New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer

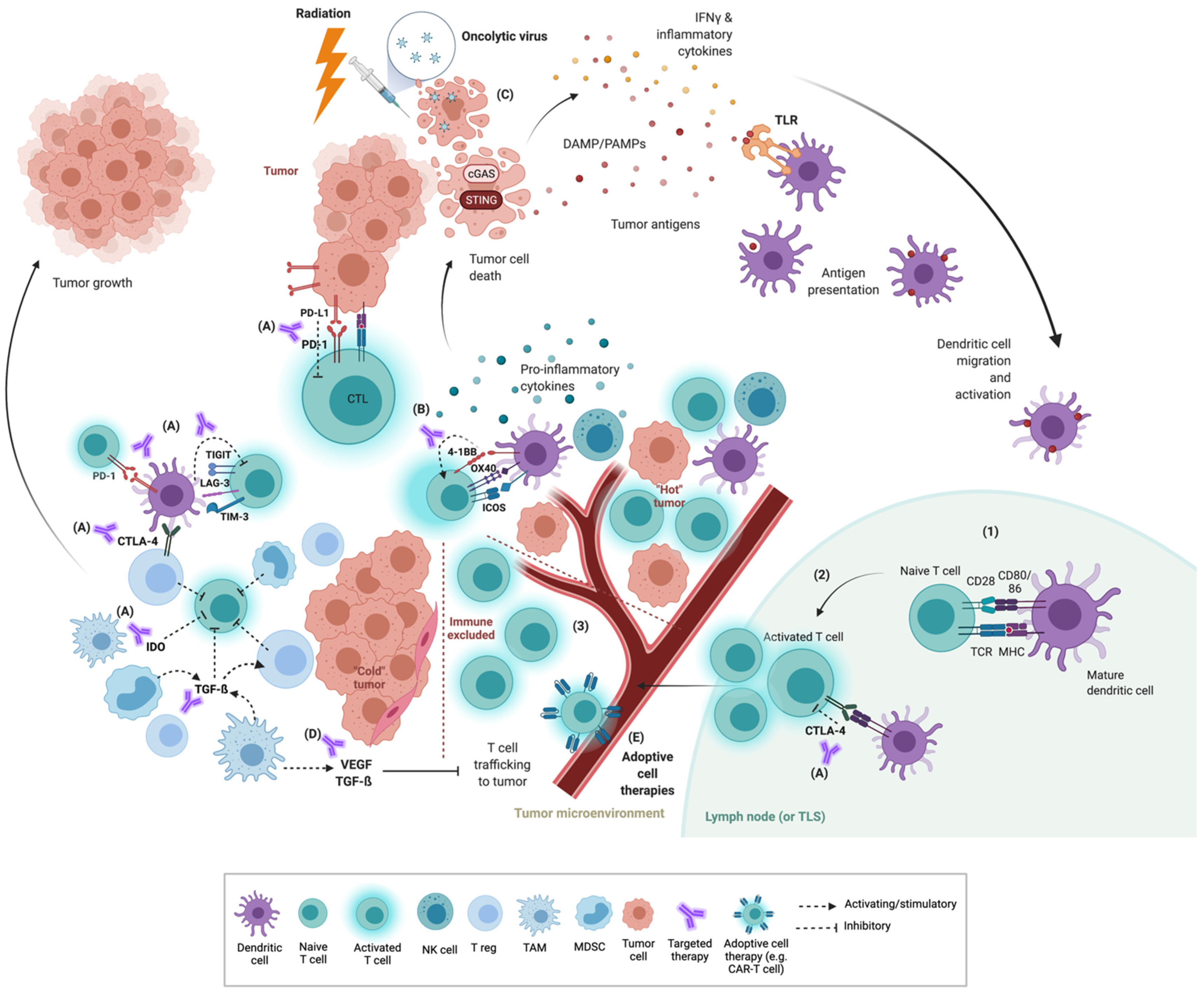
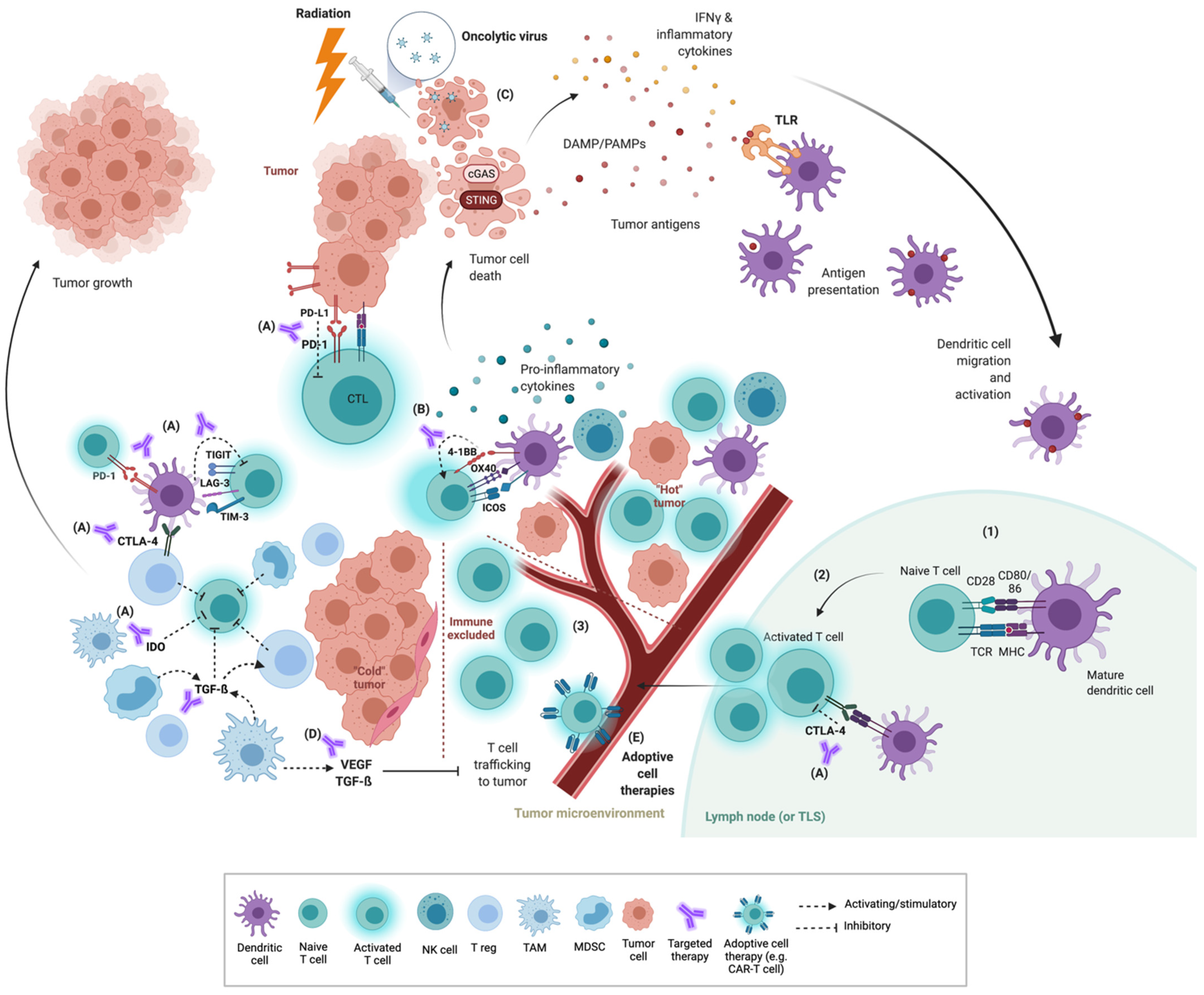{kind=link}
Abstract
:1. Introduction
2. The Tumor Immune Microenvironment (TIME)
3. Co-Inhibitory and Co-Stimulatory Checkpoints
3.1. Co-Inhibitory Molecules
3.2. Co-Stimulatory Molecules
4. Priming Strategies
4.1. Radiation
4.2. STING Agonists
4.3. TLR Agonists
4.4. Oncolytic Viruses
4.5. Cytokines
5. VEGF, Targeted Therapy and Other Immunomodulators
5.1. VEGF
5.2. TGF-β
5.3. Targeted Therapy
6. Adoptive Cell Therapy
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced Non-Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pages, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: A multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 2021, 397, 592–604. [Google Scholar] [CrossRef]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.Q.M.; Burgio, M.A.; Carpeno, J.d.C.; Pluzanski, A.; Arrieta, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes From the Randomized, Phase III Trials CheckMate 017 and 057: Nivolumab Versus Docetaxel in Previously Treated Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Pluzanski, A.; Caro, R.B.; Provencio, M.; Burgers, S.; Carcereny, E.; Park, K.; Alexandru, A.; Lupinacci, L.; Sangha, R.; et al. Abstract CT221: Nivolumab (NIVO) + ipilimumab (IPI) as first-line (1L) treatment for patients with advanced non-small cell lung cancer (NSCLC) with brain metastases: Results from CheckMate 227. Cancer Res. 2020, 80, CT221. [Google Scholar] [CrossRef]
- Boyer, M.; Şendur, M.A.N.; Rodríguez-Abreu, D.; Park, K.; Lee, D.H.; Çiçin, I.; Yumuk, P.F.; Orlandi, F.J.; Leal, T.A.; Molinier, O.; et al. Pembrolizumab Plus Ipilimumab or Placebo for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%: Randomized, Double-Blind Phase III KEYNOTE-598 Study. J. Clin. Oncol. 2021, 39, 2327–2338. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.B.; Ha, S.-J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharm. 2021, 12, 681320. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.J.; Niu, J.; Kim, D.W.; Rasco, D.; Mileham, K.F.; Chung, H.C.; Vaishampayan, U.N.; Maurice-Dror, C.; Lo Russo, P.; Golan, T.; et al. 1400P Vibostolimab, an anti-TIGIT antibody, as monotherapy and in combination with pembrolizumab in anti-PD-1/PD-L1-refractory NSCLC. Ann. Oncol. 2020, 31, S887. [Google Scholar] [CrossRef]
