasma cell-free DNA in ALK-rearranged non-small cell lung cancer (NSCLC) patients treated with ALK tyrosine kinase inhibitors. J. Clin. Oncol. 2020, 38, 9603. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | Non-Targeted First-Line Therapy | Targeted First-Line Therapy | p-Value a | |
|---|---|---|---|---|
| n | 3084 | 2219 | 865 | |
| Age (median) | 65 | 66 | 64 | |
| Female, n (%) | 1727 (56.0%) | 1162 (52.4%) | 566 (65.4%) | <0.001 |
| Histology, n (%) | ||||
| Non-squamous | 2291 (74.3%) | 1583 (71.3%) | 708 (81.8%) | <0.001 |
| Squamous | 337 (10.9%) | 322 (14.5%) | 15 (1.7%) | <0.001 |
| Not specified | 456 (14.8%) | 314 (14.2%) | 142 (16.4%) | 0.125 |
| n (%) | |
|---|---|
| n | 3084 |
| Chemotherapy ± other agents | 1943 (63.0%) |
| Immune checkpoint inhibitor ± other agents | 586 (19.0%) |
| VEGF inhibitor ± other agents | 207 (6.7%) |
| Targeted therapy ± other agents | 865 (28.0%) |
| First-/second-generation EGFR-TKI | 552 (17.9%) |
| Third-generation EGFR-TKI | 217 (7.0%) |
| EGFR monoclonal antibody | 7 (0.2%) |
| Other TKI | 89 (2.9%) |
| Alteration | All Patients | Non-Targeted First-Line Treatment | Targeted First-Line Treatment | p-Value (Non-Targeted vs. Targeted) | ||
|---|---|---|---|---|---|---|
| Any Alteration | Qualifying Alteration | Any Alteration | Qualifying Alteration | |||
| n | 2771 | 2001 | 770 | |||
| Actionable alteration a, n (%) | 1160 (41.9%) | 534 (26.7%) | 534 (26.7%) | 626 (81.3%) | 309 (40.1%) | <0.001 |
| KRAS mutation, n (%) | 416 (15.0%) | 387 (19.3%) | 131 b (6.5%) | 29 (3.8%) | 4 b (0.5%) | <0.001 |
| EGFR mutation, n (%) | 824 (29.7%) | 251 (12.5%) | 251 (12.5%) | 573 (77.4%) | 251 (32.6%) | <0.001 |
| Exon 19 deletion, n (%) | 456 (16.5%) | 131 (6.5%) | 131 (6.5%) | 325 (42.2%) | 1 (0.1%) | <0.001 |
| L858R, n (%) | 272 (9.8%) | 70 (3.5%) | 70 (3.5%) | 202 (26.2%) | 0 | <0.001 |
| T790M, n (%) | 292 (10.5%) | 41 c (2.0%) | 1 c (<0.1%) | 251 (32.6%) | 243 (31.6%) | <0.001 |
| C797S, n (%) | 24 (0.9%) | 8 (0.4%) | 0 | 16 (2.1%) | 16 (2.1%) | <0.001 |
| Other point mutation, n (%) | 62 (2.2%) | 25 (1.2%) | 25 (1.2%) | 37 (4.8%) | 0 | <0.001 |
| Exon 20 insertion, n (%) | 32 (1.2%) | 25 (1.2%) | 25 (1.2%) | 7 (0.9%) | 0 | 0.581 |
| MET alterations, n (%) | 126 (4.5%) | 68 (3.4%) | 67 (3.3%) | 58 (7.5%) | 54 (7.0%) | <0.001 |
| Amplification, n (%) | 99 (3.6%) | 46 (2.3%) | 46 (2.3%) | 53 (6.9%) | 53 (6.9%) | <0.001 |
| Exon 14 skipping, n (%) | 26 (0.9%) | 21 (1.0%) | 21 (1.0%) | 5 (0.6%) | 0 | 0.448 |
| Point mutation, n (%) | 3 (0.1%) | 1 (<0.1%) | 0 | 2 (0.3%) | 2 (0.3%) | 0.189 |
| ALK alterations, n (%) | 66 (2.4%) | 28 (1.4%) | 27 (1.3%) | 38 (4.9%) | 4 (0.5%) | <0.001 |
| Fusion, n (%) | 65 (2.3%) | 27 (1.3%) | 27 (1.3%) | 38 (4.9%) | 3 (0.4%) | <0.001 |
| Point mutation, n (%) | 7 (0.3%) | 3 (0.1%) | 0 | 4 (0.5%) | 4 (0.5%) | 0.099 |
| ERBB2 mutation, n (%) | 43 (1.6%) | 42 (2.1%) | 42 (2.1%) | 1 (0.1%) | 1 (0.1%) | <0.001 |
