Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones

 , , , , ,
, , , , ,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
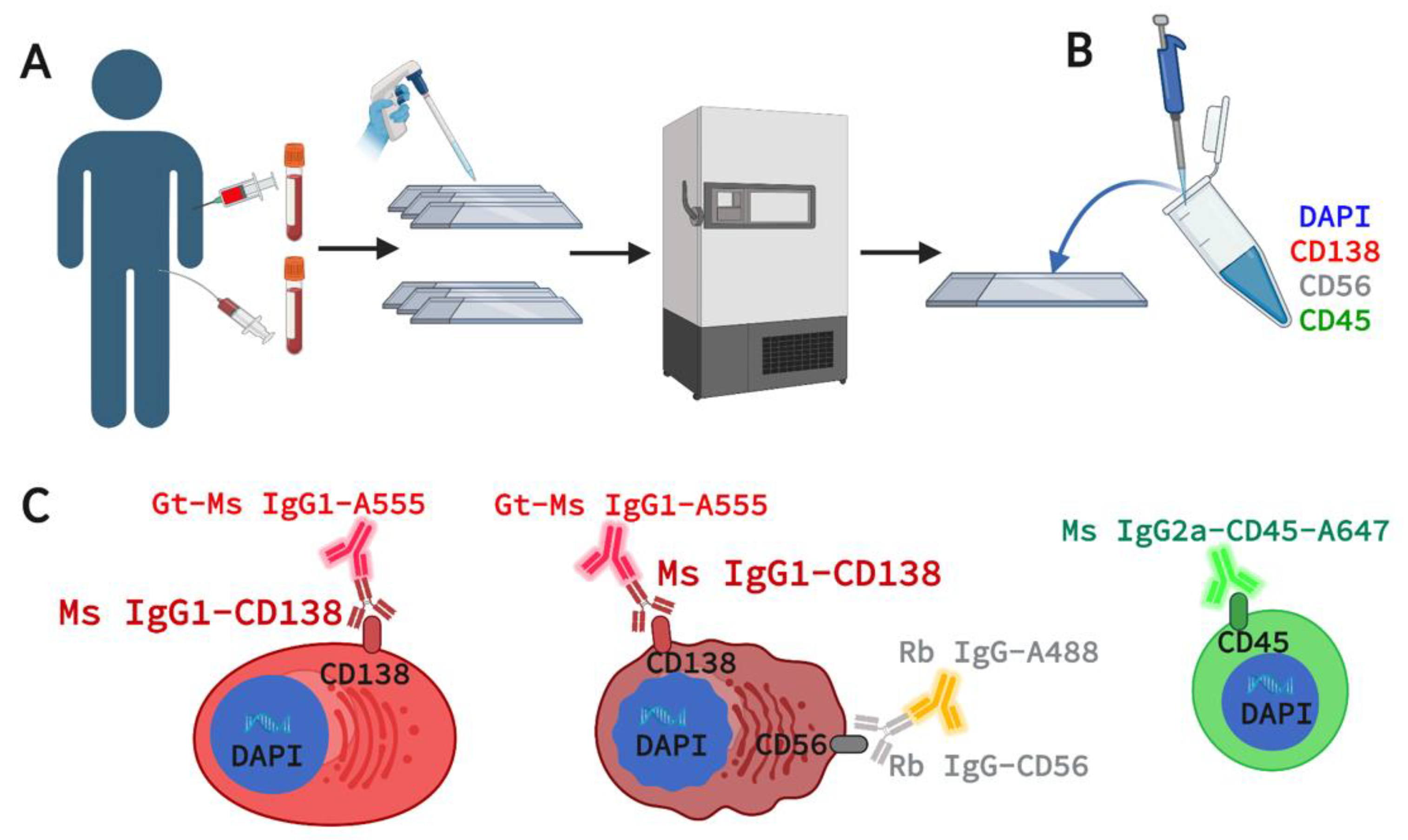
2.1. Patient Enrollment and Sample Acquisition
2.2. Marker Selection for 4-Plex Immunofluorescence Assay
2.3. Assay Staining and Validation in Cell Lines and Spiked NBD Samples
2.4. Assay Staining and Validation in Patient PB and BMA
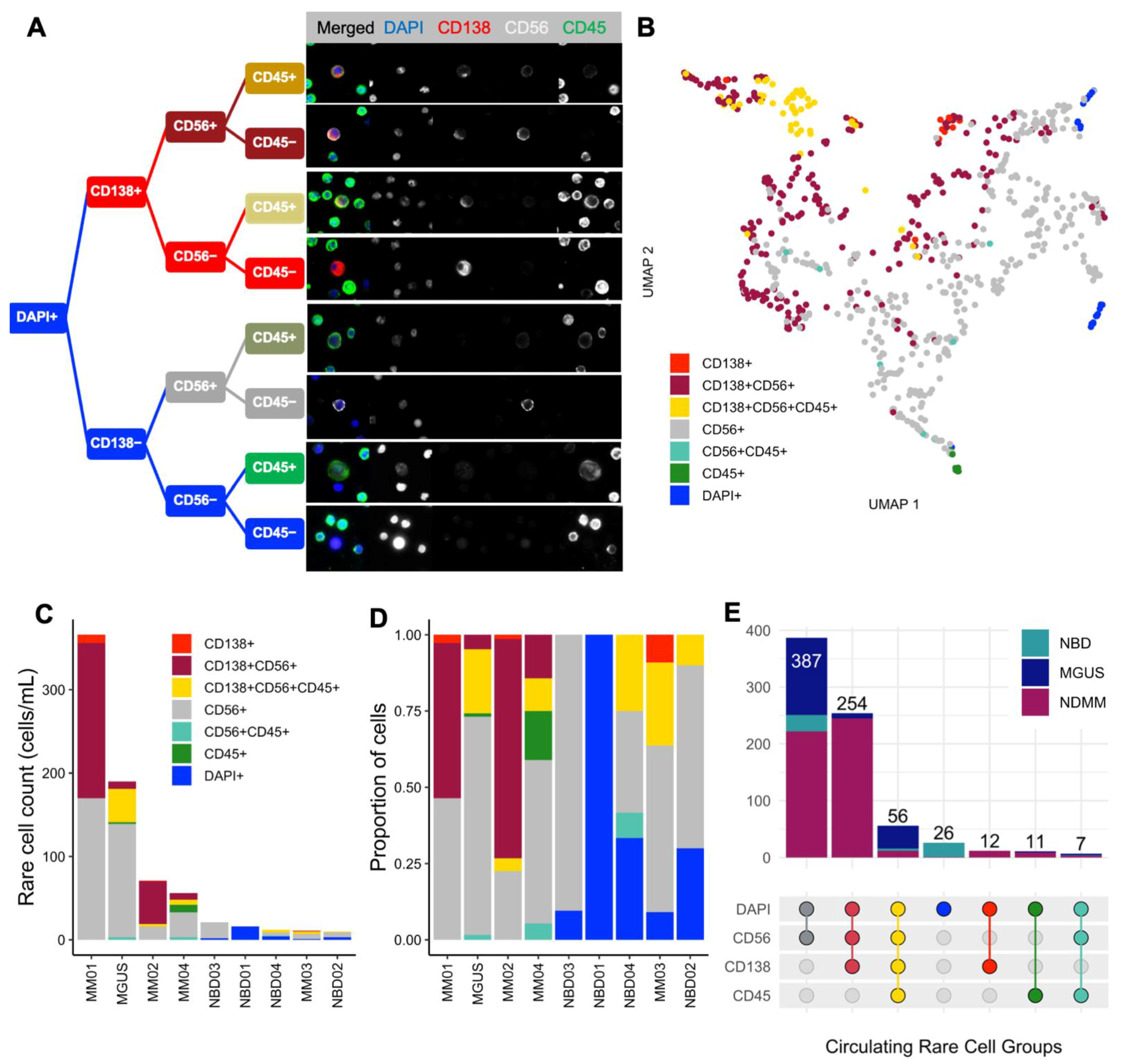
2.5. Imaging and Technical Analysis for Rare Cell Detection and Cell Classification
- CD138+
- CD138+CD56+
- CD138+CD45+
- CD138+CD56+CD45+
- CD138−: any cells larger than surrounding WBCs and have eccentric nuclei
- PC clusters: cluster consisting of two or more CD138+ cells
- Binucleated PC: CD138+ cells presenting two morphological distinguishable nuclei
2.6. Single-Cell Sequencing and CNV Analysis
2.7. Karyotyping and Fluorescent in Situ Hybridization (FISH) for Clinical Diagnosis
2.8. Correlating scCNV to Clinical Cytogenetics
2.9. Statistical Analysis
3. Results
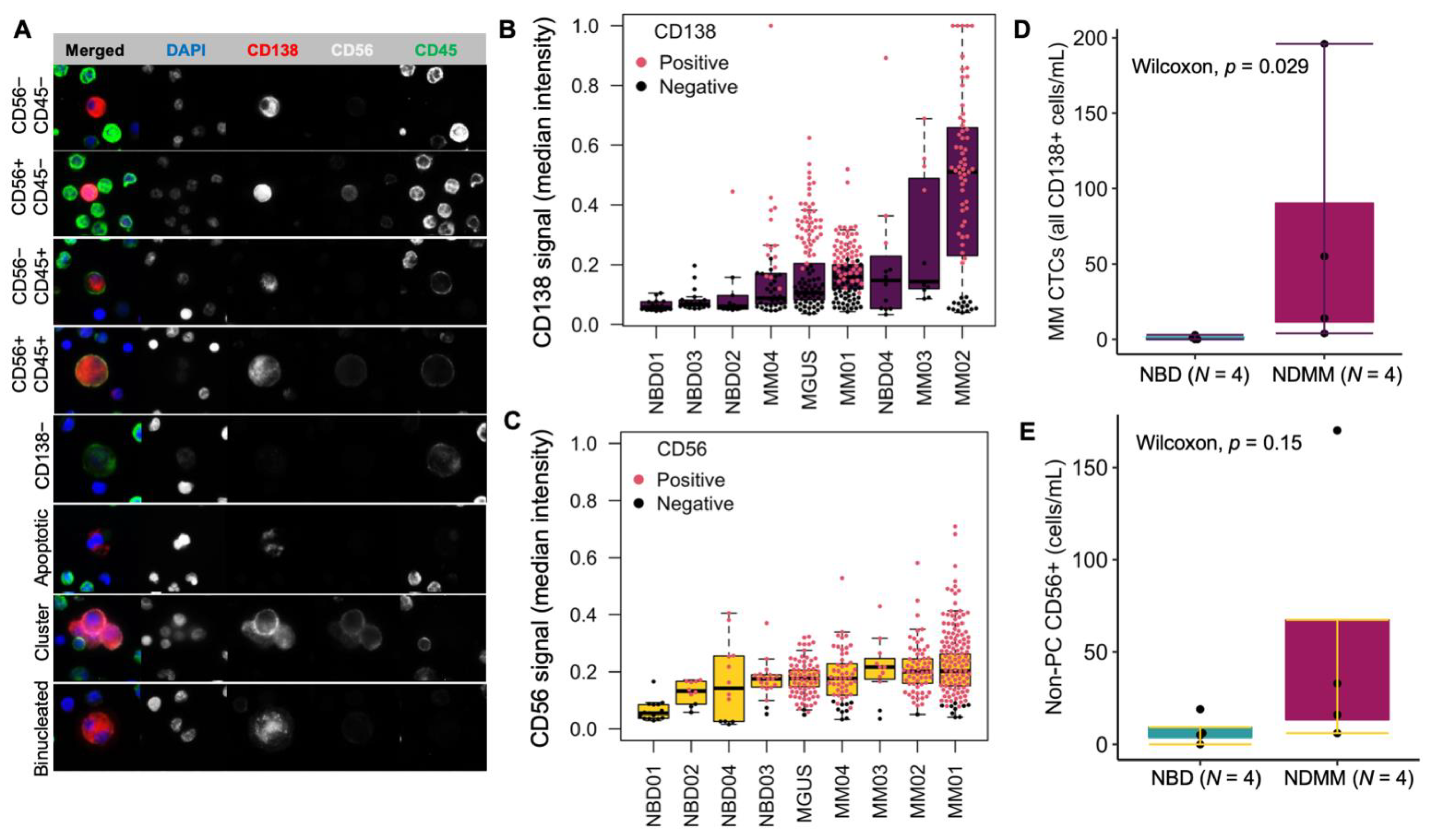
3.1. Expression of CD138 and CD56 in U266, MM.1S, and Jurkat Cells Spiked in Normal Blood
3.2. Patients and Study Cohort
3.3. Morphological Characterization, Classification, and Enumeration of MM CTCs and BMPCs
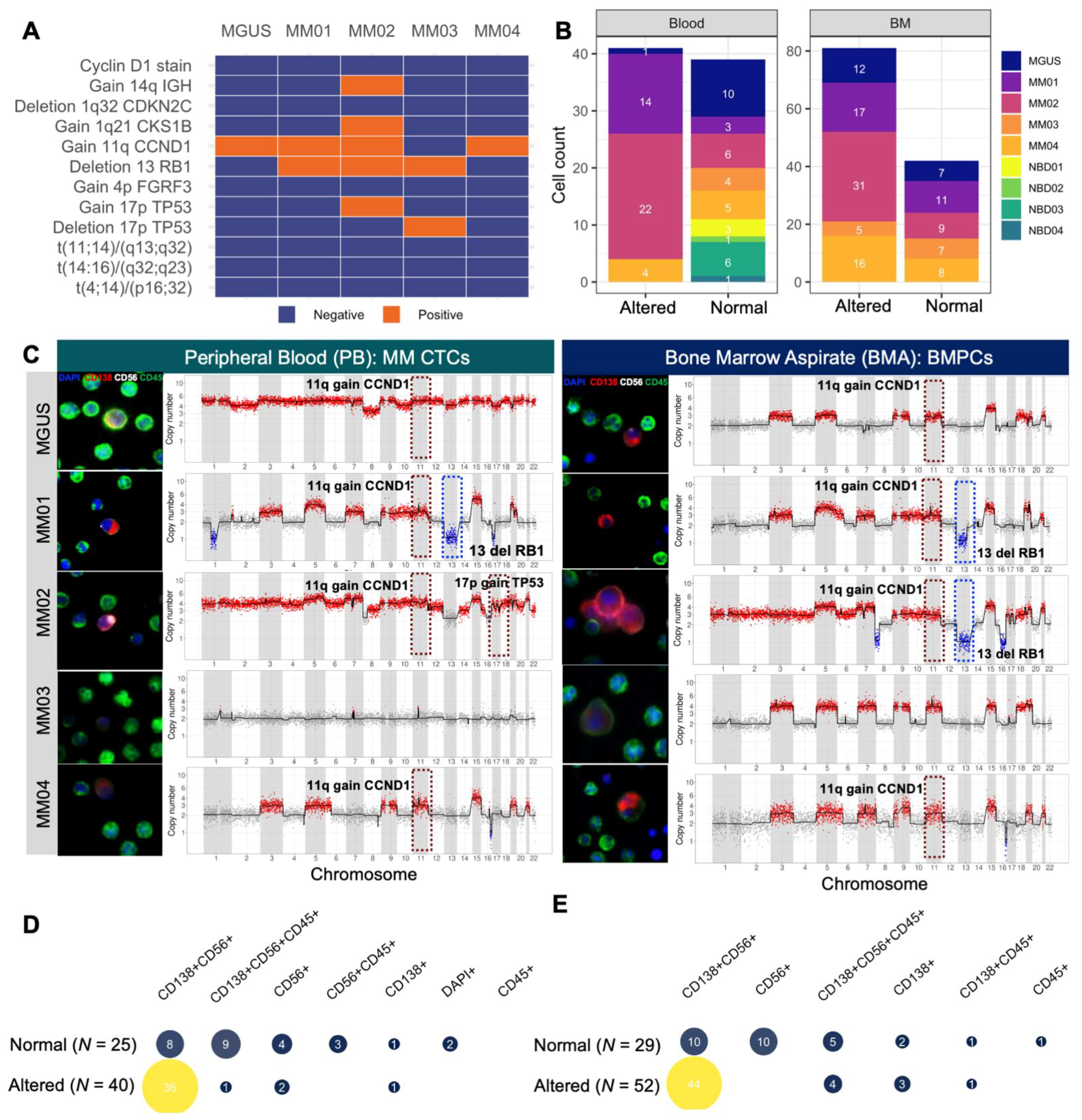
3.4. scCNV for Morphogenomic Validation of Malignant Phenotypes in Detected MM CTCs and BMPCs
3.5. Mapping scCNV Events to FISH Cytogenetics for Clinical Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of Myeloma. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 249–274. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Molecular pathogenesis of multiple myeloma. Int. J. Clin. Oncol. 2015, 20, 413–422. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia 2019, 33, 1713–1722. [Google Scholar] [CrossRef]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. 2020, 476, 337–351. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas—Current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, N.; Rajkumar, S.V.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; et al. Cytogenetic abnormalities in multiple myeloma: Association with disease characteristics and treatment response. Blood Cancer J. 2020, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Cardona-Benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. [Google Scholar] [CrossRef]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Witzig, T.E.; Dingli, D.; Tracz, M.J.; Gertz, M.A.; Lacy, M.Q.; Lust, J.A.; Dispenzieri, A.; Greipp, P.R.; Kyle, R.A.; et al. Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood 2005, 106, 2276–2279. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Morice, W.G.; Rajkumar, V.; Gupta, V.; Timm, M.M.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Singh, P.P.; Kapoor, P.; et al. Quantification of clonal circulating plasma cells in relapsed multiple myeloma. Br. J. Haematol. 2014, 167, 500–505. [Google Scholar] [CrossRef]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef]
- Sanoja-Flores, L.; Flores-Montero, J.; Garcés, J.J.; Paiva, B.; Puig, N.; García-Mateo, A.; García-Sánchez, O.; Corral-Mateos, A.; Burgos, L.; Blanco, E.; et al. Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J. 2018, 8, 117. [Google Scholar] [CrossRef]
- Paiva, B.; Vídriales, M.B.; Rosiñol, L.; Martínez-López, J.; Mateos, M.V.; Ocio, E.M.; Montalbán, M.Á.; Cordón, L.; Gutiérrez, N.C.; Corchete, L.; et al. A multiparameter flow cytometry immunophenotypic algorithm for the identification of newly diagnosed symptomatic myeloma with an MGUS-like signature and long-term disease control. Leukemia 2013, 27, 2056–2061. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Sasser, K.; Rao, C.; Foulk, B.; Gross, S.; Mallon, G.; Erb, C.; Berkowitz, A.; Mangan, P.A.; Shelly, B.K.; et al. Circulating Multiple Myeloma Cells (CMMCs): A Novel Method for Detection and Molecular Characterization of Peripheral Blood Plasma Cells in Multiple Myeloma Precursor States. Blood 2014, 124, 2031. [Google Scholar] [CrossRef]
- Foulk, B.; Schaffer, M.; Gross, S.; Rao, C.; Smirnov, D.; Connelly, M.C.; Chaturvedi, S.; Reddy, M.; Brittingham, G.; Mata, M.; et al. Enumeration and characterization of circulating multiple myeloma cells in patients with plasma cell disorders. Br. J. Haematol. 2018, 180, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Qasaimeh, M.A.; Wu, Y.C.; Bose, S.; Menachery, A.; Talluri, S.; Gonzalez, G.; Fulciniti, M.; Karp, J.M.; Prabhala, R.H.; Karnik, R. Isolation of Circulating Plasma Cells in Multiple Myeloma Using CD138 Antibody-Based Capture in a Microfluidic Device. Sci. Rep. 2017, 7, 45681. [Google Scholar] [CrossRef]
- Zhang, L.; Beasley, S.; Prigozhina, N.L.; Higgins, R.; Ikeda, S.; Lee, F.Y.; Marrinucci, D.; Jia, S. Detection and Characterization of Circulating Tumour Cells in Multiple Myeloma. J. Circ. Biomark. 2016, 5. [Google Scholar] [CrossRef]
- Marrinucci, D.; Bethel, K.; Kolatkar, A.; Luttgen, M.S.; Malchiodi, M.; Baehring, F.; Voigt, K.; Lazar, D.; Nieva, J.; Bazhenova, L.; et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 2012, 9, 016003. [Google Scholar] [CrossRef]
- Ruiz, C.; Li, J.; Luttgen, M.S.; Kolatkar, A.; Kendall, J.T.; Flores, E.; Topp, Z.; Samlowski, W.E.; McClay, E.; Bethel, K.; et al. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys. Biol. 2015, 12, 016008. [Google Scholar] [CrossRef]