- Rodriguez-Abreu, D.; Johnson, M.L.; Hussein, M.A.; Cobo, M.; Patel, A.J.; Secen, N.M.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.-H.; et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J. Clin. Oncol. 2020, 38, 9503. [Google Scholar] [CrossRef]
- Patil, N.; Cho, B.C.; Johnson, M.; Caro, R.B.; Spira, A.; Chiu, C.; Molden, N.; Pham, T.; Yang, X.; Choi, Y.; et al. P77.02 Efficacy of Tiragolumab + Atezolizumab in PD-L1 IHC and TIGIT Subgroups in the Phase II CITYSCAPE Study in First-Line NSCLC. J. Thorac. Oncol. 2021, 16, S635–S636. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; Doger, B.; Majem, M.; Carcereny, E.; Krebs, M.; Peguero, J.A.; Roxburgh, P.; Forster, M.; Bajaj, P.; Clay, T.D.; et al. Initial results from a phase II study (TACTI-002) in metastatic non-small cell lung or head and neck carcinoma patients receiving eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab. J. Clin. Oncol. 2020, 38, 3100. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Boasberg, P.; Eroglu, Z. A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in combination with TSR-042 (anti-PD-1) in patients with colorectal cancer and post-PD-1 NSCLC and melanoma. J. Immunother. Cancer 2018, 6, 114. [Google Scholar]
- Harding, J.J.; Moreno, V.; Bang, Y.J.; Hong, M.H.; Patnaik, A.; Trigo, J.; Szpurka, A.M.; Yamamoto, N.; Doi, T.; Fu, S.; et al. Blocking TIM-3 in Treatment-refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 Antibody. Clin. Cancer Res. 2021, 27, 2168–2178. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Qi, X.; Li, F.; Wu, Y.; Cheng, C.; Han, P.; Wang, J.; Yang, X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat. Commun. 2019, 10, 2141. [Google Scholar] [CrossRef] [PubMed]
