
Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review


Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Summary
3.1.1. Presenting Concerns
3.1.2. Clinical Findings
3.2. Diagnostic Tests
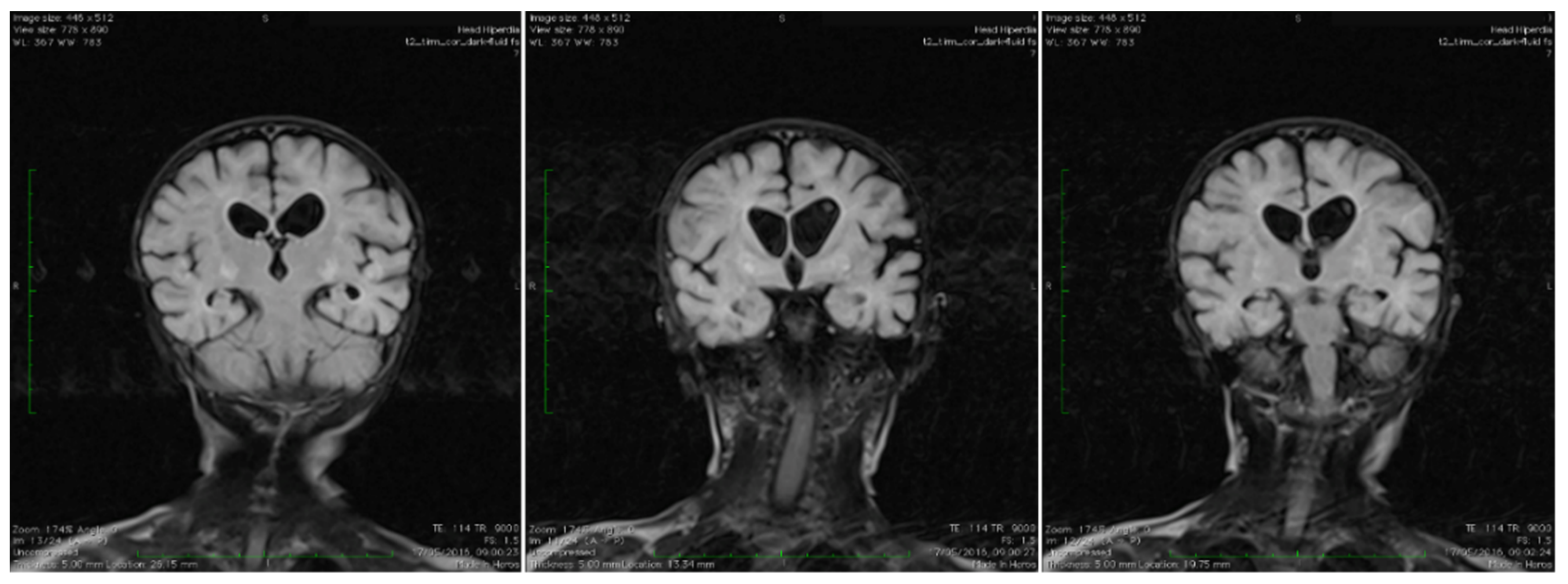
3.2.1. Biologic and Imagistic Assessment
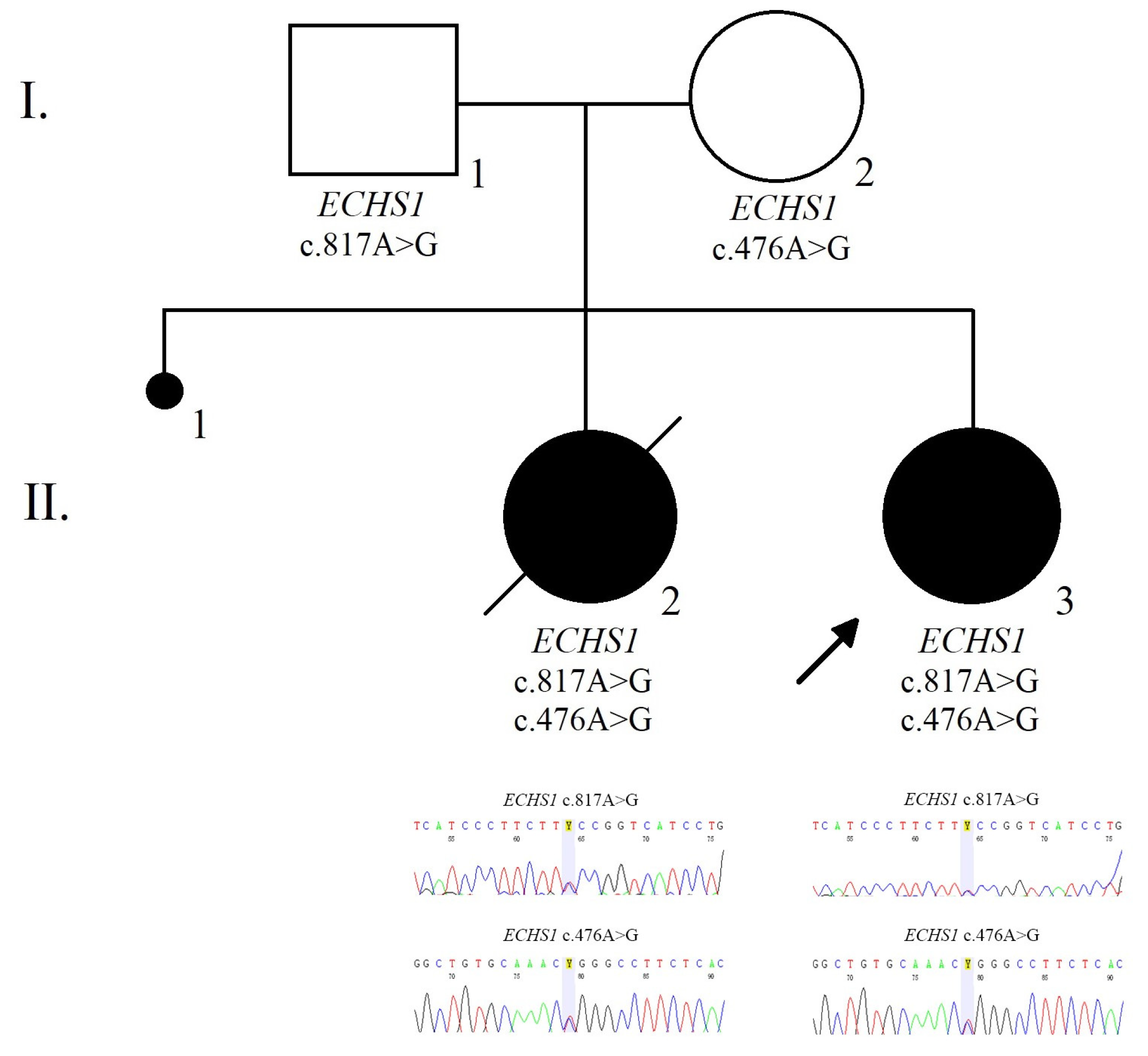
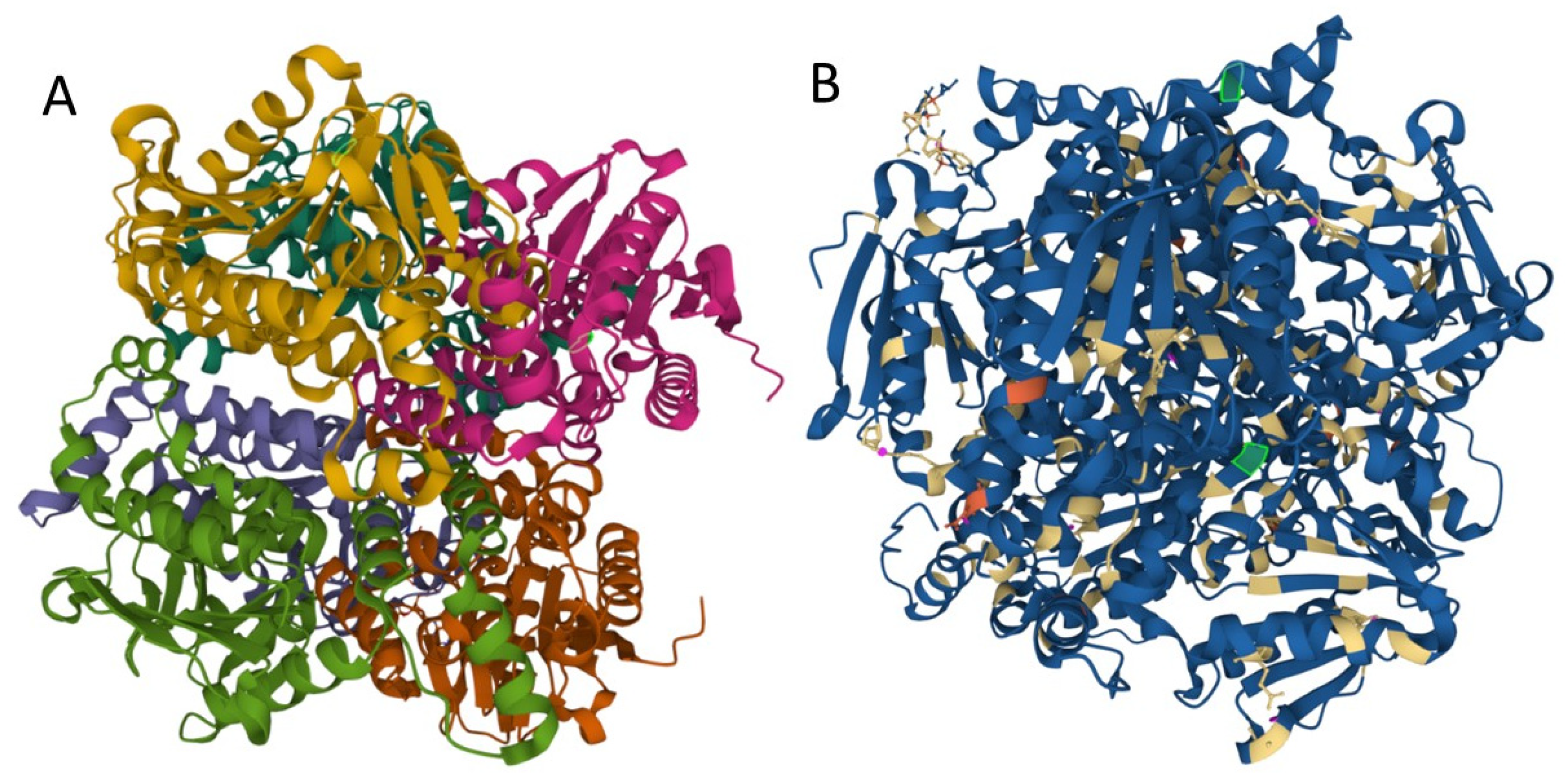
3.2.2. Genetic Analysis
3.2.3. Prenatal Testing and Genetic Diagnosis
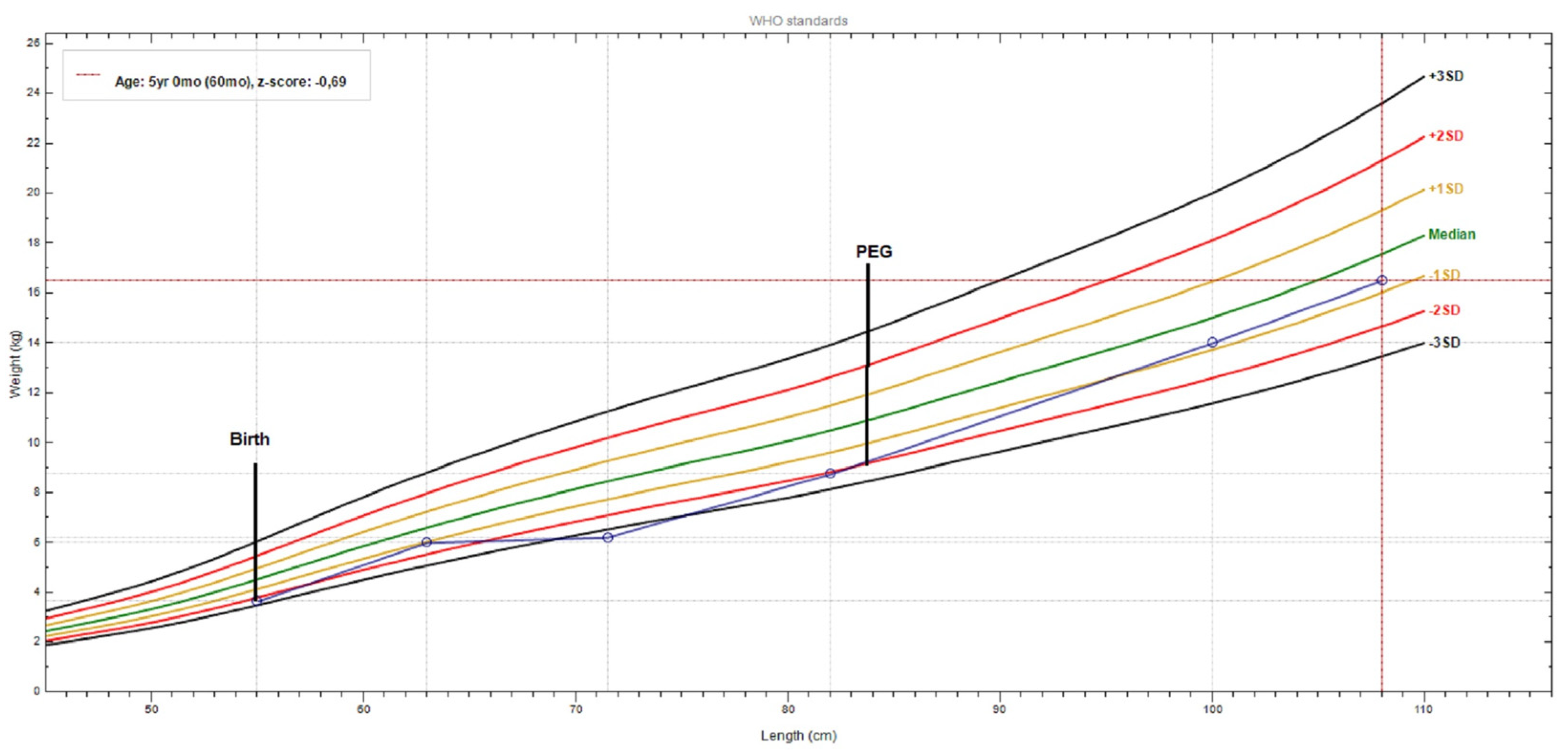
3.3. Therapeutic Focus and Assessment
3.4. Follow-Up and Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.; Yu, D. Clinical, biochemical and metabolic characterization of patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency: Two case reports and the review of the literature. BMC Pediatr. 2020, 20, 50. [Google Scholar] [CrossRef]
