Abstract
The prognosis of patients with multiple myeloma (MM) has improved dramatically with the introduction of new therapeutic drugs, but the disease eventually becomes drug-resistant, following an intractable and incurable course. A myeloma niche (MM niche) develops in the bone marrow microenvironment and plays an important role in the drug resistance mechanism of MM. In particular, adhesion between MM cells and bone marrow stromal cells mediated by adhesion molecules induces cell adhesion-mediated drug resistance (CAM-DR). Analyses of the role of mitochondria in cancer cells, including MM cells, has revealed that the mechanism leading to drug resistance involves exchange of mitochondria between cells (mitochondrial transfer) via tunneling nanotubes (TNTs) within the MM niche. Here, we describe the discovery of these drug resistance mechanisms and the identification of promising therapeutic agents primarily targeting CAM-DR, mitochondrial transfer, and TNTs.
1. Introduction
Multiple myeloma (MM) is a B-cell hematologic malignancy characterized by abnormal proliferation of plasma cells in the bone marrow microenvironment (BMM), monoclonal protein (M protein), hypercalcemia, renal dysfunction, anemia, and lytic bone lesions [1,2]. MM accounts for approximately 10% of newly diagnosed hematological malignancies [3]. With the advent of innovative analytical technologies in the 2000s, research to elucidate the molecular pathology of MM has intensified [4]. This has led to the rapid development and clinical introduction of many novel molecular-targeted therapies, including proteasome inhibitors (PI), immunomodulatory drugs, and immunotherapies, which have dramatically improved the prognosis of patients with MM [5]. However, relapse is highly likely in almost all MM patients; thus, there is an urgent need to develop next-generation therapeutic agents that could cure MM [6].
MM develops a myeloma niche (MM niche) in the BMM. Data clearly indicate that MM cells modify the microenvironment to facilitate their survival. Research also indicates that the BMM plays an important role in the drug resistance mechanism of MM. In particular, adhesion between MM cells and BM stromal cells mediated by adhesion molecules induces cell adhesion-mediated drug resistance (CAM-DR) [7]. Analyses of the role of mitochondria in cancer cells, including MM cells, has revealed that the mechanism leading to drug resistance involves the exchange of mitochondria between cells (mitochondrial transfer) via tunneling nanotubes (TNTs) within the MM niche [8,9,10,11]. Elucidation of these mechanisms has facilitated the development of many therapeutic agents targeting CAM-DR, mitochondrial transfer, and TNTs, and it is expected that more will be developed in the future.
The purpose of this review is to summarize research findings regarding the MM niche, CAM-DR, and TNTs and discuss the treatment methods targeting these mechanisms that are currently under development or have been clinically applied.
2. Hematopoietic Stem Cell (HSC) Niche and MM Niche
Various pathways and cell types have been shown to tightly control the self-renewal, proliferation, and differentiation properties of HSCs in normal hematopoiesis and development of the HSC niche [12,13,14]. In particular, the HSC niche consists of a cellular component (hematopoietic and nonhematopoietic or stromal cells, such as osteoblasts, osteoclasts, fibroblasts, adipocytes, myocytes, endothelial cells, lymphocytes, dendritic cells, and macrophages), extracellular matrix (several types of collagen, laminin, fibronectin, thrombospondin, proteoglycans, and hemonectin), and a soluble component (cytokines, growth factors, and soluble isoforms of cell adhesion molecules (e.g., serum vascular cell adhesion protein 1, serum intercellular adhesion molecule 1, sP-selectin, and sE-selectin)), and it undergoes appropriate remodeling by osteoclasts [15,16,17,18,19]. Importantly, cancer cells, including MM cells, have been shown to engraft in the endosteal HSC niche, invade bone, and induce tumor expansion and metastatic disease [12,16,20,21,22]. Especially in terms of MM, research clearly shows that osteoclasts, vascular endothelial cells, and BM stromal cells play important roles in creating the suitable environment for MM cells referred to as the MM niche [23]. In the MM niche, MM cells alter the normal HSC niche and induce the expression of specific cytokines and growth factors that promote their survival, growth, and drug resistance [12].
Interactions between MM cells and the MM niche, either directly through cell adhesion molecule-mediated interactions between MM cells and bone marrow stromal cells (BMSCs), or indirectly via the effects of growth factors released by both cell types, miRNA, or mitochondrial transfer, activate a pleiotropic proliferative and antiapoptotic cascade [24]. Importantly, the adhesion of MM cells to BMSCs and/or the extracellular matrix triggers the NF-κB-dependent transcription and secretion of cytokines such as IL-6, tumor necrosis factor-α, and osteopontin in BMSCs, which further stimulates development of drug resistance or so-called cell adhesion-mediated drug resistance (CAM-DR) [24,25,26]. Notably, CAM-DR plays a significant role in the development of drug resistance in MM [7,26,27]. Therefore, targeting CAM-DR is now thought to be a promising option to improve the prognosis of MM patients.