- Sznol, M.; Hodi, F.S.; Margolin, K.; McDermott, D.F.; Ernstoff, M.S.; Kirkwood, J.M.; Wojtaszek, C.; Feltquate, D.; Logan, T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J. Clin. Oncol. 2008, 26, 3007. [Google Scholar] [CrossRef]
- Lundqvist, A.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Sanz, L.M.; Liang, S.; Murray, S.; Larsson, O.; et al. 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer (SITC 2016): Part one. J. ImmunoTher. Cancer 2016, 4, 82. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef] [Green Version]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.-C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef]
- Goldman, J.W.; Piha-Paul, S.A.; Curti, B.D.; Pedersen, K.; Bauer, T.M.; Groenland, S.L.; Carvajal, R.D.; Chhaya, V.; Hammond, S.A.; Streicher, K.; et al. Safety and tolerability of MEDI0562 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3003. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Lam, V.K.; Ros, W.; Bauer, T.M.; Hansen, A.R.; Cho, D.C.; Hodi, F.S.; Schellens, J.H.M.; Litton, J.K.; Aspeslagh, S.; et al. Abstract CT150: A first-in-human phase I study of the OX40 agonist GSK3174998 (GSK998) +/− pembrolizumab in patients (Pts) with selected advanced solid tumors (ENGAGE-1). Cancer Res. 2020, 80, CT150. [Google Scholar] [CrossRef]
- Gutierrez, M.; Moreno, V.; Heinhuis, K.M.; Olszanski, A.J.; Spreafico, A.; Ong, M.; Chu, Q.; Carvajal, R.D.; Trigo, J.; Ochoa de Olza, M.; et al. OX40 Agonist BMS-986178 Alone or in Combination With Nivolumab and/or Ipilimumab in Patients With Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Burris, H.A.; Kummar, S.; Falchook, G.S.; Pachynski, R.K.; LoRusso, P.; Tykodi, S.S.; Gibney, G.T.; Gainor, J.F.; Rahma, O.E.; et al. ICONIC: Biologic and clinical activity of first in class ICOS agonist antibody JTX-2011 +/− nivolumab (nivo) in patients (pts) with advanced cancers. J. Clin. Oncol. 2018, 36, 3000. [Google Scholar] [CrossRef]
- Jounce Therapeutics Announces Update on Vopratelimab Program. Available online: https://ir.jouncetx.com/news-releases/news-release-details/jounce-therapeutics-announces-update-vopratelimab-program (accessed on 8 September 2021).