- Uesugi, M.; Mori, J.; Fukuhara, S.; Fujii, N.; Omae, T.; Sasai, H.; Ichimoto, K.; Murayama, K.; Osamura, T.; Hosoi, H. Short-chain enoyl-CoA hydratase deficiency causes prominent ketoacidosis with normal plasma lactate levels: A case report. Mol. Genet. Metab. Rep. 2020, 25, 100672. [Google Scholar] [CrossRef]
- Abdenur, J.E.; Sowa, M.; Simon, M.; Steenari, M.; Skaar, J.; Eftekharian, S.; Chang, R.; Ferdinandusse, S.; Pitt, J. Medical nutrition therapy in patients with HIBCH and ECHS1 defects: Clinical and biochemical response to low valine diet. Mol. Genet. Metab. Rep. 2020, 24, 100617. [Google Scholar] [CrossRef]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Klopstock, T.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimons, P.E.; Alston, C.L.; Bonnen, P.E.; Hughes, J.; Crushell, E.; Geraghty, M.T.; Tetreault, M.; O’Reilly, P.; Twomey, E.; Sheikh, Y.; et al. Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. A 2018, 176, 1115–1127. [Google Scholar] [CrossRef]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.; Boneh, A.; Pitt, J. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Waterham, H.R.; Heales, S.J.; Brown, G.K.; Hargreaves, I.P.; Taanman, J.W.; Gunny, R.; Abulhoul, L.; Wanders, R.J.; Clayton, P.T.; et al. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet. J. Rare Dis. 2013, 8, 188. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.J.; McKenzie, M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells 2018, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Loupatty, F.J.; Clayton, P.T.; Ruiter, J.P.; Ofman, R.; Ijlst, L.; Brown, G.K.; Thorburn, D.R.; Harris, R.A.; Duran, M.; Desousa, C.; et al. Mutations in the gene encoding 3-hydroxyisobutyryl-CoA hydrolase results in progressive infantile neurodegeneration. Am. J. Hum. Genet. 2007, 80, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Grier, J.; Hirano, M.; Karaa, A.; Shepard, E.; Thompson, J.L.P. Diagnostic odyssey of patients with mitochondrial disease: Results of a survey. Neurol. Genet. 2018, 4, e230. [Google Scholar] [CrossRef] [Green Version]
- Bedoyan, J.K.; Yang, S.P.; Ferdinandusse, S.; Jack, R.M.; Miron, A.; Grahame, G.; DeBrosse, S.D.; Hoppel, C.L.; Kerr, D.S.; Wanders, R.J.A. Lethal neonatal case and review of primary short-chain enoyl-CoA hydratase (SCEH) deficiency associated with secondary lymphocyte pyruvate dehydrogenase complex (PDC) deficiency. Mol. Genet. Metab. 2017, 120, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Ganetzky, R.; Stojinski, C. Mitochondrial Short-Chain Enoyl-CoA Hydratase 1 Deficiency; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; GeneReviews: Seattle, WA, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/ (accessed on 15 May 2021).