3. CAM-DR Components as Druggable Targets
The development of targeted therapies for CAM-DR is an area of growing interest [26]. CAM-DR is induced by adhesion molecules such as integrin family members [28,29], CD138 (syndecan-1) [28], CD44 [28], vascular cell adhesion molecule-1 (VCAM-1) [30], lymphocyte function-associated antigen-1 (LFA-1) [31,32], and intercellular adhesion molecule-1 (ICAM-1) [33]. Therefore, considerable research is currently focused on the development of drugs targeting these molecules (Figure 1 and Table 1) [34].
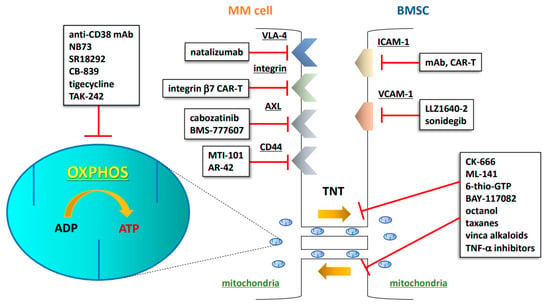
Figure 1.
Schematic illustration of CAM-DR, OXPHOS, and TNTs as druggable targets in the MM niche. MM: multiple myeloma; BMSC: bone marrow stromal cell; mAb: monoclonal antibody; CAR-T: chimeric antigen receptor T cell; TNT: tunneling nanotube; OXPHOS: oxidative phosphorylation; ADP: adenosine diphosphate; ATP: adenosine triphosphate; VLA-4: very late antigen-4; AXL: AXL receptor tyrosine kinase; ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1; TNF-α: tumor necrosis factor-α.

Table 1.
Summary of drugs/treatments targeting CAM-DR, OXPHOS, and TNTs.
Integrins play crucial roles in adhesion, migration, invasion, BM homing, survival, proliferation, and drug resistance in MM cells [37,59]. In particular, very late antigen-4 (VLA-4) (α4β1) and α4β7 play pivotal roles in the pathophysiology of MM [59], thus making these molecules attractive targets. Considering this background, natalizumab, a recombinant humanized IgG4 monoclonal antibody that binds integrin-a4, has demonstrated an ability to inhibit the adhesion of MM cells to both noncellular and cellular components of the MM niche [35,36]. Notably, Hosen et al. reported that integrin β7 is constitutively activated in MM cells, and chimeric antigen receptor (CAR) T cells targeting integrin β7 exhibit a superior anti-MM effect [37,38].
CD44 is a ubiquitous surface molecule, as well as a member of the glycoprotein family [60]. Importantly, CD44 variants are highly expressed in MM cells derived from extramedullary lesions, which play a role in the mechanism underlying the refractoriness of MM [61]. MTI-101, a first-in-class peptidomimetic, binds CD44/ITGA4-containing complexes to induce the activation of Stim1 and TRPC1 expression, triggering necrotic cell death of MM cell lines. MTI-101 and related peptidomimetics are, thus, regarded as an attractive class of compounds [40,41]. Considerable research has focused on eliciting anti-MM effects by modulating CD44 expression. Canella et al. reported that the pan-histone deacetylase inhibitor AR-42 downregulated CD44 expression and enhanced the anti-MM activity of lenalidomide in primary MM cells isolated from lenalidomide-resistant patients and cells isolated from an in vivo MM mouse model [42].
VCAM-1 is an endothelial ligand for VLA-4 (or α4β1) of the β1 subfamily of integrins, and it has been implicated as playing a role in the homing and migration of MM cells [62,63,64]. Teramachi et al. reported that inhibition of TGF-β-activated kinase-1 using LLZ1640-2 reduces VCAM-1 expression in BMSCs and impairs MM cell adhesion to BMSCs [45]. In addition, Zhang et al. reported that the Hedgehog inhibitor LDE225 (sonidegib) inhibits MM cell proliferation by blocking Hedgehog signaling and modulates stromal cells within the BMM by decreasing the expression of VCAM-1 and other adhesion molecules, suggesting that Hedgehog inhibition is a promising option for the treatment of MM [46,47].
LFA-1 is an adhesion molecule that mediates lymphocyte adhesion [65]. The LFA-1 inhibitor LFA878 exerts an anti-MM effect via inhibition of the LFA-1/FAK/PI3K/AKT axis [65]. Importantly, LFA-1 is gaining increased attention for its potential to modulate the tumor microenvironment (TME). The inability of CD8+ effector T cells in the TME is an important mechanism of immunotherapy resistance [66]. Hickman et al. elegantly demonstrated that activation of LFA-1 mediated by the small-molecule LFA-1 activator 7HP349 converts a T-cell-exclusionary TME to a T-cell-enriched TME [66]. Therefore, this activator could be a promising candidate drug for the treatment of MM.
