Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study



Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Interphase Fluorescent In Situ Hybridization
2.3. Outcome Measures
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Prognostic Factors at Diagnosis
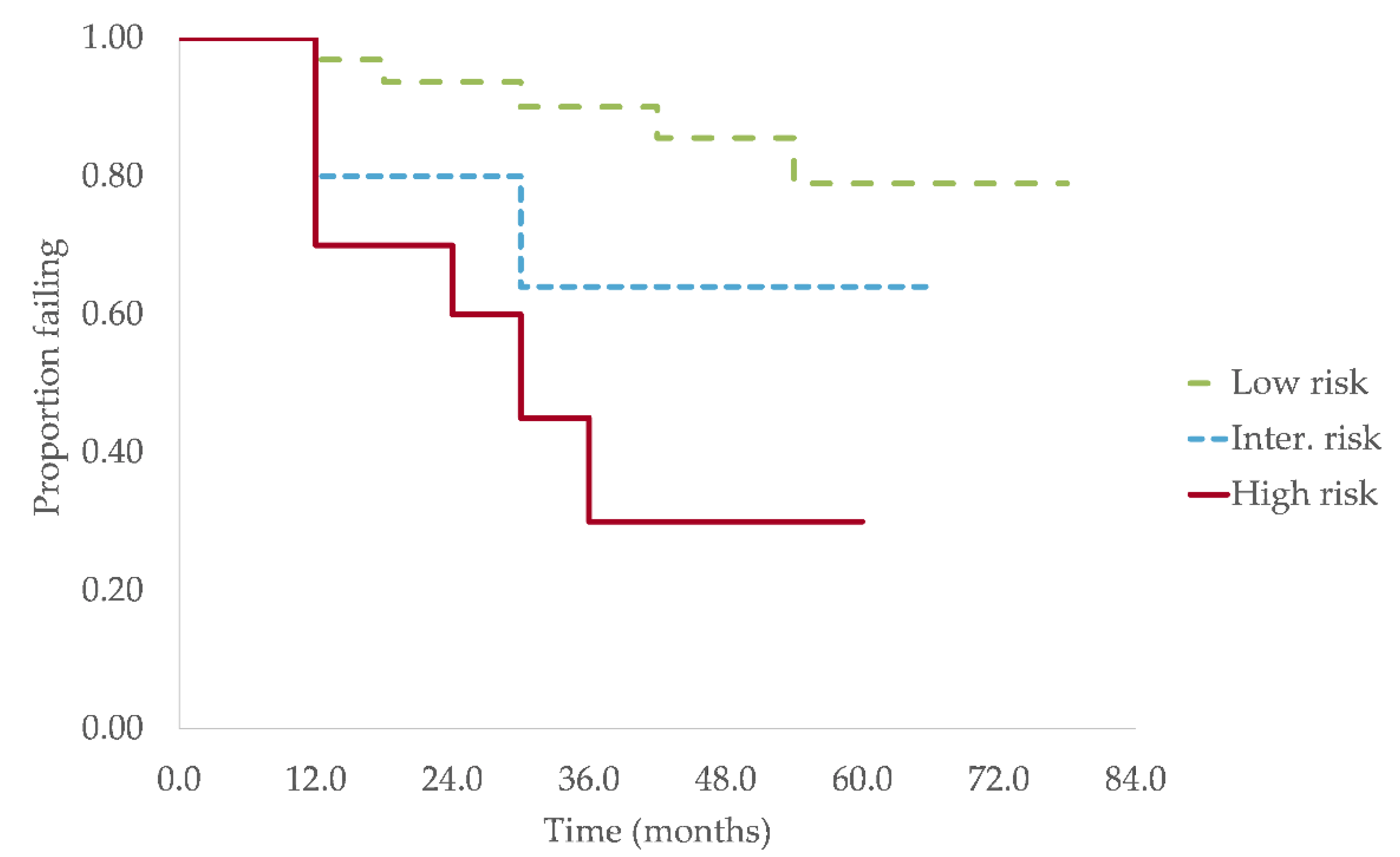
3.3. Progression and Survival Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Greipp, P.R. Smoldering multiple myeloma. N. Engl. J. Med. 1980, 302, 1347–1349. [Google Scholar] [CrossRef]
- Blum, A.; Bazou, D.; O’Gorman, P. Smoldering multiple myeloma: Prevalence and current evidence guiding treatment decisions. Blood Lymphat. Cancer 2018, 8, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.; Bartley, A.C.; Holton, S.J.; Gonsalves, W.I.; Kapoor, P.; Siddiqui, M.A.; Hashmi, S.K.; Marshall, A.L.; Ashrani, A.A.; Dispenzieri, A.; et al. Prevalence, incidence and survival of smoldering multiple myeloma in the United States. Blood Cancer J. 2016, 6, e486. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Kastritis, E.; Terpos, E.; Moulopoulos, L.; Spyropoulou-Vlachou, M.; Kanellias, N.; Eleftherakis-Papaiakovou, E.; Gkotzamanidou, M.; Migkou, M.; Gavriatopoulou, M.; Roussou, M.; et al. Extensive bone marrow infiltration and abnormal free light chain ratio identifies patients with asymptomatic myeloma at high risk for progression to symptomatic disease. Leukemia 2013, 27, 947–953. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Stewart, A.K.; Chanan-Khan, A.; Rajkumar, S.V.; Kyle, R.A.; Fonseca, R.; Kapoor, P.; Bergsagel, P.L.; McCurdy, A.; Gertz, M.A.; et al. Smoldering multiple myeloma requiring treatment: Time for a new definition? Blood 2013, 122, 4172–4181. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wheatley, K.; Clark, O.; Glasmacher, A.; Ross, H.; Djulbegovic, B. Early versus deferred treatment for early stage multiple myeloma. Cochrane Database Syst. Rev. 2003, CD004023. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, A.; Mora, O.; Tinelli, C.; Valentini, D.; Brugnatelli, S.; Spanedda, R.; De Paoli, A.; Barbarano, L.; Di Stasi, M.; Giordano, M.; et al. Long-term survival of stage I multiple myeloma given chemotherapy just after diagnosis or at progression of the disease: A multicentre randomized study. Cooperative Group of Study and Treatment of Multiple Myeloma. Br. J. Cancer 2000, 82, 1254–1260. [Google Scholar] [CrossRef]