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.S.; Hahn, S.M. Immune Priming of the Tumor Microenvironment by Radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, J.H.; Mileham, K.F.; Simone, C.B. The utilization of immunotherapy with radiation therapy in lung cancer: A narrative review. Transl. Cancer Res. 2021, 10, 2596–2608. [Google Scholar] [CrossRef]
- Ko, E.C.; Raben, D.; Formenti, S.C. The Integration of Radiotherapy with Immunotherapy for the Treatment of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5792–5806. [Google Scholar] [CrossRef] [Green Version]
- Spigel, D.R.; Faivre-Finn, C.; Gray, J.E.; Vicente, D.; Planchard, D.; Paz-Ares, L.G.; Vansteenkiste, J.F.; Garassino, M.C.; Hui, R.; Quantin, X.; et al. Five-year survival outcomes with durvalumab after chemoradiotherapy in unresectable stage III NSCLC: An update from the PACIFIC trial. J. Clin. Oncol. 2021, 39, 8511. [Google Scholar] [CrossRef]
- Gray, J.E.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; Cho, B.C.; et al. Three-Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC-Update from PACIFIC. J. Thorac. Oncol. 2020, 15, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theelen, W.; Peulen, H.M.U.; Lalezari, F.; van der Noort, V.; de Vries, J.F.; Aerts, J.; Dumoulin, D.W.; Bahce, I.; Niemeijer, A.N.; de Langen, A.J.; et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1276–1282. [Google Scholar] [CrossRef]
- Welsh, J.; Menon, H.; Chen, D.; Verma, V.; Tang, C.; Altan, M.; Hess, K.; de Groot, P.; Nguyen, Q.N.; Varghese, R.; et al. Pembrolizumab with or without radiation therapy for metastatic non-small cell lung cancer: A randomized phase I/II trial. J. Immunother. Cancer 2020, 8, e001001. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Manochakian, R.; Zhao, Y.; Lou, Y. Immunotherapies targeting stimulatory pathways and beyond. J. Hematol. Oncol. 2021, 14, 78. [Google Scholar] [CrossRef]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; Sandhu, S.K.; Hamid, O.; Spreafico, A.; Kasper, S.; Dummer, R.; Shimizu, T.; Steeghs, N.; Lewis, N.; Talluto, C.C.; et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019, 37, 2507. [Google Scholar] [CrossRef]
- Plieth, J. Chinook Marks an Ignominious End for Sting. Available online: https://www.evaluate.com/vantage/articles/news/chinook-marks-ignominious-end-sting (accessed on 7 September 2021).
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018, 29, viii712. [Google Scholar] [CrossRef]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369. [Google Scholar] [CrossRef] [PubMed]
- Babiker, H.M.; Subbiah, V.; Ali, A.; Algazi, A.; Schachter, J.; Lotem, M.; Maurice-Dror, C.; Hendler, D.; Rahimian, S.; Minderman, H.; et al. Abstract CT134: Tilsotolimod engages the TLR9 pathway to promote antigen presentation and Type-I IFN signaling in solid tumors. Cancer Res. 2020, 80, CT134. [Google Scholar] [CrossRef]
- Haymaker, C.; Andtbacka, R.H.; Johnson, D.B.; Shaheen, M.F.; Rahimian, S.; Chunduru, S.; Gabrail, N.; Doolittle, G.; Puzanov, I.; Markowitz, J.; et al. 1083MO—Final results from ILLUMINATE-204, a phase I/II trial of intratumoral tilsotolimod in combination with ipilimumab in PD-1 inhibitor refractory advanced melanoma. Ann. Oncol. 2020, 31, S672–S710. [Google Scholar] [CrossRef]
- Idera Pharmaceuticals Announces Results from ILLUMINATE-301 Trial of Tilsotolimod and Ipilimumab in Anti-PD-1 Refractory Advanced Melanoma. Available online: https://ir.iderapharma.com/news-releases/news-release-details/idera-pharmaceuticals-announces-results-illuminate-301-trial (accessed on 1 September 2021).
- Ekeke, C.N.; Russell, K.L.; Joubert, K.; Bartlett, D.L.; Luketich, J.D.; Soloff, A.C.; Guo, Z.S.; Lotze, M.T.; Dhupar, R. Fighting Fire With Fire: Oncolytic Virotherapy for Thoracic Malignancies. Ann. Surg. Oncol. 2021, 28, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Califano, R.; Lugowska, I.; Garassino, M.C. (Eds.) ESMO Handbook of Immuno-Oncology; ESMO Press: Lugano, Switzerland, 2018. [Google Scholar]
- Chen, C.Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018, 7, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Transgene Reports on the Combination Trial of TG4010, Chemotherapy and Nivolumab in Non-Small Cell Lung Cancer. Available online: https://www.businesswire.com/news/home/20191212005561/en/Transgene-Reports-on-the-Combination-Trial-of-TG4010-Chemotherapy-and-Nivolumab-in-Non-Small-Cell-Lung-Cancer (accessed on 2 September 2021).