The overexpression of ICAM-1 in MM, associated with advanced disease and poor survival, may be a potential therapeutic target even in the relapse/refractory setting [33,67,68,69]. Sherbenou et al. reported that an anti-ICAM-1 monoclonal antibody conjugated to an auristatin derivative induced potent anti-MM cytotoxicity both in vitro and in vivo. This effect was assumed to involve in part blockade of cell–cell interactions and the interaction of ICAM-1 with its ligand, thus interfering with various immune functions [33]. Furthermore, a line of anti-ICAM-1 antibody-based chimeric antigen receptor T cells was shown to exhibit significant antitumor effects both in vitro and in vivo in preclinical models of gastric cancer and thyroid cancer, suggesting they are applicable to the treatment of hematological malignancies, including MM [33,43,44].
The HSC niche can regulate the dormancy of tumor cells [21]. In MM, dormancy occurs when tumor cells enter a quiescent state (G0), in which they are under reversible growth arrest [70,71]. Importantly, dormant MM cells can be induced to re-enter the cell cycle in response to extrinsic stimuli from the microenvironment or various therapeutic agents, including bortezomib [71,72]. Drug-resistant dormant MM cells residing in skeletal endosteal niches are thought to mediate disease relapse. These cells exhibit a distinct transcriptome signature enriched in immunity-related genes and genes associated with myeloid cell differentiation, including AXL (a TAM receptor tyrosine kinase). Notably, AXL inhibition using the small-molecule inhibitors cabozatinib and BMS-777607 releases MM cells from dormancy and sensitizes them to chemotherapy [16,39]. Another study found that macrophages are the dominant cells regulating the inflammatory milieu of the MM niche; inhibition of TPL2 kinase in macrophages leads to inhibition of interleukin (IL)-1β and IL-6, ultimately resulting in myeloma progression [73]. These data could lead to the development of new therapies that improve of the outcome of MM patients.
4. Mitochondrial Transfer via TNTs: A Novel CAM-DR Concept
4.1. Mitochondrial Transfer in Cancer Cells, including MM Cells
It is now clear that the metabolic and mitochondrial functions are reprogrammed in many types of cancer cells to ensure the production of necessary molecules such as lipids, proteins, and nucleic acids and sustain the mitotic signaling that enables cell proliferation [74]. Mitochondria generate most of a cell’s energy supply, i.e., adenosine triphosphate (ATP), via oxidative phosphorylation (OXPHOS) [8]. Cancer cells tend to synthesize ATP primarily through glycolysis, even under aerobic conditions, although glycolysis is less efficient than OXPHOS in generating ATP [75]. However, cells of certain solid tumors and many hematological malignancies appear to exhibit normal or even increased OXPHOS and mitochondrial metabolism [76]. However, this remains an area of intense research, as the association between cancer cells and OXPHOS has not been fully elucidated.
Studies have clearly demonstrated the occurrence of mitochondrial transfer in hematological malignancies such as acute myeloid leukemia, acute lymphocytic leukemia, and MM [8,9,10,11,77]. Transcellular mitochondrial transfer is primarily mediated by three intercellular communication pathways: (1) TNTs, (2) extracellular vesicles, and (3) gap junctions [76]. Interestingly, cancer cells can transfer mitochondria to nonmalignant cells via mitophagy, which is a process for the clearance of damaged mitochondria [78]. Significantly, the transfer of mitochondria and/or mitochondrial DNA to cancer cells increases the mitochondrial content and enhances OXPHOS, thus favoring proliferation and invasion [8]. The transfer of mitochondria from BMSC was shown to protect mutant hematopoietic cells during chemotherapy [8]. Thus, mitochondrial exchange occurs preferentially between nonmalignant cells and cancer cells. Cancer cells that acquire mitochondria exhibit chemoresistance [79], suggesting that this process is a promising target in the treatment of various cancers, including MM.
4.2. TNT Formation in Cancer Cells, including MM Cells
TNTs are filamentous, F-actin-rich, long tubular extensions connecting the cytoplasm of adjacent and/or distant cells that mediate cell-to-cell communication [80,81,82]. TNTs are increasingly considered the primary intercellular pathway for the unidirectional and bidirectional movement of nuclear and cytoplasmic cargo, such as nucleic acids, drugs, pathogenic molecules, and organelles, including mitochondria [50,83]. Hypoxic conditions associated with the TME reportedly stimulate an increase in TNT formation [84]. Under conditions of oxidative stress, the intracellular expression of p53 is upregulated, and protein kinase B–phosphoinositide 3-kinase–mammalian target of rapamycin (AKT–PI3K–mTOR) signaling is activated, leading to TNT formation [76]. The mechanism of TNT formation is closely associated with interactions between a complex of proteins, including leukocyte specific transcript 1, M-sec, Ras-related protein A, and the exocyst complex [50,85,86,87]. TNT can rescue diseased cells and tissues by mediating the direct transfer of healthy mitochondria to compromised cells [88]. Mitochondrial transfer occurs via TNTs and partial cell fusion, and the process is significantly upregulated in the presence of chemotherapeutic drugs [89]. Importantly, transferred mitochondria were found to metabolically promote OXPHOS [11,52]. MM cells can acquire mitochondria from neighboring nonmalignant cells through TNTs. Moreover, TNT-mediated transfer from cancer cells also plays a role in drug resistance, as demonstrated by the detoxifying removal of chemotherapeutic agent-loaded lysosomal vesicles from leukemia cells [90,91]. Therefore, targeting mitochondrial transfer via TNTs is an attractive option for overcoming chemo-resistance in the treatment of cancers, including MM.