| BRAF V600X d, n (%) | 34 d (1.2%) | 19 d (0.9%) | 19 d (0.9%) | 15 e (1.9%) | 14 e (1.8%) | 0.052 |
| RET fusion, n (%) | 15 (0.5%) | 9 (0.4%) | 9 (0.4%) | 6 (0.8%) | 6 (0.8%) | 0.384 |
| ROS1 fusion, n (%) | 10 (0.4%) | 4 (0.2%) | 4 (0.2%) | 6 (0.8%) | 0 | 0.033 |
| NTRK1 fusion, n (%) | 2 (0.1%) | 0 | 0 | 2 (0.3%) | 2 (0.3%) | 0.077 |
| MSI-high, n (%) | 3 (0.1%) | 3 (0.1%) | 3 (0.1%) | 0 | 0 | 0.565 |
| n | Targeted Therapy | Non-Targeted Therapy | |||
|---|---|---|---|---|---|
| Any, n (%) | Matched, n (%) | with ICI, n (%) | without ICI, n (%) | ||
| All patients | 3084 | 928 a (30.1%) | 433 a (14.0%) | 1315 b (42.6%) | 841 (27.3%) |
| No ctDNA detected | 313 | 100 (31.9%) | NA | 145 (46.3%) | 68 (21.7%) |
| ctDNA detected | 2771 | 828 a (29.9%) | 433 a (15.6%) | 1170 b (42.2%) | 773 (27.9%) |
| First-line non-targeted therapy | 2001 | 293 a (14.6%) | 195 a (9.7%) | 1084 b (54.2%) | 624 (31.2%) |
| First-line targeted therapy | 770 | 535 (69.5%) | 238 (30.9%) | 86 (11.2%) | 149 (19.4%) |
| No qualifying alteration | 1928 | 365 (18.9%) | NA | 963 (49.9%) | 600 (31.1%) |
| Any qualifying alteration | 843 | 463 a (54.9%) | 433 a (51.4%) | 207 b (24.6%) | 173 (20.5%) |
| EGFR driver mutation c | 511 | 373 (73.0%) | 348 d (68.1%) | 50 (9.8%) | 88 (17.2%) |
| EGFR T790M c | 285 | 251 (88.1%) | 239 d (83.9%) | 10 (3.5%) | 24 (8.4%) |
| EGFR exon 20 insertion | 25 | 1 (4.0%) | 1 (4.0%) | 11 (44.0%) | 13 (52.0%) |
| KRAS G12C c | 135 | 11 (8.1%) | 8 d (5.9%) | 91 (67.4%) | 33 (24.4%) |
| MET amplificationc | 99 | 41 (41.4%) | 32 d (32.3%) | 32 (32.3%) | 26 (26.3%) |
| ERBB2 mutation | 43 | 14 (32.6%) | 14 (32.6%) | 15 (34.9%) | 14 (32.6%) |
| ALK fusion c | 36 | 24 (66.7%) | 22 d (61.1%) | 4 (11.1%) | 8 (22.2%) |
| BRAF V600X c | 33 | 19 (57.6%) | 17 (51.5%) | 9 (27.3%) | 5 (15.2%) |
| MET exon 14 skipping c | 23 | 19 (82.6%) | 19 (82.6%) | 3 (13.0%) | 1 (4.3%) |
| Other alteration c,e | 30 | 16 (53.3%) | 14 a,d (46.7%) | 7 b (23.3%) | 6 (20.0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olsen, S.; Liao, J.; Hayashi, H. Real-World Clinical Outcomes after Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer. Curr. Oncol. 2022, 29, 4811-4826. https://doi.org/10.3390/curroncol29070382
Olsen S, Liao J, Hayashi H. Real-World Clinical Outcomes after Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer. Current Oncology. 2022; 29(7):4811-4826. https://doi.org/10.3390/curroncol29070382
Chicago/Turabian StyleOlsen, Steven, Jiemin Liao, and Hidetoshi Hayashi. 2022. "Real-World Clinical Outcomes after Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer" Current Oncology 29, no. 7: 4811-4826. https://doi.org/10.3390/curroncol29070382
APA StyleOlsen, S., Liao, J., & Hayashi, H. (2022). Real-World Clinical Outcomes after Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer. Current Oncology, 29(7), 4811-4826. https://doi.org/10.3390/curroncol29070382