- Rudin, C.M.; Pandha, H.S.; Gupta, S.; Zibelman, M.R.; Akerley, W.; Day, D.; Hill, A.G.; Sanborn, R.E.; O’Day, S.J.; Clay, T.D.; et al. Phase Ib KEYNOTE-200: A study of an intravenously delivered oncolytic virus, coxsackievirus A21 in combination with pembrolizumab in advanced NSCLC and bladder cancer patients. Ann. Oncol. 2018, 29, viii732. [Google Scholar] [CrossRef]
- Bentebibel, S.-E.; Hurwitz, M.E.; Bernatchez, C.; Haymaker, C.; Hudgens, C.W.; Kluger, H.M.; Tetzlaff, M.T.; Tagliaferri, M.A.; Zalevsky, J.; Hoch, U.; et al. A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rβγ-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors. Cancer Discov. 2019, 9, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, V.; Winer, I.S.; Gilbert, L.; Vaishampayan, U.N.; Rosen, S.D.; Muzaffar, J.; Spreafico, A.; McDermott, D.F.; Chu, Q.S.; Dumas, O.; et al. ARTISTRY-1: Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2021, 39, 2513. [Google Scholar] [CrossRef]
- Diab, A.; Tannir, N.M.; Bentebibel, S.-E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.M.; Awad, M.M.; Badin, F.B.; Rubinstein, M.P.; Bhar, P.; Garner, C.; Reddy, S.K.; Soon-Shiong, P. Preliminary data from QUILT 3.055: A phase 2 multi-cohort study of N803 (IL-15 superagonist) in combination with checkpoint inhibitors (CPI). J. Clin. Oncol. 2021, 39, 2596. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGF-beta: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Mauguen, A.; Reck, M.; Sandler, A.B.; Saijo, N.; Johnson, D.H.; Burcoveanu, D.; Fukuoka, M.; Besse, B.; Pignon, J.P. Systematic review and meta-analysis of randomised, phase II/III trials adding bevacizumab to platinum-based chemotherapy as first-line treatment in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Ciuleanu, T.-E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.-Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Manegold, C.; Dingemans, A.-M.C.; Gray, J.E.; Nakagawa, K.; Nicolson, M.; Peters, S.; Reck, M.; Wu, Y.-L.; Brustugun, O.T.; Crinò, L.; et al. The Potential of Combined Immunotherapy and Antiangiogenesis for the Synergistic Treatment of Advanced NSCLC. J. Thorac. Oncol. 2017, 12, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Socinski, M.A.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; et al. IMpower150 Final Overall Survival Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J. Thorac. Oncol. 2021, 16, 1909–1924. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Hellmann, M.D.; Hamid, O.; Tsai, K.K.; Loo, K.L.; Gubens, M.A.; Rosenblum, M.; Harview, C.L.; Taube, J.M.; Handley, N.; et al. Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol. Res. 2017, 5, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Mok, T.; Socinski, M.A.; Jotte, R.M.; Lim, D.W.T.; Cappuzzo, F.; Orlandi, F.J.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; et al. 1293P IMpower150: Updated efficacy analysis in patients with EGFR mutations. Ann. Oncol. 2020, 31, S837–S838. [Google Scholar] [CrossRef]
- Neal, J.W.; Lim, F.L.; Felip, E.; Gentzler, R.D.; Patel, S.B.; Baranda, J.; Fang, B.; Squillante, C.M.; Simonelli, M.; Werneke, S.; et al. Cabozantinib in combination with atezolizumab in non-small cell lung cancer (NSCLC) patients previously treated with an immune checkpoint inhibitor: Results from cohort 7 of the COSMIC-021 study. J. Clin. Oncol. 2020, 38, 9610. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Y.; Ren, S.; Zhao, J.; Chen, G.; Chen, J.; Gu, K.; Guo, R.; Pan, Y.; Wang, Q.; et al. 1267P Efficacy of camrelizumab (SHR-1210) plus apatinib as second-line treatment for advanced squamous NSCLC. Ann. Oncol. 2020, 31, S819. [Google Scholar] [CrossRef]
- Zhou, C.; Gao, G.; Wang, Y.N.; Zhao, J.; Chen, G.; Liu, Z.; Gu, K.; Huang, M.; He, J.; Chen, J.; et al. Efficacy of PD-1 monoclonal antibody SHR-1210 plus apatinib in patients with advanced nonsquamous NSCLC with wild-type EGFR and ALK. J. Clin. Oncol. 2019, 37, 9112. [Google Scholar] [CrossRef]
- Zhao, S.; Ren, S.; Jiang, T.; Zhu, B.; Li, X.; Zhao, C.; Jia, Y.; Shi, J.; Zhang, L.; Liu, X.; et al. Low-Dose Apatinib Optimizes Tumor Microenvironment and Potentiates Antitumor Effect of PD-1/PD-L1 Blockade in Lung Cancer. Cancer Immunol. Res. 2019, 7, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β; and PD-L1, in Second-Line Treatment of Patients with NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Adams, B. GSK, German Merck’s $4.2B Bintrafusp Alfa Drug a Bust, Fails to Beat King Keytruda in Lung Cancer. Available online: https://www.fiercebiotech.com/biotech/gsk-german-merck-s-4-2b-bintrafusp-alfa-drug-a-bust-fails-to-beat-king-keytruda-lung-cancer (accessed on 15 September 2021).
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.-J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non–Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Update on the Phase III NEPTUNE Trial of Imfinzi plus Tremelimumab in Stage IV Non-Small Cell Lung Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2019/update-on-the-phase-iii-neptune-trial-of-imfinzi-plus-tremelimumab-in-stage-iv-non-small-cell-lung-cancer-21082019.html# (accessed on 4 September 2021).
- Leighl, N.B.; Laurie, S.A.; Goss, G.D.; Hughes, B.G.M.; Stockler, M.R.; Tsao, M.S.; Kulkarni, S.; Blais, N.; Joy, A.A.; Mates, M.; et al. CCTG BR.34: A randomized trial of durvalumab and tremelimumab +/- platinum-based chemotherapy in patients with metastatic (Stage IV) squamous or nonsquamous non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9502. [Google Scholar] [CrossRef]
- Johnson, M.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Vasiliev, A.; Trukhin, D.; Kim, S.; Ursol, G.; et al. Durvalumab ± Tremelimumab + Chemotherapy as First-Line Treatment for mNSCLC: Results from the Phase 3 POSEIDON Study. Presented at: World Conference on Lung Cancer 2021. Available online: https://jamanetwork.com/journals/jamaoncology/fullarticle/2763864 (accessed on 15 September 2021).