- Malihi, P.D.; Morikado, M.; Welter, L.; Liu, S.T.; Miller, E.T.; Cadaneanu, R.M.; Knudsen, B.S.; Lewis, M.S.; Carlsson, A.; Velasco, C.R.; et al. Clonal diversity revealed by morphoproteomic and copy number profiles of single prostate cancer cells at diagnosis. Converg. Sci. Phys. Oncol. 2018, 4, 015003. [Google Scholar] [CrossRef]
- Gerdtsson, E.; Pore, M.; Thiele, J.A.; Gerdtsson, A.S.; Malihi, P.D.; Nevarez, R.; Kolatkar, A.; Velasco, C.R.; Wix, S.; Singh, M.; et al. Multiplex protein detection on circulating tumor cells from liquid biopsies using imaging mass cytometry. Converg. Sci. Phys. Oncol. 2018, 4, 015002. [Google Scholar] [CrossRef]
- Rodríguez-Lee, M.; Kolatkar, A.; McCormick, M.; Dago, A.D.; Kendall, J.; Carlsson, N.A.; Bethel, K.; Greenspan, E.J.; Hwang, S.E.; Waitman, K.R.; et al. Effect of Blood Collection Tube Type and Time to Processing on the Enumeration and High-Content Characterization of Circulating Tumor Cells Using the High-Definition Single-Cell Assay. Arch. Pathol. Lab. Med. 2018, 142, 198–207. [Google Scholar] [CrossRef]
- Shishido, S.N.; Welter, L.; Rodriguez-Lee, M.; Kolatkar, A.; Xu, L.; Ruiz, C.; Gerdtsson, A.S.; Restrepo-Vassalli, S.; Carlsson, A.; Larsen, J.; et al. Preanalytical Variables for the Genomic Assessment of the Cellular and Acellular Fractions of the Liquid Biopsy in a Cohort of Breast Cancer Patients. J. Mol. Diagn. 2020, 22, 319–337. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, F.P.; Pinkus, J.L.; Pinkus, G.S. CD138 (Syndecan-1), a Plasma Cell Marker: Immunohistochemical Profile in Hematopoietic and Nonhematopoietic Neoplasms. Am. J. Clin. Pathol. 