- Pitceathly, R.D.; Keshavan, N.; Rahman, J.; Rahman, S. Moving towards clinical trials for mitochondrial diseases. J. Inherit. Metab. Dis. 2021, 44, 22–41. [Google Scholar] [CrossRef]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef]
- VarSome Database. Available online: https://varsome.com/ (accessed on 22 May 2021).
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- NTurnbull, A.P.; Salah, E.; Niesen, F.; Debreczeni, J.; Ugochukwu, E.; Pike, A.C.W.; Kavanagh, K.; Gileadi, O.; Gorrec, F.; Umeano, C.; et al. The crystal structure of human enoyl-coenzyme A (CoA) hydratase short chain 1, ECHS1.2006. Protein Data Bank 2006. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic. Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.G.; Sokol, R.J.; Hardison, R.M.; Alonso, E.M.; Squires, R.H.; Narkewicz, M.R.; Pediatric Acute Liver Failure Study Group. Lactate and Lactate: Pyruvate Ratio in the Diagnosis and Outcomes of Pediatric Acute Liver Failure. J. Pediatr. 2017, 182, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Debray, F.G.; Mitchell, G.A.; Allard, P.; Robinson, B.H.; Hanley, J.A.; Lambert, M. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin. Chem. 2007, 53, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137 Pt 11, 2903–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Mutairi, F.; Shamseldin, H.E.; Alfadhel, M.; Rodenburg, R.J.; Alkuraya, F.S. A lethal neonatal phenotype of mitochondrial short-chain enoyl-CoA hydratase-1 deficiency. Clin. Genet. 2017, 91, 629–633. [Google Scholar] [CrossRef] [PubMed]
- NCBI. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/40529478/; https://www.ncbi.nlm.nih.gov/clinvar/RCV000578268/; (accessed on 26 June 2020).
- Available online: https://gnomad.broadinstitute.org (accessed on 3 April 2021).
- Ferdinandusse, S.; Friederich, M.W.; Burlina, A.; Ruiter, J.P.; Coughlin, C.R., 2nd; Dishop, M.K.; Gallagher, R.C.; Bedoyan, J.K.; Vaz, F.M.; Waterham, H.R.; et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet. J. Rare Dis. 2015, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetreault, M.; Fahiminiya, S.; Antonicka, H.; Mitchell, G.A.; Geraghty, M.T.; Lines, M.; Boycott, K.M.; Shoubridge, E.A.; Mitchell, J.J.; Care4Rare Canada Consortium; et al. Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum. Genet. 2015, 134, 981–991. [Google Scholar] [CrossRef]
- Mahajan, A.; Constantinou, J.; Sidiropoulos, C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: Case presentation and initial benefit of intervention. J. Neurol. 2017, 264, 185–187. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Riley, L.G.; Bratkovic, D.; Ketteridge, D.; Manton, N.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Mohamed, M.; Gardeitchik, T.; et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J. Inherit. Metab. Dis. 2017, 40, 745–747. [Google Scholar] [CrossRef]
- Masnada, S.; Parazzini, C.; Bini, P.; Barbarini, M.; Alberti, L.; Valente, M.; Chiapparini, L.; De Silvestri, A.; Doneda, C.; Iascone, M.; et al. Phenotypic spectrum of short-chain enoyl-Coa hydratase-1 (ECHS1) deficiency. Eur. J. Paediatr. Neurol. 2020, 28, 151–158. [Google Scholar] [CrossRef]
- Marti-Sanchez, L.; Baide-Mairena, H.; Marcé-Grau, A.; Pons, R.; Skouma, A.; López-Laso, E.; Sigatullina, M.; Rizzo, C.; Semeraro, M.; Martinelli, D.; et al. Delineating the neurological phenotype in children with defects in the ECHS1 or HIBCH gene. J. Inherit. Metab. Dis. 2021, 44, 401–414. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R.; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef] [Green Version]