4.3. OXPHOS and TNTs as Druggable Targets in Cancer and MM Therapy
Numerous compounds have been identified that affect pathways, such as NF-κB and mTOR, or block actin polymerization, thus inducing a reduction in TNT formation. The compounds include cytochalasin D, cytarabine, latrunculin A and B, daunorubicin, everolimus, metformin, nocodazole CK-666, ML-141, 6-thio-GTP, BAY-117082, and octanol (Figure 1) [48,49]. In addition, taxanes and vinca alkaloids have the potential to partially inhibit mitochondrial transfer by inhibiting microtubule polymerization [50]. M-sec, a TNT marker and regulator of TNT formation, directly induces tumor necrosis factor (TNF)-α. These data suggest that TNF-α inhibitors could be used to indirectly reduce TNT formation, thereby inhibiting mitochondrial transfer [50,51].
Targeting mitochondrial respiration and OXPHOS is also an attractive treatment option (Figure 1). FOXM1 regulates the metabolism of myeloma cells by upregulating glycolysis and OXPHOS. NB73, a small-compound inhibitor of FOXM1, inhibits MM cell growth by promoting FOXM1 degradation, suggesting that NB73 could become a promising OXPHOS-targeted drug [53]. Xiang et al. reported that the expression of OXPHOS-associated genes is associated with higher PGC-1α expression; treatment with the PGC-1a inhibitor SR18292 was shown to significantly impair the proliferation and survival of MM cells due to dysfunction in OXPHOS metabolism [54]. Thompson et al. reported that PI-resistant MM cells exhibit an increased capacity for and reliance on mitochondrial respiration [55]. The glutaminase-1 inhibitor CB-839 inhibits mitochondrial respiration and is more cytotoxic to PI-resistant cells, suggesting that mitochondrial respiration would be a promising target in the treatment of relapsed/refractory MM [55]. The OXPHOS inhibitor tigecycline increases the sensitivity of cancer cells to bortezomib, a representative PI [56,57]. Inhibition of PGC-1a by SR18292 was shown to significantly impair the proliferation and survival of MM cells due to energy exhaustion and oxidative damage [54,56]. Toll-like receptor 4 (TLR4) induces mitochondrial biogenesis and an increase in mitochondrial mass in human MM cells. Moreover, bortezomib (BTZ) exposure activates TLR4 signaling in BTZ-resistant MM cell lines. Combining BTZ with the selective TLR4 inhibitor TAK-242 was shown to overcome drug resistance by inducing more intense and extended oxidative stress, leading to mitochondrial depolarization and severe impairment of mitochondrial fitness [58].
In the MM niche, TNT-mediated transcellular transfer of mitochondria from neighboring BMSCs to MM cells supports OXPHOS in MM cells, and this process is dependent on CD38 expression [76]. CD38 is a transmembrane glycoprotein present both on the cell membrane and in the intracellular compartment [92]. MM cells express high levels of CD38. Therefore, monoclonal antibodies against CD38 (e.g., datatumumab and isatuximab) can be used to successfully treat MM [92]. Anti-CD38 monoclonal antibodies have several mechanisms of action, including antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity, direct cellular apoptosis, and modulation of extracellular ectoenzyme activity [93]. Importantly, MM patients receiving anti-CD38 antibody therapy have shown superior survival benefit. Nevertheless, MM cells may eventually acquire resistance to anti-CD38 antibody therapy in these patients [94]. Increased CD38 expression facilitating mitochondrial transfer from BMSCs to primary MM cells is one potential resistance mechanism [50,52]. CD38 expression blockade was shown to inhibit mitochondrial transfer, reduce tumor volume, and increase overall survival in mice [50,52]. These reports suggest that TNT inhibition using anti-CD38 antibodies may be a useful anti-myeloma therapy.
5. Conclusions
With the advent of novel therapeutic drugs, especially monoclonal antibody immunotherapies and CAR-T therapies, the prognosis of patients with MM has improved dramatically. However, MM cells typically eventually develop resistance; therefore, elucidation of the mechanisms via which these cells acquire resistance is urgently needed.
