Treating Multiple Myeloma in the Context of the Bone Marrow Microenvironment


Abstract

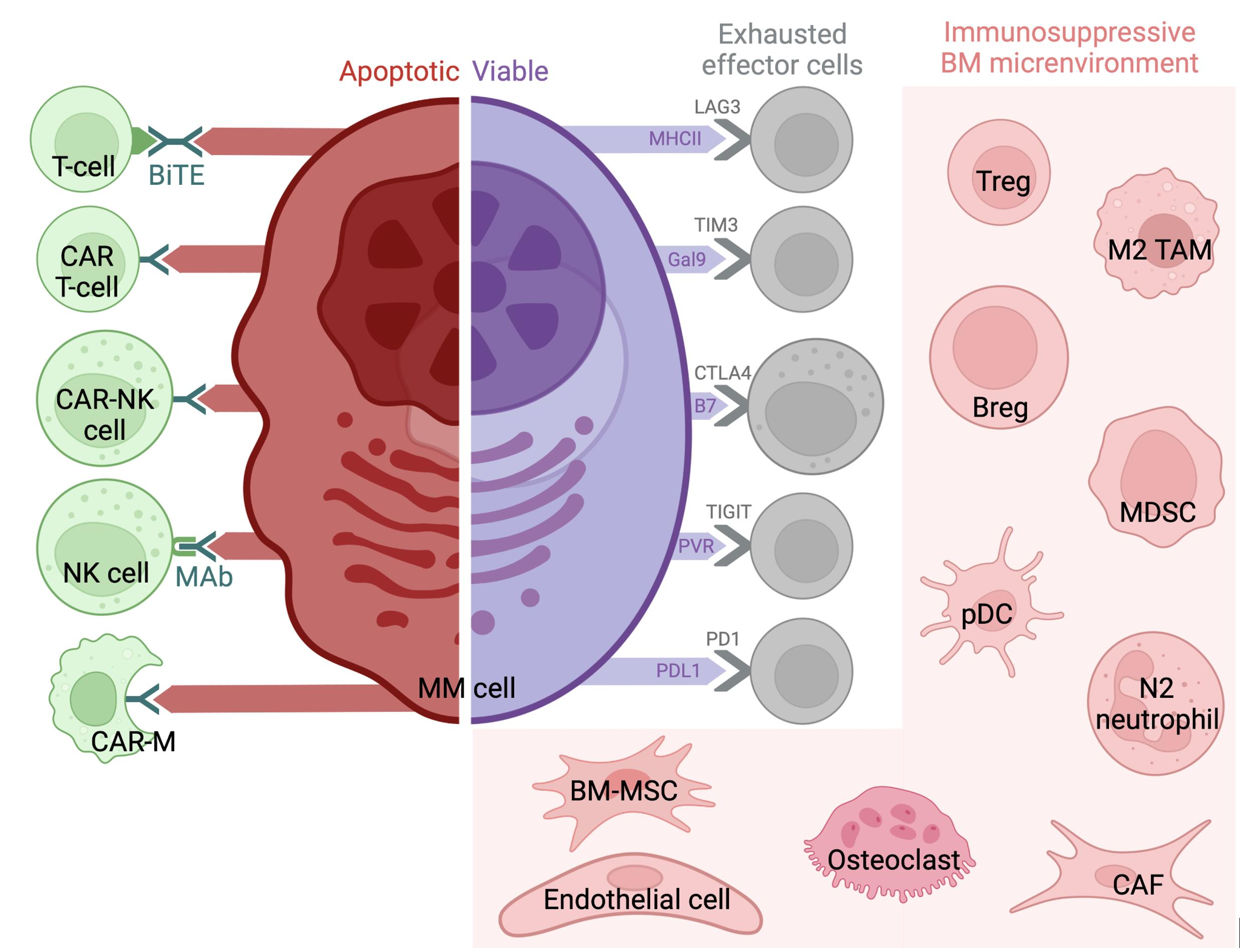
1. Introduction
2. The Role of the Tumor Microenvironment in the Initiation of the Myeloma Clone
3. The Role of the Immunosuppressive Microenvironment in the Progression of MM
3.1. Regulatory T Cells (Tregs)
3.2. Plasmacytoid Dendritic Cells (pDCs)
3.3. Myeloid-Derived Suppressor Cells (MDSCs)
3.4. Cancer-Associated Fibroblasts (CAFs)
3.5. Tumor-Associated M2-like Macrophages (M2 TAMs)
3.6. N2 Neutrophils
3.7. Regulatory B Cells (Bregs)
3.8. Impaired Immune Effector Killing in MM
4. The Era of Immunotherapy in MM
5. Chimeric Antigen Receptor T (CAR T)-Cell Therapy
5.1. Mechanisms of Resistance to CAR T-Cell Therapy
5.2. Exploiting the Tumor Microenvironment to Overcome CAR T-Cell Therapy-Related Toxicities
5.3. Beyond CAR T Cells (CAR-NK and CAR-Macrophages)
5.4. Bispecific T-Cell Engagers (BiTEs)
5.5. Beyond BiTEs (Bispecific Killer Cell Engagers, Trispecific Antibodies, and BsAbs Armed T-Cell Therapy)
6. Monoclonal Antibodies (MAbs)
6.1. CD38 MAbs
6.2. SLAMF7 MAbs
6.3. Antibody Drug Conjugates (ADCs)
6.4. MAbs against Other Targets
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef]
- Braunlin, M.; Belani, R.; Buchanan, J.; Wheeling, T.; Kim, C. Trends in the multiple myeloma treatment landscape and survival: A U.S. analysis using 2011–2019 oncology clinic electronic health record data. Leuk. Lymphoma 2020, 62, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Greil, C.; Engelhardt, M.; Ihorst, G.; Schoeller, K.; Bertz, H.; Marks, R.; Zeiser, R.; Duyster, J.; Einsele, H.; Finke, J.; et al. Allogeneic transplantation of multiple myeloma patients may allow long-term survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica 2018, 104, 370–379. [Google Scholar] [CrossRef]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef]
- Moser-Katz, T.; Joseph, N.S.; Dhodapkar, M.V.; Lee, K.P.; Boise, L.H. Game of Bones: How Myeloma Manipulates Its Microenvironment. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Ho, M.; Goh, C.Y.; Patel, A.; Staunton, S.; O’Connor, R.; Godeau, M.; Bianchi, G. Role of the Bone Marrow Milieu in Multiple Myeloma Progression and Therapeutic Resistance. Clin. Lymphoma Myeloma Leuk. 2020, 20, e752–e768. [Google Scholar] [CrossRef]
- Dhodapkar, M.V. MGUS to myeloma: A mysterious gammopathy of underexplored significance. Blood 2016, 128, 2599–2606. [Google Scholar] [CrossRef]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Barwick, B.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [PubMed]
- Armitage, R.J.; Fanslow, W.C.; Strockbine, L.; Sato, T.A.; Clifford, K.N.; Macduff, B.M.; Anderson, D.M.; Gimpel, S.D.; Davis-Smith, T.; Maliszewski, C.R.; et al. Molecular and biological characterization of a murine ligand for CD40. Nature 1992, 357, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Noelle, R.J.; Roy, M.; Shepherd, D.M.; Stamenkovic, I.; Ledbetter, J.A.; Aruffo, A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. USA 1992, 89, 6550–6554. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Snapper, C.M.; Paul, W.E. Interferon-γ and B Cell Stimulatory Factor-1 Reciprocally Regulate Ig Isotype Production. Science 1987, 236, 944–947. [Google Scholar] [CrossRef]
- Muraguchi, A.; Hirano, T.; Tang, B.; Matsuda, T.; Horii, Y.; Nakajima, K.; Kishimoto, T. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. J. Exp. Med. 1988, 167, 332–344. [Google Scholar] [CrossRef]
- Maeda, K.; Mehta, H.; Drevets, D.A.; Coggeshall, K.M. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood 2010, 115, 4699–4706. [Google Scholar] [CrossRef]
- MacLennan, I.C.M. Germinal Centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Jacob, J.; Kelsoe, G.; Rajewsky, K.; Weiss, U. Intraclonal generation of antibody mutants in germinal centres. Nature 1991, 354, 389–392. [Google Scholar] [CrossRef]
- Menu, E.; Asosingh, K.; Indraccolo, S.; De Raeve, H.; Van Riet, I.; Van Valckenborgh, E.; Van De Broek, I.; Fujii, N.; Tamamura, H.; Van Camp, B.; et al. The involvement of stromal derived factor 1alpha in homing and progression of multiple myeloma in the 5TMM model. Haematologica 2006, 91, 605–612. [Google Scholar] [PubMed]
- Ho, M.; Patel, A.; Goh, C.Y.; Moscvin, M.; Zhang, L.; Bianchi, G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia 2020, 34, 3111–3125. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.; Rihova, L.; Kralova, R.; Penka, M.; Adam, Z.; Pour, L.; Piskacek, M.; Hajek, R. Plasmacytoid Dendritic Cells in Patients with MGUS and Multiple Myeloma. J. Clin. Med. 2021, 10, 3717. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A. How do Regulatory T Cells Work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Feyler, S.; von Lilienfeld-Toal, M.; Jarmin, S.; Marles, L.; Rawstron, A.; Ashcroft, A.J.; Owen, R.G.; Selby, P.J.; Cook, G. CD4+CD25+FoxP3+regulatory T cells are increased whilst CD3+CD4−CD8−αβTCR+Double Negative T cells are decreased in the peripheral blood of patients with multiple myeloma which correlates with disease burden. Br. J. Haematol. 2009, 144, 686–695. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 Is Essential for TGF-β to Convert Naive CD4+CD25− Cells to CD25+Foxp3+ Regulatory T Cells and for Expansion of These Cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Feyler, S.; Scott, G.B.; Parrish, C.; Jarmin, S.; Evans, P.; Short, M.; McKinley, K.; Selby, P.J.; Cook, G. Tumour Cell Generation of Inducible Regulatory T-Cells in Multiple Myeloma Is Contact-Dependent and Antigen-Presenting Cell-Independent. PLoS ONE 2012, 7, e35981. [Google Scholar] [CrossRef]