- Engelstad, K.; Salazar, R.; Koenigsberger, D.; Stackowtiz, E.; Brodlie, S.; Brandabur, M.; De Vivo, D.C. Exploring triheptanoin as treatment for short chain enoyl CoA hydratase deficiency. Ann. Clin. Transl. Neurol. 2021, 8, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Shayota, B.J.; Soler-Alfonso, C.; Bekheirnia, M.R.; Mizerik, E.; Boyer, S.W.; Xiao, R.; Yang, Y.; Elsea, S.H.; Scaglia, F. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am. J. Med. Genet. A 2019, 179, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, M.; Kojima, K.; Osaka, H.; Hamada, Y.; Jimbo, E.; Watanabe, M.; Aoki, S.; Sato-Shirai, I.; Ichimoto, K.; Fushimi, T.; et al. Valine metabolites analysis in ECHS1 deficiency. Mol. Genet. Metab. Rep. 2021, 29, 100809. [Google Scholar] [CrossRef] [PubMed]
- Sato-Shirai, I.; Ogawa, E.; Arisaka, A.; Osaka, H.; Murayama, K.; Kuwajima, M.; Watanabe, M.; Ichimoto, K.; Ohtake, A.; Kumada, S. Valine-restricted diet for patients with ECHS1 deficiency: Divergent clinical outcomes in two Japanese siblings. Brain Dev. 2021, 43, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Calvert, S.; Barwick, K.; Par, M.; Ni Tan, K.; Borges, K. A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur. J. Paediatr. Neurol. 2018, 22, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Vockley, J.; Burton, B.; Berry, G.T.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; Humphrey, R.; et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: Safety and efficacy in children and adults following 24weeks of treatment. Mol. Genet. Metab. 2017, 120, 370–377. [Google Scholar] [CrossRef]
- Ronchi, D.; Monfrini, E.; Bonato, S.; Mancinelli, V.; Cinnante, C.; Salani, S.; Bordoni, A.; Ciscato, P.; Fortunato, F.; Villa, M.; et al. Dystonia-ataxia syndrome with permanent torsional nystagmus caused by ECHS1 deficiency. Ann. Clin. Transl. Neurol. 2020, 7, 839–845. [Google Scholar] [CrossRef]
- Simon, M.T.; Eftekharian, S.S.; Ferdinandusse, S.; Tang, S.; Naseri, T.; Reupena, M.S.; McGarvey, S.T.; Minster, R.L.; Weeks, D.E.; Samoan Obesity, Lifestyle, and Genetic Adaptations (OLaGA) Study Group; et al. ECHS1 disease in two unrelated families of Samoan descent: Common variant—Rare disorder. Am. J. Med. Genet. A 2021, 185, 157–167. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum Parameters | Measured Value | Normal Range |
|---|---|---|
| lactate | 43.07 mmol/L | <2.5 mmol/L |
| ammonia | 1238 μg/L | 187–869 μg/L |
| lactic acid | 3.88 mmol/L | 0.5–2.2 mmol/L |
| pyruvate | 0.7 mmol/L | 0.41–0.67 mmol/L |
| Plasma Amino Acid | Value | Urinary Organic Acid | Value |
|---|---|---|---|
| Cystine | ↑ | 3-hydroxyisovaleric acid | ↑↑ |
| Valine | ↑ | pyruvic acid | ↑↑ |
| Alpha alanine | N | 2-methyl-2,3-dihydroxybutyric | ↑ |
| Acyl-carnitine | N |
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Romanian parents | Iraqi-Turkish Jewish/mother Iraqi -Libyan Jewish | Japanese and American parents | Netherlands parents | German parents | French-Canadian parents | NL | NL | Italian parents | Belgian parents | Greek parents |
| Genetic mutation | c.476A>G/c.817A>G | c.433C>T/c.476A>G | c.176A>G/ c.476A>G | c.161G>A/c.817A>G | c.229G>C/c.476A>G | c.538A>C/c.476A>G | c.518C>T/c.817A>G | c.476A>G/c.538A>G; | c.476A>G/c.139G>A | c.239C>T/c.817A>C | c.476A>C/ c.542G>T |
| Protein effect | p.Gln159Arg/p.Lys273Glu | p.Leu145Phe/ p.Gln159Arg | p.Asn59Ser/ p.Gln159Arg | p.Arg54His/ p.Lys273Glu | p.Glu77Gln/ p.Gln159Arg | p.Thr180Ala/p.Gln159Arg | p.Ala173Val/ p.Lys273Glu | p.Gln159Arg/ p.Thr180Ala | p.Gln159Arg/p.Val47Met | p.Pro80Leu/p.Lys273Glu | p.Gln159Arg/p.Arg181Leu |