- Hjorth, M.; Hellquist, L.; Holmberg, E.; Magnusson, B.; Rodjer, S.; Westin, J. Initial versus deferred melphalan-prednisone therapy for asymptomatic multiple myeloma stage I—A randomized study. Myeloma Group of Western Sweden. Eur. J. Haematol. 1993, 50, 95–102. [Google Scholar] [CrossRef]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Study of Siltuximab in High-Risk Smoldering Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Mateos, M.V.; Kumar, S.; Dimopoulos, M.A.; Gonzalez-Calle, V.; Kastritis, E.; Hajek, R.; De Larrea, C.F.; Morgan, G.J.; Merlini, G.; Goldschmidt, H.; et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Aljama, M.A.; Sidiqi, M.H.; Lakshman, A.; Dispenzieri, A.; Jevremovic, D.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Dingli, D.; Muchtar, E.; et al. Plasma cell proliferative index is an independent predictor of progression in smoldering multiple myeloma. Blood Adv. 2018, 2, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Larrea, C.; Isola, I.; Pereira, A.; Cibeira, M.T.; Magnano, L.; Tovar, N.; Rodriguez-Lobato, L.G.; Calvo, X.; Arostegui, J.I.; Diaz, T.; et al. Evolving M-protein pattern in patients with smoldering multiple myeloma: Impact on early progression. Leukemia 2018, 32, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Calle, V.; Davila, J.; Escalante, F.; de Coca, A.G.; Aguilera, C.; Lopez, R.; Barez, A.; Alonso, J.M.; Hernandez, R.; Hernandez, J.M.; et al. Bence Jones proteinuria in smoldering multiple myeloma as a predictor marker of progression to symptomatic multiple myeloma. Leukemia 2016, 30, 2026–2031. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.T.; Kumar, S.K.; Dispenzieri, A.; Kyle, R.A.; Katzmann, J.A.; Rajkumar, S.V. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia 2013, 27, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Corral, L.; Gutierrez, N.C.; Vidriales, M.B.; Mateos, M.V.; Rasillo, A.; Garcia-Sanz, R.; Paiva, B.; San Miguel, J.F. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin. Cancer Res. 2011, 17, 1692–1700. [Google Scholar] [CrossRef]
- Mullikin, T.C.; Rajkumar, S.V.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Lin, Y.; Dingli, D.; Go, R.S.; Hayman, S.R.; Zeldenrust, S.R.; et al. Clinical characteristics and outcomes in biclonal gammopathies. Am. J. Hematol. 2016, 91, 473–475. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Perez-Persona, E.; Vidriales, M.B.; Mateo, G.; Garcia-Sanz, R.; Mateos, M.V.; de Coca, A.G.; Galende, J.; Martin-Nunez, G.; Alonso, J.M.; de Las Heras, N.; et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef]
- Rosinol, L.; Blade, J.; Esteve, J.; Aymerich, M.; Rozman, M.; Montoto, S.; Gine, E.; Nadal, E.; Filella, X.; Queralt, R.; et al. Smoldering multiple myeloma: Natural history and recognition of an evolving type. Br. J. Haematol. 2003, 123, 631–636. [Google Scholar] [CrossRef]