- Scott, G.B.; Carter, C.; Parrish, C.; Wood, P.M.; Cook, G. Downregulation of myeloma-induced ICOS-L and regulatory T cell generation by lenalidomide and dexamethasone therapy. Cell. Immunol. 2015, 297, 1–9. [Google Scholar] [CrossRef]
- Minnema, M.C.; Van Der Veer, M.S.; Aarts, T.; Emmelot, M.; Mutis, T.; Lokhorst, H.M. Lenalidomide alone or in combination with dexamethasone is highly effective in patients with relapsed multiple myeloma following allogeneic stem cell transplantation and increases the frequency of CD4+Foxp3+ T cells. Leukemia 2008, 23, 605–607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, B.-N.; Gao, H.; Cohen, E.N.; Badoux, X.; Wierda, W.G.; Estrov, Z.; Faderl, S.H.; Keating, M.J.; Ferrajoli, A.; Reuben, J.M.; et al. Treatment with lenalidomide modulates T-cell immunophenotype and cytokine production in patients with chronic lymphocytic leukemia. Cancer 2011, 117, 3999–4008. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Ganeshan, P.; Hakim, M.; Verma, R.; Sharma, A.; Kumar, L. Significantly reduced regulatory T cell population in patients with untreated multiple myeloma. Leuk. Res. 2011, 35, 874–878. [Google Scholar] [CrossRef]
- Raja, K.R.M.; Kovářová, L.; Hajek, R. Induction by lenalidomide and dexamethasone combination increases regulatory cells of patients with previously untreated multiple myeloma. Leuk. Lymphoma 2012, 53, 1406–1408. [Google Scholar] [CrossRef]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2009, 24, 22–32. [Google Scholar] [CrossRef]
- Lutsiak, M.E.C.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.S.; Schlom, J.; Sabzevari, H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Sharabi, A.; Laronne-Bar-On, A.; Meshorer, A.; Haran-Ghera, N. Chemoimmunotherapy Reduces the Progression of Multiple Myeloma in a Mouse Model. Cancer Prev. Res. 2010, 3, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; Van De Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.-T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef]
- Kitadate, A.; Kobayashi, H.; Abe, Y.; Narita, K.; Miura, D.; Takeuchi, M.; Matsue, K. Pre-treatment CD38-positive regulatory T cells affect the durable response to daratumumab in relapsed/refractory multiple myeloma patients. Haematologica 2019, 105, e37–e40. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef]
- Bonanno, G.; Mariotti, A.; Procoli, A.; Folgiero, V.; Natale, D.; De Rosa, L.; Majolino, I.; Novarese, L.; Rocci, A.; Gambella, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity correlates with immune system abnormalities in multiple myeloma. J. Transl. Med. 2012, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.M.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 Axis Converts Human T H 1 Cells into Regulatory T Cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef]
- Matthes, T.; Dunand-Sauthier, I.; Santiago-Raber, M.-L.; Krause, K.-H.; Donze, O.; Passweg, J.; McKee, T.; Huard, B. Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood 2011, 118, 1838–1844. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Lin, L.; Xing, L.; Cho, S.-F.; Yu, T.; Acharya, C.; Wen, K.; Hsieh, P.A.; Dulos, J.; Van Elsas, A.; et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: Therapeutic implications. Leukemia 2018, 33, 426–438. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; Van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Raja, K.R.M.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLOS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: Detailed characterisation of immune status in multiple myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Brimnes, M.K.; Vangsted, A.J.; Knudsen, L.M.; Gimsing, P.; Gang, A.O.; Johnsen, H.E.; Svane, I.M. Increased Level of both CD4+FOXP3+ Regulatory T Cells and CD14+HLA-DR−/low Myeloid-Derived Suppressor Cells and Decreased Level of Dendritic Cells in Patients with Multiple Myeloma. Scand. J. Immunol. 2010, 72, 540–547. [Google Scholar] [CrossRef]
- Beyer, M.; Kochanek, M.; Giese, T.; Endl, E.; Weihrauch, M.R.; Knolle, P.A.; Classen, S.; Schultze, J.L. In vivo peripheral expansion of naive CD4+CD25highFoxP3+ regulatory T cells in patients with multiple myeloma. Blood 2006, 107, 3940–3949. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T regulatory cells in multiple myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, M.; Castella, B.; Mariani, S.; Coscia, M.; Godio, L.; Ferracini, R.; Ruggeri, M.; Muccio, V.; Omedé, P.; Palumbo, A.; et al. The bone marrow of myeloma patients is steadily inhabited by a normal-sized pool of functional regulatory T cells irrespectiveof the disease status. Haematologica 2014, 99, 1605–1610. [Google Scholar] [CrossRef]
- D’Arena, G.; Rossi, G.; Laurenti, L.; Statuto, T.; D’Auria, F.; Valvano, L.; Simeon, V.; Giudice, A.; Innocenti, I.; De Feo, V.; et al. Circulating Regulatory T-Cells in Monoclonal Gammopathies of Uncertain Significance and Multiple Myeloma: In Search of a Role. J. Immunol. Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Lad, D.; Huang, Q.; Hoeppli, R.; Garcia, R.; Xu, L.; Levings, M.; Song, K.; Broady, R. Evaluating the role of Tregs in the progression of multiple myeloma. Leuk. Lymphoma 2019, 60, 2134–2142. [Google Scholar] [CrossRef]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2013, 55, 1090–1098. [Google Scholar] [CrossRef]