Research has shown that MM cells acquire drug resistance via contact with BMM constituents such as BMSCs. In particular, CAM-DR plays an important role in this process, and therapeutic agents that overcome resistance mediated by CAM-DR are, thus, being developed. Furthermore, recent data indicate that drug resistance is dynamically induced through mitochondrial transfer between MM cells and other BMM cells via TNTs, providing additional new therapeutic targets. Successful application of these therapies in clinical practice could bring us one step closer to making MM a curable disease.
Author Contributions
All authors contributed to writing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Grants-in-Aid for Scientific Research of the Japan Society for the Promotion of Science KAKENHI, as well as a Tokai University Tokuda Memorial Cancer/Genome Basic Research Grant for Young Investigators.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple Myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Ludwig, H.; Novis Durie, S.; Meckl, A.; Hinke, A.; Durie, B. Multiple Myeloma Incidence and Mortality around the Globe; Interrelations between Health Access and Quality, Economic Resources, and Patient Empowerment. Oncologist 2020, 25, e1406–e1413. [Google Scholar] [CrossRef]
- Soliman, A.M.; Das, S.; Teoh, S.L. Next-Generation Biomarkers in Multiple Myeloma: Understanding the Molecular Basis for Potential Use in Diagnosis and Prognosis. Int. J. Mol. Sci. 2021, 22, 7470. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; Morè, S.; Olivieri, A. Novel Experimental Drugs for Treatment of Multiple Myeloma. J. Exp. Pharmacol. 2021, 13, 245–264. [Google Scholar] [CrossRef]
- Dimopoulos, M.-A.; Richardson, P.; Lonial, S. DUPLICATE: Treatment Options for Patients With Heavily Pretreated Relapsed and Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk 2022, 22, 460–473. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Tang, J.; Qin, S.; Shen, X.; He, S.; Ju, S. CAM-DR: Mechanisms, Roles and Clinical Application in Tumors. Front. Cell Dev. Biol. 2021, 9, 698047. [Google Scholar] [CrossRef]
- Mohammadalipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Noll, J.E.; Williams, S.A.; Purton, L.E.; Zannettino, A.C. Tug of War in the Haematopoietic Stem Cell Niche: Do Myeloma Plasma Cells Compete for the Hsc Niche? Blood Cancer J. 2012, 2, e91. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P. Bone and the hematopoietic niche: A tale of two stem cells. Blood 2011, 117, 5281–5288. [Google Scholar] [CrossRef]
- Marchand, T.; Pinho, S. Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front. Immunol. 2021, 12, 775128. [Google Scholar] [CrossRef]
- Giannakoulas, N.; Ntanasis-Stathopoulos, I.; Terpos, E. The Role of Marrow Microenvironment in the Growth and Development of Malignant Plasma Cells in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 4462. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts Degrade Endosteal Components and Promote Mobilization of Hematopoietic Pro-genitor Cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Boyd, A.L.; Campbell, C.J.; Hopkins, C.I.; Fiebig-Comyn, A.; Russell, J.; Ulemek, J.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche Displacement of Human Leukemic Stem Cells Uniquely Allows Their Competitive Re-placement with Healthy Hspcs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human Prostate Cancer Me-tastases Target the Hematopoietic Stem Cell Niche to Establish Footholds in Mouse Bone Marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Basak, G.W.; Srivastava, A.S.; Malhotra, R.; Carrier, E. Multiple myeloma bone marrow niche. Curr. Pharm. Biotechnol. 2009, 10, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding Multiple Myeloma Path-ogenesis in the Bone Marrow to Identify New Therapeutic Targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef]
- Katz, B.-Z. Adhesion molecules—The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to Fibronectin Via Beta1 Integrins Regulates P27kip1 Levels and Contributes to Cell Adhesion Mediated Drug Resistance (Cam-Dr). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef]