2004, 121, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Yang, Y. Syndecan-1: A dynamic regulator of the myeloma microenvironment. Clin. Exp. Metastasis 2008, 25, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Samiee, S.; Yi, Q.L. Prognostic relevance of CD56 expression in multiple myeloma: A study including 107 cases treated with high-dose melphalan-based chemotherapy and autologous stem cell transplant. Leuk. Lymphoma 2006, 47, 43–47. [Google Scholar] [CrossRef]
- Fujino, M. The histopathology of myeloma in the bone marrow. J. Clin. Exp. Hematop. 2018, 58, 61–67. [Google Scholar] [CrossRef]
- Paiva, B.; Almeida, J.; Pérez-Andrés, M.; Mateo, G.; López, A.; Rasillo, A.; Vídriales, M.-B.; López-Berges, M.-C.; Miguel, J.F.S.; Orfao, A. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytom. Part B Clin. Cytom. 2010, 78B, 239–252. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Robillard, N.; Wuillème, S.; Moreau, P.; Béné, M.C. Immunophenotype of Normal and Myelomatous Plasma-Cell Subsets. Front. Immunol. 2014, 5, 137. [Google Scholar] [CrossRef]
- Sanoja-Flores, L.; Flores-Montero, J.; Pérez-Andrés, M.; Puig, N.; Orfao, A. Detection of Circulating Tumor Plasma Cells in Monoclonal Gammopathies: Methods, Pathogenic Role, and Clinical Implications. Cancers 2020, 12, 1499. [Google Scholar] [CrossRef]
- Welter, L.; Xu, L.; McKinley, D.; Dago, A.E.; Prabakar, R.K.; Restrepo-Vassalli, S.; Xu, K.; Rodriguez-Lee, M.; Kolatkar, A.; Nevarez, R.; et al. Treatment response and tumor evolution: Lessons from an extended series of multianalyte liquid biopsies in a metastatic breast cancer patient. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005819. [Google Scholar] [CrossRef]