- Plieth, J. Sanofi does a spring clean. Available online: https://www.evaluate.com/vantage/articles/news/corporate-strategy/sanofi-does-spring-clean (accessed on 5 September 2021).
- Shim, B.Y.; Lee, S.; de Castro Carpeño, J.; Chiu, C.H.; Cobo, M.; Kim, H.R.; Ryu, J.S.; Tarruella, M.M.; Summers, Y.; Thomas, C.A.; et al. 1269P EMPOWER-lung 4: Phase II, randomized, open-label high dose or standard dose cemiplimab alone/plus ipilimumab in the second-line treatment of advanced non-small cell lung cancer (NSCLC). Ann. Oncol. 2020, 31, S820. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Coward, J.; Ganju, V.; Behzadigohar, R.; Kwong, K.; Xu, J.; Van, H.; Kong, P.; Yang, F.; Chen, L.; Guo, K.; et al. Preliminary safety, efficacy, and pharmacokinetics (PK) results of KN046 (bispecific anti-PD-L1/CTLA4) from a first-in-human study in subjects with advanced solid tumors. J. Clin. Oncol. 2019, 37, 2554. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Sumrow, B.J.; Stahl, K.; Shah, K.; Liu, D.; Li, J.; Hao, S.S.; De Costa, A.; Kaul, S.; Bendell, J.; et al. Development and Preliminary Clinical Activity of PD-1-Guided CTLA-4 Blocking Bispecific DART Molecule. Cell Rep. Med. 2020, 1, 100163. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Bar, J.; Rasco, D.W.; Ahn, M.J.; Yoh, K.; Kim, D.W.; Nagrial, A.; Satouchi, M.; Lee, D.H.; Spigel, D.R.; et al. Safety and efficacy of quavonlimab, a novel anti-CTLA-4 antibody (MK-1308), in combination with pembrolizumab in first-line advanced non-small-cell lung cancer. Ann. Oncol. 2021, 32, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Bedard, P.; Bang, Y.-J.; LoRusso, P.; Hodi, S.; Gordon, M.; D’Angelo, S.; D’Angelo, S.; Desai, J.; Garralda, E.; et al. Abstract CT302: Phase Ia/Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. Cancer Res. 2020, 80, CT302. [Google Scholar] [CrossRef]
- Johnson, M.L.; Patel, M.R.; Cherry, M.; Kang, Y.-K.; Yamaguchi, K.; Oh, D.-Y.; Hussein, M.A.; Kitano, S.; Kondo, S.; Hansen, A.R.; et al. Safety of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb, in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3063. [Google Scholar] [CrossRef]
- Hong, D.S.; Schoffski, P.; Calvo, A.; Sarantopoulos, J.; Olza, M.O.D.; Carvajal, R.D.; Prawira, A.; Kyi, C.; Esaki, T.; Akerley, W.L.; et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J. Clin. Oncol. 2018, 36, 3012. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Lakhani, N.J.; Johnson, M.L.; Park, H.; Wang, D.; Yap, T.A.; Dowlati, A.; Maki, R.G.; Lynce, F.; Ulahannan, S.V.; et al. First-in-human study of REGN3767 (R3767), a human LAG-3 monoclonal antibody (mAb), ± cemiplimab in patients (pts) with advanced malignancies. J. Clin. Oncol. 2019, 37, 2508. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.M.; Larkin, J.; Gomez-Roca, C.; Schadendorf, D.; Dummer, R.; et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann. Oncol. 2017, 28, v611–v612. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Hamilton, E.P.; Chmielowski, B.; Ulahannan, S.V.; Kindler, H.L.; Bahadur, S.W.; Clingan, P.R.; Mallesara, G.; Weickhardt, A.J.; et al. A phase I, first-in-human, open-label, dose-escalation study of MGD013, a bispecific DART molecule binding PD-1 and LAG-3, in patients with unresectable or metastatic neoplasms. J. Clin. Oncol. 2020, 38, 3004. [Google Scholar] [CrossRef]
- Harding, J.J.; Patnaik, A.; Moreno, V.; Stein, M.; Jankowska, A.M.; Mendizabal, N.V.d.; Liu, Z.T.; Koneru, M.; Calvo, E. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J. Clin. Oncol. 2019, 37, 12. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, W.M.D.; Forde, P.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, S.; et al. Abstract CT183: Phase (Ph) I/II study of MBG453± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019, 79, CT183. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Shitara, K.; Shimizu, T.; Yonemori, K.; Matsubara, N.; Ohno, I.; Kogawa, T.; Naito, Y.; Leopold, L.; et al. The safety and tolerability of epacadostat alone and in combination with pembrolizumab in patients with advanced solid tumors: Results from a first-in-Japanese phase I study (KEYNOTE-434). Investig. New Drugs 2021, 39, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Gettinger, S.; Chow, L.Q.M.; Gordon, M.; Awad, M.M.; Cha, E.; Gong, X.; Zhou, G.; Walker, C.; Leopold, L.; et al. Phase 1 study of epacadostat in combination with atezolizumab for patients with previously treated advanced nonsmall cell lung cancer. Int. J. Cancer 2020, 147, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Plieth, J.; Armstrong, M. Incyte’s Epacadostat Blow-Up Leaves a Trail of Destruction. Available online: https://www.evaluate.com/vantage/articles/news/incytes-epacadostat-blow-leaves-trail-destruction (accessed on 6 September 2021).