- Abe, M.; Hiura, K.; Ozaki, S.; Kido, S.; Matsumoto, T. Vicious Cycle between Myeloma Cell Binding to Bone Marrow Stromal Cells Via Vla-4-Vcam-1 Adhesion and Macrophage Inflammatory Protein-1alpha and Mip-1beta Production. J. Bone Miner. Metab. 2009, 27, 16–23. [Google Scholar] [CrossRef]
- Barker, H.F.; Ball, J.; Drew, M.; Hamilton, M.S.; Franklin, I.M. The Role of Adhesion Molecules in Multiple Myeloma. Leuk. Lymphoma 1992, 8, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Di Loreto, M.; Ribatti, D.; Di Stefano, R.; Gadaleta-Caldarola, G.; Iodice, G.; Caloro, D.; Dammacco, F. Bone Marrow of Patients with Active Multiple Myeloma: Angiogenesis and Plasma Cell Adhesion Molecules Lfa-1, Vla-4, Lam-1, and Cd44. Am. J. Hematol. 1995, 50, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Sherbenou, D.W.; Su, Y.; Behrens, C.R.; Aftab, B.T.; de Acha, O.P.; Murnane, M.; Bearrows, S.C.; Hann, B.C.; Wolf, J.L.; Martin, T.G.; et al. Potent Activity of an Anti-ICAM1 Antibody–Drug Conjugate against Multiple Myeloma. Clin. Cancer Res. 2020, 26, 6028–6038. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Mu, Q.; Yu, J.; Griffin, J.I.; Xu, X.; Ho, R.J.Y. ICAM-1 Targeted Drug Combination Nanoparticles Enhanced Gemcitabine-Paclitaxel Exposure and Breast Cancer Suppression in Mouse Models. Pharmaceutics 2021, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Savino, F.D.; Scaringella, A.; Potenza, M.A.; Nacci, C.; Frassanito, M.A.; Vacca, A.; Montagnani, M. The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value. J. Clin. Med. 2022, 11, 2513. [Google Scholar] [CrossRef]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.T.; Vallet, S.; Halama, N.; Jager, D.; Olson, D.L.; et al. The Selective Adhesion Molecule Inhibitor Natalizumab Decreases Multiple Myeloma Cell Growth in the Bone Marrow Microenvironment: Therapeutic Implications. Br. J. Haematol 2011, 155, 438–448. [Google Scholar] [CrossRef]
- Hosen, N.; Yoshihara, S.; Takamatsu, S.; Ri, M.; Nagata, Y.; Kosugi, H.; Shimomura, Y.; Hanamura, I.; Fuji, S.; Minauchi, K.; et al. Expression of Activated Integrin Beta7 in Multiple Myeloma Patients. Int. J. Hematol. 2021, 114, 3–7. [Google Scholar] [CrossRef]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The Activated Conformation of Integrin Beta7 Is a Novel Multiple Myelo-ma-Specific Target for Car T Cell Therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Gebhard, A.W.; Jain, P.; Nair, R.R.; Emmons, M.F.; Argilagos, R.F.; Koomen, J.M.; McLaughlin, M.L.; Hazlehurst, L.A. MTI-101 (Cyclized HYD1) Binds a CD44 Containing Complex and Induces Necrotic Cell Death in Multiple Myeloma. Mol. Cancer Ther. 2013, 12, 2446–2458. [Google Scholar] [CrossRef]
- Emmons, M.F.; Anreddy, N.; Cuevas, J.; Steinberger, K.; Yang, S.; McLaughlin, M.; Silva, A.; Hazlehurst, L.A. MTI-101 treatment inducing activation of Stim1 and TRPC1 expression is a determinant of response in multiple myeloma. Sci. Rep. 2017, 7, 2685. [Google Scholar] [CrossRef] [PubMed]
- Canella, A.; Nieves, H.C.; Sborov, D.W.; Cascione, L.; Radomska, H.S.; Smith, E.; Stiff, A.; Consiglio, J.; Caserta, E.; Rizzotto, L.; et al. HDAC inhibitor AR-42 decreases CD44 expression and sensitizes myeloma cells to lenalidomide. Oncotarget 2015, 6, 31134–31150. [Google Scholar] [CrossRef] [PubMed]
- Vedvyas, Y.; McCloskey, J.E.; Yang, Y.; Min, I.M.; Fahey, T.J.; Zarnegar, R.; Hsu, Y.-M.S.; Hsu, J.-M.; Van Besien, K.; Gaudet, I.; et al. Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci. Rep. 2019, 9, 10634. [Google Scholar] [CrossRef]
- Jung, M.; Yang, Y.; McCloskey, J.E.; Zaman, M.; Vedvyas, Y.; Zhang, X.; Stefanova, D.; Gray, K.D.; Min, I.M.; Zarnegar, R.; et al. Chimeric Antigen Receptor T Cell Therapy Targeting ICAM-1 in Gastric Cancer. Mol. Ther. Oncolytics 2020, 18, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Teramachi, J.; Tenshin, H.; Hiasa, M.; Oda, A.; Bat-Erdene, A.; Harada, T.; Nakamura, S.; Ashtar, M.; Shimizu, S.; Iwasa, M.; et al. TAK1 is a pivotal therapeutic target for tumor progression and bone destruction in myeloma. Haematologica 2020, 106, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Kizaki, M.; Tabayashi, T. The Role of Intracellular Signaling Pathways in the Pathogenesis of Multiple Myeloma and Novel Therapeutic Approaches. J. Clin. Exp. Hematop. 2016, 56, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.; Neelapu, S.S.; Romaguera, J.; McCarty, N. Hedgehog inhibitors selectively target cell migration and adhesion of mantle cell lymphoma in bone marrow microenvironment. Oncotarget 2016, 7, 14350–14365. [Google Scholar] [CrossRef]