- Braga, W.M.T.; Da Silva, B.R.; de Carvalho, A.C.; Maekawa, Y.H.; Bortoluzzo, A.; Gil Rizzatti, E.; Atanackovic, D.; Colleoni, G.W.B. FOXP3 and CTLA4 overexpression in multiple myeloma bone marrow as a sign of accumulation of CD4+ T regulatory cells. Cancer Immunol. Immunother. 2014, 63, 1189–1197. [Google Scholar] [CrossRef]
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.-J.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, V.C.; Khaiboullina, S.F.; Rizvanov, A.A. Plasmacytoid dendritic cells, a role in neoplastic prevention and progression. Eur. J. Clin. Investig. 2014, 45, 1–8. [Google Scholar] [CrossRef]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.-T.; Mitsiades, C.; et al. Functional Interaction of Plasmacytoid Dendritic Cells with Multiple Myeloma Cells: A Therapeutic Target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef]
- Ray, A.; Das, D.S.; Song, Y.; Richardson, P.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Targeting PD1–PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia 2015, 29, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Stocker, N.; Gaugler, B.; Ricard, L.; De Vassoigne, F.; Marjanovic, Z.; Mohty, M.; Malard, F. Daratumumab prevents programmed death ligand-1 expression on antigen-presenting cells in de novo multiple myeloma. Cancer Med. 2020, 9, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Malek, E.; de Lima, M.; Letterio, J.J.; Kim, B.-G.; Finke, J.H.; Driscoll, J.J.; Giralt, S.A. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016, 30, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Wang, H.; Xiong, S.; Li, Y.; Tao, Q.; Xiao, W.; Qin, H.; Wang, Y.; Zhai, Z. Tumor-induced CD14+HLA-DR−/low myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol. Immunother. 2014, 64, 389–399. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Hausladen, A.; Liu, L.; Hess, D.T.; Zeng, M.; Miao, Q.X.; Kane, L.S.; Gow, A.J.; Stamler, J.S. Fas-Induced Caspase Denitrosylation. Science 1999, 284, 651–654. [Google Scholar] [CrossRef]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G.; et al. IL-4-Induced Arginase 1 Suppresses Alloreactive T Cells in Tumor-Bearing Mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Van Ginderachter, J.A.; Lub, S.; De Beule, N.; Thielemans, K.; Bautmans, I.; Oyajobi, B.O.; De Bruyne, E.; Menu, E.; Lemaire, M.; et al. Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget 2015, 6, 10532–10547. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara-Ota, S.; Shimura, Y.; Steinebach, C.; Isa, R.; Yamaguchi, J.; Nishiyama, D.; Fujibayashi, Y.; Takimoto-Shimomura, T.; Mizuno, Y.; Matsumura-Kimoto, Y.; et al. Lenalidomide and pomalidomide potently interfere with induction of myeloid-derived suppressor cells in multiple myeloma. Br. J. Haematol. 2020, 191, 784–795. [Google Scholar] [CrossRef]
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma. Cancers 2014, 6, 1363–1381. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2013, 28, 904–916. [Google Scholar] [CrossRef]
- Sun, J.; Park, C.; Guenthner, N.; Gurley, S.; Zhang, L.; Lubben, B.; Adebayo, O.; Bash, H.; Chen, Y.; Maksimos, M.; et al. Tumor-associated macrophages in multiple myeloma: Advances in biology and therapy. J. Immunother. Cancer 2022, 10, e003975. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2018, 224, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of Wound Healing by Growth Factors and Cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef]
- Ahmad, A. Epigenetic regulation of immunosuppressive tumor-associated macrophages through dysregulated microRNAs. Semin. Cell Dev. Biol. 2021, 124, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Salah, A.; Li, Y.; Wang, H.; Qi, N.; Wu, Y. Macrophages as a Double-Edged Weapon: The Use of Macrophages in Cancer Immunotherapy and Understanding the Cross-Talk Between Macrophages and Cancer. DNA Cell Biol. 2021, 40, 429–440. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells 2019, 8, 1526. [Google Scholar] [CrossRef]
- Ribatti, D.; Moschetta, M.; Vacca, A. Macrophages in multiple myeloma. Immunol. Lett. 2014, 161, 241–244. [Google Scholar] [CrossRef]
- Papadimitriou, K.; Tsakirakis, N.; Malandrakis, P.; Vitsos, P.; Metousis, A.; Orologas-Stavrou, N.; Ntanasis-Stathopoulos, I.; Kanellias, N.; Eleutherakis-Papaiakovou, E.; Pothos, P.; et al. Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers 2020, 12, 3245. [Google Scholar] [CrossRef]
- Wang, H.; Hu, W.-M.; Xia, Z.-J.; Liang, Y.; Lu, Y.; Lin, S.-X.; Tang, H. High numbers of CD163+ tumor-associated macrophages correlate with poor prognosis in multiple myeloma patients receiving bortezomib-based regimens. J. Cancer 2019, 10, 3239–3245. [Google Scholar] [CrossRef]
- Suyanı, E.; Sucak, G.T.; Akyürek, N.; Şahin, S.; Baysal, N.A.; Yağcı, M.; Haznedar, R. Tumor-associated macrophages as a prognostic parameter in multiple myeloma. Ann. Hematol. 2013, 92, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Muz, B.; Alhallak, K.; Park, C.; Lubben, B.; Fiala, M.; Vij, R.; Azab, A.K. P-079: IL10R inhibition reprograms tumor-associated macrophages and reverses drug resistance in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, S82. [Google Scholar] [CrossRef]