- Chai, S.; Matsumoto, N.; Storgard, R.; Peng, C.C.; Aparicio, A.; Ormseth, B.; Rappard, K.; Cunningham, K.; Kolatkar, A.; Nevarez, R.; et al. Platelet-Coated Circulating Tumor Cells Are a Predictive Biomarker in Patients with Metastatic Castrate-Resistant Prostate Cancer. Mol. Cancer Res. 2021, 19, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Pau, G.; Fuchs, F.; Sklyar, O.; Boutros, M.; Huber, W. EBImage—an R package for image processing with applications to cellular phenotypes. Bioinformatics 2010, 26, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Dago, A.E.; Stepansky, A.; Carlsson, A.; Luttgen, M.; Kendall, J.; Baslan, T.; Kolatkar, A.; Wigler, M.; Bethel, K.; Gross, M.E.; et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS ONE 2014, 9, e101777. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.A.; Pitule, P.; Hicks, J.; Kuhn, P. Single-Cell Analysis of Circulating Tumor Cells. Methods Mol. Biol. 2019, 1908, 243–264. [Google Scholar] [CrossRef]
- Baslan, T.; Kendall, J.; Ward, B.; Cox, H.; Leotta, A.; Rodgers, L.; Riggs, M.; D’Italia, S.; Sun, G.; Yong, M.; et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015, 25, 714–724. [Google Scholar] [CrossRef]
- Pérez-Persona, E.; Vidriales, M.B.; Mateo, G.; García-Sanz, R.; Mateos, M.V.; de Coca, A.G.; Galende, J.; Martín-Nuñez, G.; Alonso, J.M.; de Las Heras, N.; et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Horst, A.; Hunzelmann, N.; Arce, S.; Herber, M.; Manz, R.A.; Radbruch, A.; Nischt, R.; Schmitz, J.; Assenmacher, M. Detection and characterization of plasma cells in peripheral blood: Correlation of IgE+ plasma cell frequency with IgE serum titre. Clin. Exp. Immunol. 2002, 130, 370–378. [Google Scholar] [CrossRef]
- Caraux, A.; Klein, B.; Paiva, B.; Bret, C.; Schmitz, A.; Fuhler, G.M.; Bos, N.A.; Johnsen, H.E.; Orfao, A.; Perez-Andres, M.; et al. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica 2010, 95, 1016–1020. [Google Scholar] [CrossRef]