- Allegra, A.; Di Gioacchino, M.; Cancemi, G.; Casciaro, M.; Petrarca, C.; Musolino, C.; Gangemi, S. Specialized Inter-cellular Communications Via Tunnelling Nanotubes in Acute and Chronic Leukemia. Cancers 2022, 14, 659. [Google Scholar] [CrossRef]
- Mittal, R.; Karhu, E.; Wang, J.-S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef]
- Zampieri, L.; Silva-Almeida, C.; Rondeau, J.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Sarma, V.; Wolf, F.W.; Marks, R.M.; Shows, T.B.; Dixit, V.M. Cloning of a Novel Tumor Necrosis Fac-tor-Alpha-Inducible Primary Response Gene That Is Differentially Expressed in Development and Capillary Tube-Like Formation in Vitro. J. Immunol. 1992, 148, 3302–3312. [Google Scholar] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Sun, F.; Thornton, K.; Jing, X.; Dong, J.; Yun, G.; Pisano, M.; Zhan, F.; Kim, S.H.; Katzenellenbogen, J.A.; et al. FOXM1 regulates glycolysis and energy production in multiple myeloma. Oncogene 2022, 41, 3899–3911. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef]
- Ma, R.; Zhang, Y.; Wang, W.; Wu, J.; Yang, Q.; Xu, W.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Inhibition of Autophagy En-hances the Antitumour Activity of Tigecycline in Multiple Myeloma. J. Cell Mol. Med. 2018, 22, 5955–5963. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Hosen, N. Integrins in multiple myeloma. Inflamm. Regen. 2020, 40, 1–4. [Google Scholar] [CrossRef]
- Mackay, C.; Terpe, H.J.; Stauder, R.; Marston, W.L.; Stark, H.; Günthert, U. Expression and modulation of CD44 variant isoforms in humans. J. Cell Biol. 1994, 124, 71–82. [Google Scholar] [CrossRef]
- Kikuchi, J.; Kodama, N.; Takeshita, M.; Ikeda, S.; Kobayashi, T.; Kuroda, Y.; Uchiyama, M.; Osada, N.; Bogen, B.; Yasui, H.; et al. EMD originates from hyaluronan-induced homophilic interactions of CD44 variant-expressing MM cells under shear stress. Blood Adv. 2022. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Migkou, M.; Christoulas, D.; Gavriatopoulou, M.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Iakovaki, M.; Panagiotidis, I.; Ziogas, D.C.; Fotiou, D.; et al. Increased circulating VCAM-1 correlates with advanced disease and poor survival in patients with multiple myeloma: Reduction by post-bortezomib and lenalidomide treatment. Blood Cancer J. 2016, 6, e428. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M.; Bendas, G. Vascular Cell Adhesion Molecule-1 (Vcam-1)--an Increasing Insight into Its Role in Tu-morigenicity and Metastasis. Int. J. Cancer 2015, 136, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Hawley, R.G.; Kodaka, M.; Okuno, H. Significance of Vla-4-Vcam-1 Interaction and Cd44 for Transendo-thelial Invasion in a Bone Marrow Metastatic Myeloma Model. Clin. Exp. Metastasis 1999, 17, 623–629. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Mandl-Weber, S.; Gaul, L.; Baumann, P.; Bumeder, I.; Straka, C.; Emmerich, B. Inhibition of Lym-phocyte Function Associated Antigen 1 by Lfa878 Induces Apoptosis in Multiple Myeloma Cells and Is Associated with Downregulation of the Focal Adhesion Kinase/Phosphatidylinositol 3 Kinase/Akt Pathway. Int. J. Oncol. 2007, 31, 969–976. [Google Scholar]
- Hickman, A.; Koetsier, J.; Kurtanich, T.; Nielsen, M.C.; Winn, G.; Wang, Y.; Bentebibel, S.-E.; Shi, L.; Punt, S.; Williams, L.; et al. LFA-1 activation enriches tumor-specific T cells in a cold tumor model and synergizes with CTLA-4 blockade. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Veitonmäki, N.; Hansson, M.; Zhan, F.; Sundberg, A.; Löfstedt, T.; Ljungars, A.; Li, Z.-C.; Martinsson-Niskanen, T.; Zeng, M.; Yang, Y.; et al. A Human ICAM-1 Antibody Isolated by a Function-First Approach Has Potent Macrophage-Dependent Antimyeloma Activity In Vivo. Cancer Cell 2013, 23, 502–515. [Google Scholar] [CrossRef]
- Sampaio, M.S.S.; Vettore, A.L.; Yamamoto, M.; Chauffaille, M.D.L.L.F.; Zago, M.A.; Colleoni, G.W.B. Expression of eight genes of nuclear factor-kappa B pathway in multiple myeloma using bone marrow aspirates obtained at diagnosis. Histol. Histopathol. 2009, 24, 991. [Google Scholar]