- Kim, J.; Denu, R.A.; Dollar, B.A.; Escalante, L.E.; Kuether, J.P.; Callander, N.; Asimakopoulos, F.; Hematti, P. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br. J. Haematol. 2012, 158, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Dong, M.; Liu, X.; Shen, Q.; He, D.; Huang, X.; Zhang, E.; Lin, X.; Chen, Q.; Guo, X.; et al. Multiple myeloma cell-derived IL-32γ increases the immunosuppressive function of macrophages by promoting indoleamine 2,3-dioxygenase (IDO) expression. Cancer Lett. 2019, 446, 38–48. [Google Scholar] [CrossRef]
- Opperman, K.S.; Vandyke, K.; Clark, K.C.; Coulter, E.A.; Hewett, D.R.; Mrozik, K.M.; Schwarz, N.; Evdokiou, A.; Croucher, P.I.; Psaltis, P.J.; et al. Clodronate-Liposome Mediated Macrophage Depletion Abrogates Multiple Myeloma Tumor Establishment In Vivo. Neoplasia 2019, 21, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2015, 66, 157–167. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Y.; Li, R.; Jiang, Y.; Zheng, Y.; Qian, J.; Bi, E.; Zheng, C.; Hou, J.; Wang, S.; et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 2017, 32, 176–183. [Google Scholar] [CrossRef]
- Chen, H.; Li, M.; Sanchez, E.; Soof, C.M.; Bujarski, S.; Ng, N.; Cao, J.; Hekmati, T.; Zahab, B.; Nosrati, J.D.; et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br. J. Haematol. 2019, 188, 283–294. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Romano, A.; Parrinello, N.L.; Simeon, V.; Puglisi, F.; La Cava, P.; Bellofiore, C.; Giallongo, C.; Camiolo, G.; D’Auria, F.; Grieco, V.; et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Petersson, J.; Askman, S.; Pettersson, Å.; Wichert, S.; Hellmark, T.; Johansson, C.M.; Hansson, M. Bone Marrow Neutrophils of Multiple Myeloma Patients Exhibit Myeloid-Derived Suppressor Cell Activity. J. Immunol. Res. 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tai, Y.-T.; Ho, M.; Xing, L.; Chauhan, D.; Gang, A.; Qiu, L.; Anderson, K.C. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017, 7, e547. [Google Scholar] [CrossRef]
- Zou, Z.; Guo, T.; Cui, J.; Zhang, L.; Pan, L. Onset of Regulatory B Cells Occurs at Initial Stage of B Cell Dysfunction in Multiple Myeloma. Blood 2019, 134, 1780. [Google Scholar] [CrossRef]
- Papoutselis, M.; Spanoudakis, E. Navigating the Role of CD1d/Invariant Natural Killer T-cell/Glycolipid Immune Axis in Multiple Myeloma Evolution: Therapeutic Implications. Clin. Lymphoma Myeloma Leuk. 2020, 20, 358–365. [Google Scholar] [CrossRef]
- Mozaffari, F.; Hansson, L.; Kiaii, S.; Ju, X.; Rossmann, E.D.; Rabbani, H.; Mellstedt, H.; Österborg, A. Signalling molecules and cytokine production in T cells of multiple myeloma-increased abnormalities with advancing stage. Br. J. Haematol. 2004, 124, 315–324. [Google Scholar] [CrossRef]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Luetkens, T.; Kröger, N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia 2013, 28, 993–1000. [Google Scholar] [CrossRef]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.K.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2012, 27, 464–472. [Google Scholar] [CrossRef]
- Tan, J.; Chen, S.; Huang, J.; Chen, Y.; Yang, L.; Wang, C.; Zhong, J.; Lu, Y.; Wang, L.; Zhu, K.; et al. Increased exhausted CD8+T cells with programmed death-1, T-cell immunoglobulin and mucin-domain-containing-3 phenotype in patients with multiple myeloma. Asia-Pac. J. Clin. Oncol. 2018, 14, e266–e274. [Google Scholar] [CrossRef]
- Jing, W.; Gershan, J.A.; Weber, J.; Tlomak, D.; McOlash, L.; Sabatos-Peyton, C.; Johnson, B.D. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J. Immunother. Cancer 2015, 3, 2. [Google Scholar] [CrossRef]
- Bae, J.; Accardi, F.; Hideshima, T.; Tai, Y.-T.; Prabhala, R.; Shambley, A.; Wen, K.; Rowell, S.; Richardson, P.G.; Munshi, N.C.; et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia 2021, 36, 138–154. [Google Scholar] [CrossRef]
- Tremblay-Lemay, R.; Rastgoo, N.; Chang, H. Modulating PD-L1 expression in multiple myeloma: An alternative strategy to target the PD-1/PD-L1 pathway. J. Hematol. Oncol. 2018, 11, 46. [Google Scholar] [CrossRef]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Sheykhhasan, M.; Manoochehri, H.; Dama, P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: A review study. Cancer Gene Ther. 2022, 29, 1080–1096. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen–Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Cremer, F.W.; Reme, T.; Raab, M.; Mahtouk, K.; Kaukel, P.; Pantesco, V.; De Vos, J.; Jourdan, E.; Jauch, A.; et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005, 106, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas-Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Campbell, T.B.; Petrocca, F.; Hege, K.; Kaiser, S.; Anderson, K.; et al. Biallelic Loss of BCMA Triggers Resistance to Anti-BCMA CAR T Cell Therapy in Multiple Myeloma. Blood 2020, 136, 14. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Manasanch, E.E.; Lam, B.N.; Vanasse, B.D.; Brudno, J.N.; Maric, M.I.; Rose, M.J.J.; Stetler-Stevenson, M.; Wang, H.-W.; Yuan, C.M.; et al. T Cells Expressing an Anti-B-Cell Maturation Antigen (BCMA) Chimeric Antigen Receptor with a Fully-Human Heavy-Chain-Only Antigen Recognition Domain Induce Remissions in Patients with Relapsed Multiple Myeloma. Blood 2019, 134, 3230. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.I.; Chu, Y.; Berneman, Z.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Steiner, N.; Gunsilius, E. CAR-T cells in multiple myeloma: Current status. memo - Mag. Eur. Med Oncol. 2020, 13, 43–49. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Rodríguez-Lobato, L.G.; Danhof, S.; Sierro-Martínez, B.; Goetz, R.; Sauer, M.; Perez-Simon, J.A.; Einsele, H.; Hudecek, M.; Prommersbe, S. ATRA Augments BCMA Expression on Myeloma Cells and Enhances Recognition By BCMA-CAR T-Cells. Blood 2020, 136, 13–14. [Google Scholar] [CrossRef]