- Siragusa, S.; Morice, W.; Gertz, M.A.; Kyle, R.A.; Greipp, P.R.; Lust, J.A.; Witzig, T.E.; Lacy, M.Q.; Zeldenrust, S.R.; Rajkumar, S.V.; et al. Asymptomatic immunoglobulin light chain amyloidosis (AL) at the time of diagnostic bone marrow biopsy in newly diagnosed patients with multiple myeloma and smoldering myeloma. A series of 144 cases and a review of the literature. Ann. Hematol. 2011, 90, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Melton, L.J., 3rd; Benson, J.T.; Kumar, S.; Rajkumar, S.V. Clinical course of light-chain smouldering multiple myeloma (idiopathic Bence Jones proteinuria): A retrospective cohort study. Lancet Haematol. 2014, 1, e28–e36. [Google Scholar] [CrossRef]
- Rossi, D.; Fangazio, M.; De Paoli, L.; Puma, A.; Riccomagno, P.; Pinto, V.; Zigrossi, P.; Ramponi, A.; Monga, G.; Gaidano, G. Beta-2-microglobulin is an independent predictor of progression in asymptomatic multiple myeloma. Cancer 2010, 116, 2188–2200. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Iida, S.; Matsue, K.; Sunami, K.; Isoda, J.; Harada, N.; Saburi, Y.; Okamura, S.; Kumagae, K.; Watanabe, J.; et al. Predictive Significance of Serum Beta 2-Microglobulin Levels and M-Protein Velocity for Symptomatic Progression of Smoldering Multiple Myeloma. Blood 2014, 124, 3379. [Google Scholar] [CrossRef]
- Sorrig, R.; Klausen, T.W.; Salomo, M.; Vangsted, A.J.; Ostergaard, B.; Gregersen, H.; Frolund, U.C.; Andersen, N.F.; Helleberg, C.; Andersen, K.T.; et al. Smoldering multiple myeloma risk factors for progression: A Danish population-based cohort study. Eur. J. Haematol. 2016, 97, 303–309. [Google Scholar] [CrossRef]
- Hajek, R.; Sandecka, V.; Spicka, I.; Raab, M.; Goldschmidt, H.; Beck, S.; Minarik, J.; Pavlicek, P.; Radocha, J.; Heindorfer, A.; et al. Identification of patients with smouldering multiple myeloma at ultra-high risk of progression using serum parameters: The Czech Myeloma Group model. Br. J. Haematol. 2020, 190, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Hielscher, T.; Schult, D.; Mai, E.K.; Raab, M.S.; Hillengass, J.; Seckinger, A.; Hose, D.; Granzow, M.; Jauch, A.; et al. Cytogenetic subclone formation and evolution in progressive smoldering multiple myeloma. Leukemia 2020, 34, 1192–1196. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Risk Stratification Model | Number of Risk Factors | 2 Years PFS (%) |
|---|---|---|
| 20/20/20 model (Lakshman et al.) | ||
| Low risk | 0 | 90.3 |
| Intermediate risk | 1 | 73.7 |
| High risk | 2–3 | 52.6 |