- Schmidmaier, R.; Mörsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for Cell Adhesion-Mediated Drug Resistance of Multiple Myeloma Cells in Vivo. Int. J. Biol. Markers 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Dadzie, T.G.; Green, A.C. The role of the bone microenvironment in regulating myeloma residual disease and treatment. Front. Oncol. 2022, 12, 999939. [Google Scholar] [CrossRef] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. Inhibition of eIF2α Dephosphorylation Maximizes Bortezomib Efficiency and Eliminates Quiescent Multiple Myeloma Cells Surviving Proteasome Inhibitor Therapy. Cancer Res. 2009, 69, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.; Ollar, S.J.; Heninger, E.; Hebron, E.; Jensen, J.L.; Kim, J.; Maroulakou, I.; Miyamoto, S.; Leith, C.; Yang, D.T.; et al. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood 2014, 123, 3305–3315. [Google Scholar] [CrossRef]
- Murphy, M.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Song, I.; Kim, H.; Lee, S.; Jeong, S.; Kim, N.; Ko, K.; Rhee, B.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell Adhesion-Mediated Mitochondria Transfer Contributes to Mesenchymal Stem Cell-Induced Chemo-resistance on T Cell Acute Lymphoblastic Leukemia Cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Zampieri, L.X.; Sboarina, M.; Cacace, A.; Grasso, D.; Thabault, L.; Hamelin, L.; Vazeille, T.; Dumon, E.; Rossignol, R.; Frederick, R.; et al. Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Te-mozolomide-Resistant Human Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 11938. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Tiwari, V.; Koganti, R.; Russell, G.; Sharma, A.; Shukla, D. Role of Tunneling Nanotubes in Viral Infection, Neuro-degenerative Disease, and Cancer. Front. Immunol. 2021, 12, 680891. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.; Subramanian, S. Tunneling Nanotubes: A New Paradigm for Studying Intercellular Communication and Therapeutics in Cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.; Gondaliya, P.; Patel, T. Tunneling Nanotube-Mediated Communication: A Mechanism of Intercellular Nucleic Acid Transfer. Int. J. Mol. Sci. 2022, 23, 5487. [Google Scholar] [CrossRef]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Schiller, C.; Diakopoulos, K.; Rohwedder, I.; Kremmer, E.; von Toerne, C.; Ueffing, M.; Weidle, U.H.; Ohno, H.; Weiss, E.H. LST1 promotes the assembly of a molecular machinery responsible for tunneling nanotube formation. J. Cell Sci. 2012, 126, 767–777. [Google Scholar] [CrossRef]
- Eugenin, E.; Camporesi, E.; Peracchia, C. Direct Cell-Cell Communication via Membrane Pores, Gap Junction Channels, and Tunneling Nanotubes: Medical Relevance of Mitochondrial Exchange. Int. J. Mol. Sci. 2022, 23, 6133. [Google Scholar] [CrossRef]
- Matula, Z.; Mikala, G.; Lukácsi, S.; Matkó, J.; Kovács, T.; Monostori, É.; Uher, F.; Vályi-Nagy, I. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers 2021, 13, 3461. [Google Scholar] [CrossRef]
- Hekmatshoar, Y.; Nakhle, J.; Galloni, M.; Vignais, M.L. The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem. J. 2018, 475, 2305–2328. [Google Scholar] [CrossRef]
- Omsland, M.; Bruserud, O.; Gjertsen, B.T.; Andresen, V. Tunneling Nanotube (Tnt) Formation Is Downregulated by Cytarabine and Nf-Kappab Inhibition in Acute Myeloid Leukemia (Aml). Oncotarget 2017, 8, 7946–7963. [Google Scholar] [CrossRef] [PubMed]
- Szlasa, W.; Czarny, J.; Sauer, N.; Rakoczy, K.; Szymanska, N.; Stecko, J.; Kolodziej, M.; Kazmierczak, M.; Barg, E. Tar-geting Cd38 in Neoplasms and Non-Cancer Diseases. Cancers 2022, 14, 4169. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Mina, R.; Gay, F. Anti-Cd38 Monoclonal Antibodies in Multiple Myeloma: Another Cook in the Kitchen? Lancet Haematol. 2020, 7, e355–e357. [Google Scholar] [CrossRef]
- Leleu, X.; Martin, T.; Weisel, K.; Schjesvold, F.; Iida, S.; Malavasi, F.; Manier, S.; Chang-Ki, M.; Ocio, E.M.; Pawlyn, C.; et al. Anti-Cd38 Antibody Therapy for Patients with Re-lapsed/Refractory Multiple Myeloma: Differential Mechanisms of Action and Recent Clinical Trial Outcomes. Ann. Hematol. 2022, 101, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).