- Van Der Schans, J.J.; Van De Donk, N.W.C.J.; Mutis, T. Dual Targeting to Overcome Current Challenges in Multiple Myeloma CAR T-Cell Treatment. Front. Oncol. 2020, 10, 1362. [Google Scholar] [CrossRef]
- Sakemura, R.; Hefazi, M.; Siegler, E.L.; Cox, M.J.; Larson, D.P.; Hansen, M.J.; Roman, C.M.; Schick, K.J.; Can, I.; Tapper, E.E.; et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022, 139, 3708–3721. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; de Larrea, C.F.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef]
- Dar, H.; Henderson, D.; Padalia, Z.; Porras, A.; Mu, D.; Kyungah, M.; Police, S.; Kalaitzidis, D.; Terrett, J.; Sagert, J. Preclinical Development of CTX120, an Allogeneic CAR-T Cell Targeting Bcma. Blood 2018, 132, 1921. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nahi, H.; Chrobok, M.; Meinke, S.; Gran, C.; Marquardt, N.; Afram, G.; Sutlu, T.; Gilljam, M.; Stellan, B.; Wagner, A.K.; et al. Autologous NK cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep. Med. 2022, 3. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Karvouni, M.; Wagner, A.K.; Baloch, A.H.; Gilljam, M.; Lundqvist, A.; Alici, E. A novel antiCD38-CAR construct increases the in vitro responsiveness of NK cells against Multiple Myeloma cells. Clin. Lymphoma Myeloma Leuk. 2019, 19, e163–e164. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2021, 29, 475–483. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 1–18. [Google Scholar] [CrossRef]
- Subklewe, M. BiTEs better than CAR T cells. Blood Adv. 2021, 5, 607–612. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Raje, N.S.; Costello, C.; Dholaria, B.R.; Solh, M.M.; Levy, M.Y.; Tomasson, M.H.; Dube, H.; Liu, F.; Liao, K.H.; et al. Efficacy and safety of elranatamab (PF-06863135), a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MM). J. Clin. Oncol. 2021, 39, 8006. [Google Scholar] [CrossRef]
- Rodríguez-Otero, P.; D’Souza, A.; Reece, D.E.; van de Donk, N.W.; Chari, A.; Krishnan, A.Y.; Martin, T.G.; Mateos, M.-V.; Morillo, D.; Hurd, D.D.; et al. A novel, immunotherapy-based approach for the treatment of relapsed/refractory multiple myeloma (RRMM): Updated phase 1b results for daratumumab in combination with teclistamab (a BCMA x CD3 bispecific antibody). J. Clin. Oncol. 2022, 40, 8032. [Google Scholar] [CrossRef]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.-X.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor Burden Limits Bispecific Antibody Efficacy through T-cell Exhaustion Averted by Concurrent Cytotoxic Therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hänel, G.; Nixdorf, D.; et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Caraccio, C.; Krishna, S.; Phillips, D.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Gantke, T.; Reusch, U.; Kellner, C.; Ellwanger, K.; Fucek, I.; Weichel, M.; Kerber, A.; Peipp, M.; Treder, M. AFM26 is a novel, highly potent BCMA/CD16A-directed bispecific antibody for high affinity NK-cell engagement in multiple myeloma. J. Clin. Oncol. 2017, 35, 8045. [Google Scholar] [CrossRef]
- Watkins-Yoon, J.; Guzman, W.; Oliphant, A.; Haserlat, S.; Leung, A.; Chottin, C.; Ophir, M.; Vekeria, J.; Nelson, A.P.; Frye, Z.; et al. CTX-8573, an Innate-Cell Engager Targeting BCMA, is a Highly Potent Multispecific Antibody for the Treatment of Multiple Myeloma. Blood 2019, 134, 3182. [Google Scholar] [CrossRef]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef]
- Lancman, G.; Sastow, D.L.; Cho, H.J.; Jagannath, S.; Madduri, D.; Parekh, S.S.; Richard, S.; Richter, J.; Sanchez, L.; Chari, A. Bispecific Antibodies in Multiple Myeloma: Present and Future. Blood Cancer Discov. 2021, 2, 423–433. [Google Scholar] [CrossRef]
- Gantke, T.; Weichel, M.; Herbrecht, C.; Reusch, U.; Ellwanger, K.; Fucek, I.; Eser, M.; Müller, T.; Griep, R.; Molkenthin, V.; et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng. Des. Sel. 2017, 30, 673–684. [Google Scholar] [CrossRef]
- Park, A.J.; Santich, B.H.; Xu, H.; Lum, L.G.; Cheung, N.-K.V. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J. Immunother. Cancer 2021, 9, e002222. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Wang, Y.; Siegel, D.S.; Wang, M.L. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.; Themeli, M.; Korlaar, R.D.J.; Ruiter, R.W.; Poddighe, P.J.; Yuan, H.; De Bruijn, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica 2018, 104, e100–e103. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Jansen, J.H.M.; Nederend, M.; Van Bueren, J.J.L.; Groen, R.; Parren, P.W.H.I.; Leusen, J.H.W.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor–Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo–Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef]