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; Ochoa de Olza, M.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Hansen, A.R.; Pishvaian, M.J.; Chow, L.Q.M.; McArthur, G.A.; Bauer, T.M.; Liu, S.V.; Sandhu, S.K.; Tsai, F.Y.-C.; Kim, J.; et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 101. [Google Scholar] [CrossRef]
- Massarelli, E.; Balmanoukian, A.S.; Vieito, M.; Tourneau, C.L.; Hernandez-Guerrero, T.; Trigo, J.M.; Aljumaily, R.; Chisamore, M.J.; Rogan, D.; Sung, R.; et al. INDUCE-1: Report on safety run-in cohorts combining Inducible T-cell co-stimulatory receptor (ICOS) agonist GSK3359609 (GSK609) with platinum+5-FU chemotherapy (5-FU/plat), with or without pembrolizumab (PE), for the treatment of advanced solid tumors. J. Clin. Oncol. 2020, 38, 6544. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Bozorgmehr, F.; Fischer, J.R.; Bischof, M.; Atmaca, A.; Wetzel, S.; Faehling, M.; Bottke, D.; Wermke, M.; Troost, E.G.C.; Schmidtke-Schrezenmeier, G.; et al. LBA58 ORR in patients receiving nivolumab plus radiotherapy in advanced non-small cell lung cancer: First results from the FORCE trial. Ann. Oncol. 2020, 31, S1187. [Google Scholar] [CrossRef]
- Tang, C.; Welsh, J.W.; de Groot, P.; Massarelli, E.; Chang, J.Y.; Hess, K.R.; Basu, S.; Curran, M.A.; Cabanillas, M.E.; Subbiah, V.; et al. Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin. Cancer Res. 2017, 23, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Kreimer, A.; Wei, J.; Wu, J.; Corum, L.; Reusch, E.; Woodward, J.; Cohen, D.; Bondada, S.; Adams, V.R.; et al. Priming immunotherapy with radiotherapy (RT) in advanced non-small cell lung cancer (NSCLC) and head and neck squamous cell cancer (HNSCC): Interim analysis of phase II clinical trial. J. Clin. Oncol. 2021, 39, 2628. [Google Scholar] [CrossRef]
- Bassetti, M.F.; Sethakorn, N.; Lang, J.M.; Schehr, J.L.; Schultz, Z.; Morris, Z.S.; Matkowskyj, K.A.; Eickhoff, J.C.; Morris, B.; Traynor, A.M.; et al. Outcomes and safety analysis of a phase IB trial of stereotactic body radiotherapy (SBRT) to all sites of oligometastatic non-small cell lung cancer combined with durvalumab and tremelimumab. J. Clin. Oncol. 2021, 39, e21212. [Google Scholar] [CrossRef]
- Lin, J.; Hoffman-Censits, J.H.; Kelly, W.K.; Tuluc, M.; Shaw, C.; Philipose, S.; Leiby, B.E.; Louie, J.; Harshyne, L.; Kean, R.; et al. An exploratory study to investigate the immunomodulatory activity of radiation therapy in combination with pembrolizumab in patients with renal cell cancer. J. Clin. Oncol. 2017, 35, e16058. [Google Scholar] [CrossRef]
- Patel, M.R.; Tolcher, A.W.; Rasco, D.W.; Johnson, M.L.; Alistar, A.T.; Li, L.; Chung, A.H.; Andtbacka, R.H.I. BDB001, an intravenously administered toll-like receptor 7 and 8 (TLR7/8) agonist, in combination with pembrolizumab in advanced solid tumors: Phase 1 safety and efficacy results. J. Clin. Oncol. 2021, 39, 2512. [Google Scholar] [CrossRef]
- Garon, E.B.; Spira, A.I.; Johnson, M.; Bazhenova, L.; Leach, J.; Cummings, A.L.; Candia, A.; Coffman, R.L.; Janatpour, M.J.; Janssen, R.; et al. A Phase Ib Open-Label, Multicenter Study of Inhaled DV281, a TLR9 Agonist, in Combination with Nivolumab in Patients with Advanced or Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 4566–4573. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Jimenez, M.M.; Shimizu, T.; Keam, B.; Meric-Bernstam, F.; Rutten, A.; Glaspy, J.; Parikh, N.S.; Ising, M.; Hassounah, N.; et al. Abstract CT103: Phase I study of LHC165 ± spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2021, 81, CT103. [Google Scholar] [CrossRef]
- Siu, L.; Brody, J.; Gupta, S.; Marabelle, A.; Jimeno, A.; Munster, P.; Grilley-Olson, J.; Rook, A.H.; Hollebecque, A.; Wong, R.K.S.; et al. Safety and clinical activity of intratumoral MEDI9197 alone and in combination with durvalumab and/or palliative radiation therapy in patients with advanced solid tumors. J. ImmunoTher. Cancer 2020, 8, e001095. [Google Scholar] [CrossRef]
- Merchan, J.R.; Patel, M.; Cripe, T.P.; Old, M.O.; Strauss, J.F.; Thomassen, A.; Diaz, R.M.; Peng, K.W.; Russell, S.J.; Russell, L.; et al. Relationship of infusion duration to safety, efficacy, and pharmacodynamics (PD): Second part of a phase I-II study using VSV-IFNβ-NIS (VV1) oncolytic virus in patients with refractory solid tumors. J. Clin. Oncol. 2020, 38, 3090. [Google Scholar] [CrossRef]