| 20/20/20 model with high-risk cytogenetics (Mateos et al.) | ||
| Low risk | 0 | 94.0 |
| Low-intermediate risk | 1 | 77.2 |
| Intermediate risk | 2 | 54.5 |
| High risk | 3–4 | 36.9 |
| Parameters | Study Cohort | Mayo Clinic Cohort | Other Cohorts 1 |
|---|---|---|---|
| Age, years, median (range) | 70.0 (39.0–86.9) | 64.9 (30.2–92.1) | 63–70 years [3,4,18,25] |
| Sex | |||
| Male, n (%) | 54 (60.7) | 58.2% | 53–64% [4,17,18] |
| Evolution of gammopathy | |||
| MGUS prior to SMM, n (%) | 21 (23.6) | n/d | 28% [25] |
| New Mayo Clinic model markers | |||
| BMPC (n = 89), %, median (range) | 15.0 (7.5–50.0) | 20 (5–50) | 15–20% [14,16,17,18] |
| BMPC ≥ 20%, n (%) | 21 (23.6) | ||
| Serum M-protein (n = 89), g/L, median (range) | 11.7 (0–35.2) | 20 (0–50) | 16–25 g/L [6,14,17,18] |
| M-protein ≥ 20 g/L, n (%) | 8 (9.0) | ||
| FLCr (n = 52), median (range) | 7.4 (1.1–76.4) | 7.8 (0.3–281.5) | 6.9–11.6 [14,19,24] |
| FLCr ≥ 20, n (%) | 14 (26.9) | ||
| Heavy chain isotype (n = 89), n (%) | |||
| IgG | 52 (58.4) | 75.8% | 69–75% [4,14,17,18] |
| IgA | 32 (36.0) | 19.7% | 19–31% [4,16,17,18] |
| IgM | 0 (0.0) | 0.9% | 0.9–1% [14,16,19] |
| Light chain | 5 (5.6) | 3.6% | 1–4% [14,16,17,19] |
| Nb of M-protein spike (n = 89), n (%) | |||
| 0 | 4 (4.5) | n/d | n/a |
| 1 | 73 (82.0) | n/d | n/a |
| ≥2 | 12 (13.5) | n/d | 2–12% [19,21,26] |
| Immunoparesis (n = 86), n (%) | |||
| Present | 52 (60.5) | 70.4% | 50–83% [4,14,17,18] |
| Reduction of 1 Ig | 17 (19.8) | n/d | 18–31% [4,23,27] |
| Reduction of ≥ 2 Ig | 35 (40.7) | n/d | 29–52% [4,23,27] |
| Abnormal FISH (n = 54), n (%) | |||
| 2008–2014 (n = 15) | 1 (6.7) | n/a | |
| 2015–2020 (n = 39) | 14 (35.9) | 79.8% | |
| Deletion 17p | 2 (5.1) | 2.4% | 1.7–6.1% [14,15,22,24] |
| Translocation t(4;14) | 3 (7.7) | 11.1% | 8.9–11.1% [14,15,22,24] |
| Hyperdiploidy | 8 (20.5) | 43.4% | 31.4–43.9% [14,15,22,24] |
| Other biomarkers, n (%) | |||
| Positive Bence-Jones (n = 73) | 33 (45.2) | n/d | 22–40% [17,18,23] |
| LDH ≥ 250 UI/L (n = 63) | 4 (6.3) | 8.6% | n/a 2 |
| Albumin < 35 g/L (n = 61) | 3 (4.9) | 30.8% | n/a 2 |
| β2-microglobuline ≥ 186 μmol/L (n = 73) | 46 (63.0) | n/d | n/a 2 |
| Possible Prognosis Factors | TTP, Months, Median (IQR) | Univariable Analysis | Multivariable Analysis | ||
|---|---|---|---|---|---|
| HR (95% C.I.) | p Value | HR (95% C.I.) | p Value | ||
| Overall | 19.3 (9.73–35.6) | --- | --- | --- | --- |
| Sex | |||||
| Male | 24.9 (11.0–40.3) | 0.95 (0.46–1.96) | 0.885 | --- | --- |
| Female | 18.0 (9.3–33.0) | ||||
| Evolution of gammopathy | |||||
| MGUS prior to SMM | 18.0 (9.5–26.9) | 1.61 (0.74–3.50) | 0.231 | --- | --- |