- Vitkon, R.; Netanely, D.; Levi, S.; Ziv-Baran, T.; Ben-Yzak, R.; Katz, B.-Z.; Benyamini, N.; Trestman, S.; Mittelman, M.; Cohen, Y.; et al. Daratumumab in combination with proteasome inhibitors, rapidly decreases polyclonal immunoglobulins and increases infection risk among relapsed multiple myeloma patients: A single center retrospective study. Ther. Adv. Hematol. 2021, 12. [Google Scholar] [CrossRef]
- Terpos, E.; Gavriatopoulou, M.; Ntanasis-Stathopoulos, I.; Briasoulis, A.; Gumeni, S.; Malandrakis, P.; Fotiou, D.; Papanagnou, E.-D.; Migkou, M.; Theodorakakou, F.; et al. The neutralizing antibody response post COVID-19 vaccination in patients with myeloma is highly dependent on the type of anti-myeloma treatment. Blood Cancer J. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Sonneveld, P.; Hungria, V.; Nooka, A.K.; Estell, J.A.; Barreto, W.; Corradini, P.; Min, C.-K.; Medvedova, E.; Weisel, K.; et al. Daratumumab, Bortezomib, and Dexamethasone Versus Bortezomib and Dexamethasone in Patients With Previously Treated Multiple Myeloma: Three-year Follow-up of CASTOR. Clin. Lymphoma Myeloma Leuk. 2019, 20, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lúcio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef]
- Van Bueren, J.L.; Jakobs, D.; Kaldenhoven, N.; Roza, M.; Hiddingh, S.; Meesters, J.; Voorhorst, M.; Gresnigt, E.; Wiegman, L.; Buijsse, A.O.; et al. Direct in Vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood 2014, 124, 3474. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.R.; An, G.; Zhong, M.; Feng, X.; Wang, L.; DaSilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2015, 30, 399–408. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.-F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.; Efebera, Y.A.; Kwon, H.; Starling, G.; Ciarlariello, D.; Bhaskar, S.S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Pazina, T.; James, A.M.; Colby, K.B.; Yang, Y.; Gale, A.; Jhatakia, A.; Kearney, A.Y.; Graziano, R.F.; Bezman, N.A.; Robbins, M.D.; et al. Enhanced SLAMF7 Homotypic Interactions by Elotuzumab Improves NK Cell Killing of Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 1633–1646. [Google Scholar] [CrossRef]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer Ther. 2018, 17, 1454–1463. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a Potential New Therapeutic Antibody Target for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front. Immunol. 2018, 9, 2551. [Google Scholar] [CrossRef]
- Awwad, M.H.S.; Mahmoud, A.; Bruns, H.; Echchannaoui, H.; Kriegsmann, K.; Lutz, R.; Raab, M.S.; Bertsch, U.; Munder, M.; Jauch, A.; et al. Selective elimination of immunosuppressive T cells in patients with multiple myeloma. Leukemia 2021, 35, 2602–2615. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Hori, M.; Iha, H.; Toyama-Sorimachi, N.; Hagiwara, S.; Kuroda, Y.; Koyama, D.; Izumi, T.; Yasui, H.; Suzuki, A.; et al. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia 2019, 34, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; Leblanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Chung, C. Role of Immunotherapy in Targeting the Bone Marrow Microenvironment in Multiple Myeloma: An Evolving Therapeutic Strategy. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 129–143. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2019, 21, 207–221. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Ailawadhi, S.; Niesvizky, R.; Wolf, J.L.; Zildjian, S.H.; O’Leary, J.; Chanan-Khan, A. Phase I Study of Lorvotuzumab Mertansine (IMGN901) In Combination with Lenalidomide and Dexamethasone In Patients with CD56-Positive Relapsed or Relapsed/Refractory Mulitple Myeloma - A Preliminary Safety and Efficacy Analysis of the Combination. Blood 2010, 116, 1934. [Google Scholar] [CrossRef]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 1–17. [Google Scholar] [CrossRef]
- Kelly, K.R.; Siegel, D.S.; Chanan-Khan, A.A.; Somlo, G.; Heffner, L.T.; Jagannath, S.; Zimmerman, T.; Munshi, N.C.; Madan, S.; Mohrbacher, A.; et al. Indatuximab Ravtansine (BT062) in Combination with Low-Dose Dexamethasone and Lenalidomide or Pomalidomide: Clinical Activity in Patients with Relapsed / Refractory Multiple Myeloma. Blood 2016, 128, 4486. [Google Scholar] [CrossRef]
- Iida, S.; Ogiya, D.; Abe, Y.; Taniwaki, M.; Asou, H.; Maeda, K.; Uenaka, K.; Nagaoka, S.; Ishiki, T.; Conti, I.; et al. Dose-escalation study of tabalumab with bortezomib and dexamethasone in Japanese patients with multiple myeloma. Cancer Sci. 2016, 107, 1281–1289. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Manges, R.F.; Sonneveld, P.; Jagannath, S.; Somlo, G.; Krishnan, A.; Lentzsch, S.; Frank, R.C.; Zweegman, S.; Wijermans, P.W.; et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 161, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.; E Gutierrez, M.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-McKiver, M.; Farsaci, B.; Zhu, M.L.; Lesokhin, A.M.; et al. A Phase 1 Study of Nivolumab in Combination with Ipilimumab for Relapsed or Refractory Hematologic Malignancies (CheckMate 039). Blood 2016, 128, 183. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Cho, H.J.; Costa, L.J.; Davies, F.E.; Neparidze, N.; Vij, R.; Feng, Y.; Teterina, A.; Fritsch, E.W.; Wenger, M.; Kaufman, J.L. Atezolizumab in Combination with Daratumumab with or without Lenalidomide or Pomalidomide: A Phase Ib Study in Patients with Multiple Myeloma. Blood 2018, 132, 597. [Google Scholar] [CrossRef]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; McKiver, M.P.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2020, 35, 777–786. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Product (Trial Name) | Target (Costim Domain) | Phase | Study Population | Response and Outcomes | Grade ≥3 CRS (Grade ≥3 NTX) | PMID (NCT) |