- Galffy, G.; Lugowska, I.; Poddubskaya, E.; Cho, B.C.; Ahn, M.-J.; Han, J.-Y.; Su, W.-C.; Hauke, R.; Dyar, S.; Lee, D.H.; et al. 281 JAVELIN Medley VEGF: Phase 2 study of avelumab + axitinib in patients with previously treated non-small cell lung cancer (NSCLC) or treatment naive, cisplatin-ineligible urothelial cancer (UC). J. ImmunoTher. Cancer 2020, 8, A171–A172. [Google Scholar] [CrossRef]
- Solomon, B.; Callejo, A.; Bar, J.; Berchem, G.; Bazhenova, L.; Saintigny, P.; Raymond, E.; Girard, N.; Sulaiman, R.; Bresson, C.; et al. 1574P—Survival prolongation by rationale innovative genomics (SPRING): An international WIN consortium phase I study exploring safety and efficacy of avelumab, palbociclib, and axitinib in advanced non-small cell lung cancer (NSCLC) with integrated genomic and transcriptomic correlates. Ann. Oncol. 2019, 30, v648. [Google Scholar] [CrossRef]
- Ardeshir-Larijani, F.; Althouse, S.K.; Leal, T.; Feldman, L.E.; Hejleh, T.A.; Patel, M.; Gentzler, R.D.; Miller, A.R.; Hanna, N.H. Phase II trial of atezolizumab (A) + carboplatin (C) + pemetrexed (P) + bevacizumab (B) in pts with stage IV non-squamous non-small cell lung cancer (NSq-NSCLC): Big Ten Cancer Research Consortium Study LUN 17-139. J. Clin. Oncol. 2021, 39, 9034. [Google Scholar] [CrossRef]
- Nishio, M.; Peled, N.; Zer, A.; Houghton, B.; Bar, J.; Drew, D.; Herbst, R.; Rodriguez-Abreu, D.; Talpur, R.; Golden, L.; et al. 1313P Phase III LEAP-006 safety run-in (Part 1): 1L pembrolizumab (Pembro) + chemotherapy (Chemo) with lenvatinib (Len) for metastatic NSCLC. Ann. Oncol. 2020, 31, S848–S849. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Golan, T.; Lin, C.-C.; Dahan, L.; Fu, S.; Moreno, V.; Geva, R.; Reck, M.; Wasserstrom, H.A.; Mi, G.; et al. Ramucirumab (Ram) and durvalumab (Durva) treatment of metastatic non-small cell lung cancer (NSCLC), gastric/gastroesophageal junction (G/GEJ) adenocarcinoma, and hepatocellular carcinoma (HCC) following progression on systemic treatment(s). J. Clin. Oncol. 2019, 37, 2528. [Google Scholar] [CrossRef]
- Herbst, R.S.; Arkenau, H.T.; Santana-Davila, R.; Calvo, E.; Paz-Ares, L.; Cassier, P.A.; Bendell, J.; Penel, N.; Krebs, M.G.; Martin-Liberal, J.; et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non-small-cell lung cancer, gastro-oesophageal cancer, or urothelial carcinomas (JVDF): A multicohort, non-randomised, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1109–1123. [Google Scholar] [CrossRef]
- Herbst, R.S.; Arkenau, H.T.; Bendell, J.; Arrowsmith, E.; Wermke, M.; Soriano, A.; Penel, N.; Santana-Davila, R.; Bischoff, H.; Chau, I.; et al. Phase 1 Expansion Cohort of Ramucirumab Plus Pembrolizumab in Advanced Treatment-Naive NSCLC. J. Thorac. Oncol. 2021, 16, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.; Guenther, K.; Alpert, A.; Andersson, B.; Coughlin, Z.; Fritsche, J.; Hilf, N.; Hwu, P.; Kalra, M.; Kuttruff-Coqui, S.; et al. 293 Resultsof the first-in-human clinical trial with personalized multi-target adoptive cell therapy (ACTolog IMA101). J. ImmunoTher. Cancer 2020, 8, A179. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Borch, T.H.; Chamberlain, C.; Lorentzen, C.L.; Nielsen, M.; Kjeldsen, J.W.; Moerk, S.K.; Donia, M.; Svane, I.M. 1022MO Clinical potential of adoptive cell therapy with tumour infiltrating lymphocytes therapy in combination with checkpoint inhibitors in non-melanoma patients. Ann. Oncol. 2020, 31, S706. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corke, L.; Sacher, A. New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer. Curr. Oncol. 2022, 29, 38-55. https://doi.org/10.3390/curroncol29010004
Corke L, Sacher A. New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer. Current Oncology. 2022; 29(1):38-55. https://doi.org/10.3390/curroncol29010004
Chicago/Turabian StyleCorke, Lucy, and Adrian Sacher. 2022. "New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer" Current Oncology 29, no. 1: 38-55. https://doi.org/10.3390/curroncol29010004
APA StyleCorke, L., & Sacher, A. (2022). New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer. Current Oncology, 29(1), 38-55. https://doi.org/10.3390/curroncol29010004