| No previous MGUS | 24.5 (9.9–37.2) | ||||
| New Mayo Clinic model | |||||
| BMPC percentage | |||||
| BMPC ≥ 20% | 13.9 (7.7–28.9) | 3.91 (1.90–8.01) | <0.001 | 4.28 (1.90–9.61) | <0.001 |
| BMPC < 20% | 24.9 (14.9–43.9) | ||||
| Serum M-protein | |||||
| M-protein ≥ 20 g/L | 7.5 (2.6–11.4) | 2.64 (0.92–7.62) | 0.072 | 4.20 (1.13–15.53) | 0.032 |
| M-protein < 20 g/L | 24.9 (11.0–36.1) | ||||
| FLCr | |||||
| FLCr ≥ 20 | 14.9 (9.1–30.7) | 3.25 (1.09–9.71) | 0.035 | --- | --- |
| FLCr < 20 | 21.4 (9.8–33.5) | ||||
| Heavy chain isotype | |||||
| IgG | 24.9 (11.4–41.3) | 0.91 (0.42–1.99) | 0.815 | --- | --- |
| IgA | 18.5 (8.6–26.4) | ||||
| Light chain | 19.2 (9.0–33.0) | 2.42 (1.35–4.35) | 0.003 | 1.46 (0.33–6.42) | 0.619 |
| Number of M-protein spike | |||||
| >1 | 18.0 (6.9–27.4) | 2.39 (1.10–5.19) | 0.028 | 2.40 (0.88–6.53) | 0.086 |
| 1 | 24.5 (10.8–40.8) | ||||
| Immunoparesis | |||||
| Present | 19.2 (9.6–28.9) | 2.99 (1.27–7.03) | 0.012 | 2.61 (1.07–6.41) | 0.036 |
| Absent | 36.1 (19.0–47.6) | ||||
| FISH | |||||
| Normal and low-risk | 21.9 (8.6–34.0) | 1.57 (0.54–4.56) | 0.403 | --- | --- |
| Inter. and high-risk | 18.0 (13.6–24.9) | ||||
| High-risk only | 21.1 (11.4–25.3) | 2.22 (0.71–6.96) | 0.169 | --- | --- |
| Biomarkers, n (%) | |||||
| Bence-Jones | |||||
| Positive | 29.6 (9.9–39.2) | 1.15 (0.52–2.53) | 0.732 | --- | --- |
| Negative | 19.0 (10.0–38.2) | ||||
| β2-microglobuline | |||||
| ≥186 μmol/L | 18.5 (9.0–40.8) | 0.98 (0.44–2.22) | 0.970 | --- | --- |
| <186 μmol/L | 24.5 (10.7–39.8) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessier, C.; Allard, T.; Boudreault, J.-S.; Kaedbey, R.; Éthier, V.; Fortin, F.; Pavic, M. Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study. Curr. Oncol. 2021, 28, 2029-2039. https://doi.org/10.3390/curroncol28030188
Tessier C, Allard T, Boudreault J-S, Kaedbey R, Éthier V, Fortin F, Pavic M. Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study. Current Oncology. 2021; 28(3):2029-2039. https://doi.org/10.3390/curroncol28030188
Chicago/Turabian StyleTessier, Camille, Thomas Allard, Jean-Samuel Boudreault, Rayan Kaedbey, Vincent Éthier, Fléchère Fortin, and Michel Pavic. 2021. "Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study" Current Oncology 28, no. 3: 2029-2039. https://doi.org/10.3390/curroncol28030188
APA StyleTessier, C., Allard, T., Boudreault, J.-S., Kaedbey, R., Éthier, V., Fortin, F., & Pavic, M. (2021). Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study. Current Oncology, 28(3), 2029-2039. https://doi.org/10.3390/curroncol28030188