|---|---|---|---|---|---|---|
| FDA approved | ||||||
| Ide-cel (KarMMa) | BCMA (4-1BB) | 2; FDA approved | RRMM | ORR: 73%; mPFS: 12.1 months at 450 × 106 target dose | 6% (3%) | PMID: 33626253 (NCT03361748) |
| Cilta-cel (CARTITUDE-1) | BCMA (4-1BB) | 1b/2; FDA approved | RRMM | ORR: 97%; mPFS and mOS not reached | 5.1% (12.3%) | PMID: 35658469 (NCT04133636) |
| BCMA CAR T-cell trials with reported data | ||||||
| CAR-BCMA first-in-human trial | BCMA (CD28) | 1 | RRMM | ORR: 81%; mPFS: 7.8 months | 25% (4%) | PMID: 27412889 (NCT02215967) |
| CART-BCMA | BCMA (4-1BB) | 1 | RRMM | ORR: 48%; mPFS: 2.7 months | 32% (12%) | PMID: 30896447 (NCT02546167) |
| Ide-cel + PI3K inhibitor (CRB-402) | BCMA (4-1BB) | 1 | RRMM | ORR: 55% (≥CR: 18%) | 4.3% (6.5%) | (NCT03274219) |
| Cilta-cel (CARTITUDE-2) | BCMA (4-1BB) | 2 | RRMM and NDMM | ORR: 88.9% (≥ CR: 27.8%) | NA | (NCT4133636) |
| Zevor-cel (LUMMICAR-1) | BCMA (4-1BB) | 1 | RRMM | ORR: 87.5%; mDOR: 21.8 months | 0% (0%) | (NCT03975907) |
| Zevor-cel (LUMMICAR-2) | BCMA (4-1BB) | 1b/2 | RRMM | ORR: 100% | 0% (0%) | (NCT03915184) |
| Zevor-cel | BCMA (4-1BB) | 1 | RRMM | ORR: 87.5%; mPFS not reached | 0% (4%) | (NCT03302403) (NCT03716856) (NCT03380039) |
| C-CAR088 | BCMA (4-1BB) | 1 | RRMM | ORR: 95.2%; mPFS not reached | 5% (0%) | (NCT04322292) (NCT03815383) (NCT03751293) (NCT04295018) |
| Single-domain antibody CAR | BCMA (4-1BB) | 1 | RRMM | ORR: 88.2%; mPFS 12.1 months; | 2.9% (0%) | (NCT03661554) |
| FCARH143 | BCMA (4-1BB) | 1 | RRMM | ORR: 100%; mPFS not reached | 0% (0%) | (NCT03338972) |
| Orva-cel (EVOLVE) | BCMA (4-1BB) | 1/2 | RRMM | ORR: 91%; mPFS not reached | 2% (4%) | (NCT03430011) |
| MCARH171 | BCMA (4-1BB) | 1 | RRMM | ORR: 64%; mPFS not reached | 20% (0%) | (NCT03070327) |
| FHVH33 | BCMA (4-1BB) | 1 | RRMM | ORR: 80% | 7% | (NCT03602612) |
| LCAR-B38M | BCMA (4-1BB) | 1 | RRMM | ORR: 88.2%; 12-month PFS: 52.9% | 41.2% (0%) | (NCT03090660) |
| LCAR-B38M (LEGEND-2) | BCMA (4-1BB) | 1/2 | RRMM | ORR: 88%; mPFS: 15 months | 7% (0%) | PMID: 30572922 (NCT03090659) |
| tEGFR suicide switch CAR | BCMA (4-1BB) | 1 | RRMM | ORR: 86%; mPFS not reached | 0% (0%) | (NCT03093168) |
| KITE-585 | BCMA (CD28) | 1 | RRMM | ORR: 100%; mPFS 1 month at 1 × 109 dose | 0% (0%) | PMID: 34249462 (NCT03318861) |
| Equecabtagene Autoleucel (FUMANBA-1) | BCMA | 1/2 | RRMM | ORR: 94.9% (≥CR: 68.4%) | 0% (0%) | ChiCTR1800018137 (NCT05066646) |
| P-BCMA-101 (PRIME) | BCMA (4-1BB) | 1/2 | RRMM | ORR: 57%; mPFS not reached | 2% (0%) | (NCT03288493) |
| CTL119 | BCMA, CD19 (4-1BB) | 1 | High-risk MM | ORR: 80% | 0% | (NCT03549442) |
| CART-19/BCMA (cocktail) | BCMA, CD19 (OX40/CD28) | 1/2 | High-risk NDMM | ORR: 100%; mPFS not reached | 0% (0%) | PMID: 35114022 (NCT03455972) |
| SZ-MM-CART01 | BCMA, CD19, CD138 (OX40/CD28) | 1 | RRMM | ORR: 100% | 34% | (NCT03196414) |
| ARC-101 | BCMA (4-1BB) | 1 | RRMM | ORR: 100% | 4.8% (0%) | (NCT04155749) |
| ALLO-715 | BCMA | 1 | RRMM | ORR: 60% in 320 × 106 dose | 0% (0%) | (NCT04093596) |
| CAR T cells against other targets | ||||||
| CTL019 | CD19 (4-1BB) | 1 | RRMM | ORR: 80% | 0% (0%) | PMID: 29669947 (NCT02135406) |
| Tisa-cel | CD19 | 2 | RRMM | NA | NA | (NCT02794246) |
| Various | SLAMF7 | 1/2 | RRMM | NA | NA | (NCT03710421) (NCT04142619) (NCT04541368) (NCT03958656) (NCT04499339) |
| MCARH109 | GPRC5D | 1 | RRMM | ORR: 71%; mPFS not reached | 6% (6%) | PMID: 36170501 (NCT04555551) |
| Various | GPRC5D | 1 | RRMM | NA | NA | (NCT05219721) (NCT05016778) |
| CAR T-138 | CD138 (4-1BB) | 1/2 | RRMM | ORR: 80% | 0% (0%) | (NCT01886976) |
| ALTCAR.CD138 | CD138 | 1 | RRMM | NA | NA | (NCT03672318) |
| CAR2 Anti-CD38 A2 CAR T Cells | CD38 | 1 | RRMM | NA | NA | (NCT03464916) |
| CM-CS1 T cell infusion | NKG2D (Dap10) | 1 | RRMM | ORR: 0% | 0% (0%) | PMID: 30396908 (NCT02203825) |
| NKR-2 (CYAD-01) (THINK) | NKG2D | 1/2 | RRMM | ORR: 0% | NA | (NCT03018405) |
| CAR T-TnMUC1 | Tn-MUC1 | 1 | RRMM | NA | NA | (NCT04025216) |
| MLM-CAR44.1 T cells | CD44v6 | 1/2 | RRMM | NA | NA | (NCT04097301) |
| OPC-415 | MMG49 | 1/2 | RRMM | NA | NA | (NCT04649073) |
| Dual-Targeting CAR T cells | ||||||
| GC012F | BCMA and CD19 | 1 | RRMM | ORR: 93.8% at 3 × 105/kg dose | 7.1% | (NCT04236011) (NCT04182581) |
| Various | BCMA and CD19 | 1 | RRMM | NA | NA | (NCT04412889) (NCT04795882) (NCT04162353) (NCT03706547) (NCT04714827) |
| AUTO2 | BCMA and TACI | 1/2 | RRMM | ORR: 43% | 0% (0%) | (NCT03287804) |
| Various | BCMA and TACI | 1 | RRMM | NA | NA | (NCT05020444) (NCT04657861) |
| Various | BCMA and SLAMF7 | 1 | RRMM | NA | NA | (NCT04662099) (NCT04156269) |
| NA | BCMA and CD38 | 1/2 | RRMM | NA | NA | (NCT03767751) |
| Product (Trial Name) | Target | Phase | Study Population | Response and Outcomes | Grade ≥3 CRS (Grade ≥3 NTX) | PMID (NCT) |
|---|---|---|---|---|---|---|
| BCMA BiTE trials with reported data | ||||||