- Peterson, J.F.; Rowsey, R.A.; Marcou, C.A.; Pearce, K.E.; Williamson, C.M.; Frederick, L.A.; Greipp, P.T.; Ketterling, R.P.; Kumar, S.; Viswanatha, D.S.; et al. Hyperhaploid plasma cell myeloma characterized by poor outcome and monosomy 17 with frequently co-occurring TP53 mutations. Blood Cancer J. 2019, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Owens, R.; Tricot, G.; Wilson, C.S. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol. 2004, 121, 482–488. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
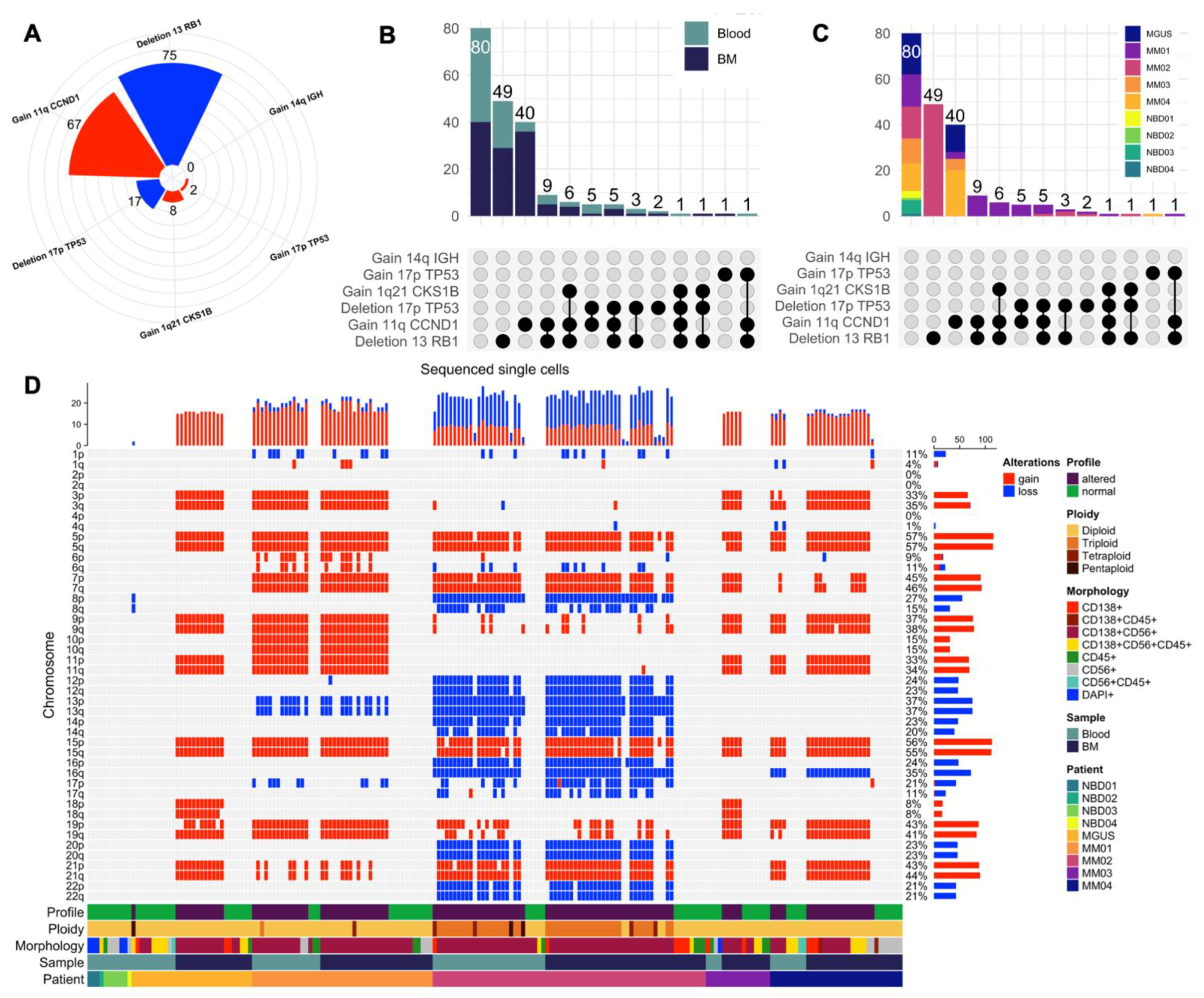{kind=link}
| MGUS | MM01 | MM02 | MM03 | MM04 | |
|---|---|---|---|---|---|
| Age | 78 | 80 | 63 | 54 | 66 |
| Sex | Male | Male | Female | Male | Male |
| Diagnosis | MGUS | NDMM | NDMM | NDMM | NDMM |
| Ig Isotype | IgGk | IgGk | IgGk | IgGk | IgAk |
| Percent BMPC in the aspirate | 1 | 14 | 15 | 30 | 8 |
| Percent Aberrant PC from the total PC BM compartment | 92 | 64.5 | 95.2 | 98.8 | 98.2 |
| Flow CD138 | Positive | Positive | Positive | Positive | Positive |
| Flow CD56 | Positive | Positive | Positive | Negative | Positive |
| Flow CD45 | Positive | Positive | Negative | Positive (dim) | Negative |
| M-Spike (g/dL) | 0.7 | 1.6 | 0.4 | 2.9 | 4.3 |
| sFLC ratio | 8.14 | 93.43 | 186.84 | 17.28 | 6.17 |
| Karyotype | Normal | NA | Normal | Hypodiploid | Normal |
| FISH (Positive) | Three copies of CCND1 | Three copies of CCND1; Monosomy 13 | Three copies of EGFR3 and CCND1; trisomies 1 and 17; monosomy 13 | Monosomies 1, 13, and 17; loss of one copy of IGH | Three copies of CCND1 |
| Clinical Presentation | Low-risk MGUS for progression to MM by PETHEMA [46] criteria | Patient with standard-risk myeloma achieved complete remission after initial therapy with carfilzomib, lenalidomide, dexamethasone | Patient with standard-risk myeloma achieved a partial