| Elranatamab (MagnetisMM-1) | BCMA | 1 | RRMM | ORR:75%; mPFS: not published | 0% (0%) | (NCT03269136) |
| Teclistamab or talquetamab + daratumumab (TRIMM-2) | BCMA or GPRC5D | 1 | RRMM | ORR: 78%; mPFS: not published | 0% (0%) | (NCT04108195) |
| Pacanalotamab | BCMA | 1 | RRMM | ORR 70; mPFS: 23.5 | 2% (4%) | PMID: 31895611 (NCT02514239) |
| Alnuctamab | BCMA | 1 | RRMM | ORR 83%; mPFS: not published | 5% (0%) | NCT03486067 |
| TNB-383B | BCMA | 1 | RRMM | ORR 52%; mPFS: not published | 0% (0%) | NCT03933735 |
| REGN5458 (LINKER-MM1) | BCMA | 1/2 | RRMM | ORR 60%; mPFS: not published | 0% (0%) | NCT03761108 |
| Pavurutamab + pomalidomide (ParadigMM-1B) | BCMA | 1/2 | RRMM | ORR 82%; mPFS: not published | 7% (0%) | NCT03287908 |
| Teclistamab (MajesTEC-1) | BCMA | 2 | RRMM | ORR 65%; mPFS: not published | 0% (0%) | NCT04557098 |
| Non-BCMA BiTE trials | ||||||
| Talquetamab (MonumenTAL-1) | GPRC5D | 1 | RRMM | OR: 63%; mPFS not Published | 4% (0%) | NCT03399799 |
| Talquetamab + anticancer drugs (MonumenTAL-2) | GPRC5D | 1 | RRMM | NA | NA | NCT05050097 |
| Talquetamab | GPRC5D | 1 | RRMM | NA | NA | NCT04773522 |
| Talquetamab | GPRC5D | 2 | RRMM | NA | NA | NCT04634552 |
| Blinatumomab | CD19 | 1 | RRMM | NA | NA | NCT03173430 |
| AMG 424 | CD38 | 1 | RRMM | NA | NA | NCT03445663 |
| Y150 | CD38 | 1 | RRMM | NA | NA | NCT05011097 |
| GBR1342 | CD38 | 1 | RRMM | NA | NA | NCT03309111 |
| Cevostamab (CAMMA 1) | FcRH5 | 1 | RRMM | NA | NA | NCT04910568 |
| Cevostamab | FcRH5 | 1 | RRMM | ORR: 52%; mPFS not reached | 2% (0%) | NCT03275103 |
| Trial Name | Intervention | Study Population | Median Progression Free Survival (Months) | Overall Response Rate (Percent) | Median Overall Survival |
|---|---|---|---|---|---|
| Daratumumab for Treatment of Relapsed or Refractory Multiple Myeloma | |||||
| SIRIUS | Dara monotherapy | ≥2–3 lines of therapy including PI and IMiD | 3.7 | 29.2 | 1-year: 65% |
| CASTOR | Dara-Vd versus Vd | ≥1 lines of therapy with response and progression | 16.7 versus 7.1 | 83.0 versus 63.0 | NR (at 3 years: 61% vs. 51%) |
| POLLUX | Dara-Rd versus Rd | ≥1 lines of therapy with response and progression | 44.5 versus 17.5 | 92.9 versus 76.4 | NR (at 42 months: 65% vs. 57%) |
| APOLLO | Dara-Pd versus Pd | ≥1 lines of therapy including lenalidomide and a proteasome inhibitor | 12.4 versus 6.9 | N/A (at 16.9 months 69 versus 46) | Overall survival data were not mature |
| CANDOR | Dara-Kd versus Kd | 1–3 lines of prior therapy | NR versus 15.8 | 84.3 versus 74.7 | NR |
| Daratumumab for Treatment of Newly Diagnosed Multiple Myeloma | |||||
| MAIA | Dara-Rd versus Rd | Newly diagnosed transplant-ineligible multiple myeloma | NR versus 33.8 | 92.9 versus 81.3 | NR |
| ALCYONE | Dara-VMP versus VMP | Newly diagnosed transplant-ineligible multiple myeloma | 36.9 versus 19.3 | 90.9 versus 73.9 | NR |
| CASSIOPEIA | Dara-VTd versus VTd | Newly diagnosed transplant eligible multiple myeloma | NR vs. 46.7 months | sCR 29 vs 20 | NR |
| GRIFFIN | Dara-VRd versus VRd | Newly diagnosed transplant eligible multiple myeloma | NR (at 24 months: 95.8 vs. 89.8) | sCR 42 vs 34 | Overall survival data were not mature |
| Trial Name | Intervention | Study Population | Median Progression Free Survival (Months) | Overall Response Rate (Percent) | Median Overall Survival |
|---|---|---|---|---|---|
| ICARIA-MM | IPd versus Pd | ≥2 prior lines including IMid and PI | 11.5 vs. 6.5 | 60.4 versus 35.3 | At 12 months: 72% versus 63% |
| IKEMA | IKd versus Kd | 1–3 lines of prior therapy, no prior carfilzomib | NR vs. 19.2 | 87 versus 83 | Overall survival data were not mature |
| Trial Name | Intervention | Study Population | Median Progression Free Survival (Months) | Overall Response Rate (Percent) | Median Overall Survival |
|---|---|---|---|---|---|
| ELOQUENT-2 | ERd versus Rd | ≥1 lines of therapy | 19.4 vs. 14.9 | 79% vs. 66% | NR |
| ELOQUENT-3 | EPd versus Pd | ≥2 lines of therapy therapies, including lenalidomide and a proteasome inhibitor | 10.25 vs. 4.67 | 53.3% vs. 26.3% | NR |
| Trial Name | Intervention | Study Population | Median Progression Free Survival (Months) | Overall Response Rate (Percent) | Median Overall Survival |
|---|---|---|---|---|---|
| Dreamm-2 | Belantamab | ≥4 prior therapies, including an anti-CD38 monoclonal antibody, a proteasome inhibitor, and an immunomodulatory agent | 5.7 | 31 | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, M.; Xiao, A.; Yi, D.; Zanwar, S.; Bianchi, G. Treating Multiple Myeloma in the Context of the Bone Marrow Microenvironment. Curr. Oncol. 2022, 29, 8975-9005. https://doi.org/10.3390/curroncol29110705
Ho M, Xiao A, Yi D, Zanwar S, Bianchi G. Treating Multiple Myeloma in the Context of the Bone Marrow Microenvironment. Current Oncology. 2022; 29(11):8975-9005. https://doi.org/10.3390/curroncol29110705
Chicago/Turabian StyleHo, Matthew, Alexander Xiao, Dongni Yi, Saurabh Zanwar, and Giada Bianchi. 2022. "Treating Multiple Myeloma in the Context of the Bone Marrow Microenvironment" Current Oncology 29, no. 11: 8975-9005. https://doi.org/10.3390/curroncol29110705
APA StyleHo, M., Xiao, A., Yi, D., Zanwar, S., & Bianchi, G. (2022). Treating Multiple Myeloma in the Context of the Bone Marrow Microenvironment. Current Oncology, 29(11), 8975-9005. https://doi.org/10.3390/curroncol29110705