response after initial therapy with carfilzomib, lenalidomide, dexamethasone | Patient with high-risk myeloma achieved complete remission after therapy with carfilzomib, lenalidomide, dexamethasone but passed away with myeloma progressive disease 21 months after diagnosis | Patient with standard-risk myeloma achieved complete remission after initial therapy with carfilzomib, lenalidomide, dexamethasone |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndacayisaba, L.J.; Rappard, K.E.; Shishido, S.N.; Ruiz Velasco, C.; Matsumoto, N.; Navarez, R.; Tang, G.; Lin, P.; Setayesh, S.M.; Naghdloo, A.; et al. Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones. Curr. Oncol. 2022, 29, 2954-2972. https://doi.org/10.3390/curroncol29050242
Ndacayisaba LJ, Rappard KE, Shishido SN, Ruiz Velasco C, Matsumoto N, Navarez R, Tang G, Lin P, Setayesh SM, Naghdloo A, et al. Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones. Current Oncology. 2022; 29(5):2954-2972. https://doi.org/10.3390/curroncol29050242
Chicago/Turabian StyleNdacayisaba, Libere J., Kate E. Rappard, Stephanie N. Shishido, Carmen Ruiz Velasco, Nicholas Matsumoto, Rafael Navarez, Guilin Tang, Pei Lin, Sonia M. Setayesh, Amin Naghdloo, and et al. 2022. "Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones" Current Oncology 29, no. 5: 2954-2972. https://doi.org/10.3390/curroncol29050242
APA StyleNdacayisaba, L. J., Rappard, K. E., Shishido, S. N., Ruiz Velasco, C., Matsumoto, N., Navarez, R., Tang, G., Lin, P., Setayesh, S. M., Naghdloo, A., Hsu, C.-J., Maney, C., Symer, D., Bethel, K., Kelly, K., Merchant, A., Orlowski, R., Hicks, J., Mason, J., ... Kuhn, P. (2022). Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones. Current Oncology, 29(5), 2954-2972. https://doi.org/10.3390/curroncol29050242