| Allele source | c.476A>G mother c.817A>G father | NL | c.476A>G mother c.176A>G father | ND | c.476A>G mother father- ND | c.538A>C mother father NA | c.518C>T father c.817A>G mother | Parents- heterozygous for variants | c.476A > G father c.139G > A mother | NL | NL |
| Sibling (sex, mutation) | F, dead/c.476A>G/c.817A>G | No | F, dead in 1st day of life: respiratory failure, severe lactic acidosis; mutation NL | NL | M, alive, c.229G>C/= (p.Glu77Gln/=) (heterozygous carrier) | NL | NL | NL | NL | NL | NL |
| Sex | F | F | F | M | F | F | M | F | M | M | F |
| Age of onset | 3 mo | early infancy | birth | birth | 11 mo | 1 mo | 8 ys | 17 mo | birth | 35 mo | 8 mo |
| First neurological signs/symptoms | hypotonia | hypotonia | hypotonia | hypotonia | fist-clenching, teeth-gnashing, horizontal nystagmus | development delay or regression | dyskinesias | hypotonia, lower limbs-flexed on the trunk | lower limb paroxysmal dystonia | encephalopathy | |
| Neurologic involvement/characteristics | microcephaly, hypotonia dystonia, spasticity, nystagmus, seizure, inconsolable crying, optic atrophy, global developmental delay, hearing loss? | microcephaly, hypotonia, global developmental delay, optic atrophy, hearing loss | deafness | microcephaly, hypotonia, dystonia, spasticity, inconsolable crying, global developmental regress | dystonia, developmental delay, spastic, nystagmus, hearing loss, optic atrophy | microcephaly, hypotonia, dystonia, nystagmus, optic atrophy, hearing loss | dyskinesias, dystonia | hypotonia, dystonia global developmental delay | hypotonia, limbs spasticity, hearing loss | dystonia, spasticity, mild developmental delay | microcephaly, nystagmus, severe development delay optic atrophy, hearing loss |
| Seizure, age of onset | Yes, 6 mo | No | Yes | Yes, 1.3 ys | Yes | Yes / + | No | No | No | No | No |
| Feeding difficulties | Yes | Yes | NL | yes | NL | NL | No | NL | NL | No | Yes |
| Cardiomyopathy/Cardiac involvement | HCM | transient hypertrophy of the interventricular septum | HCM, cardiac failure | No | No | NL | NL | No | LVH | No | No |
| Outcome alive/death (age) | Alive 6 ys | Alive at 7 ys | Death at 4 mo | Death at 7.5 ys | Alive at 31 ys | Alive at 12 ys | Alive at 8 ys | Alive at 4.5 ys | Death at 62 d | Alive 6 ys | Death 9 ys |
| Metabolic workup | |||||||||||
| Plasma Lactate | High | High | High | ND | High | N | N | High | High | N | High |
| Plasma Pyruvate | High | NL | NL | NL | N | NL | N | N | |||
| Alanine | N | High | NL | NL | NL | NL | N | NL | High | N | N |
| Urinary organic acids | NL | NL | metabolic profiling (amino acid analysis, urine organic acid analysis, acylcarnitine analysis) was unremarkable | NL | NL | severe ketosis and hyperlactaturia (resolved with treatment) | N | normal plasma amino acids, urine organic, and amino acid analysis | NL | NL | NL |
| 2-methyl-2,3-dihydroxybutyric acid | High | High | N | NL | N | NL | N | NL | High | N | N |
| MRI changes | brain atrophy, hyperintensity within putamen, cavitation at this level, extensive diffuse white matter changes | atrophy of the cerebellum and brain stem, mild ventricular dilatation, generalized atrophy of the grey matter, thinning of the corpus callosum | moderate brain atrophy, low intensity in cerebral white matter | Extensive brain atrophy, widening of the subarachnoid space and of the ventricular system | no atrophy, hyperintensity in nucleus caudatus and putamen | cerebellar atrophy, hyperintense T2-weighted images in the putamen, globus palidus, caudate nuclei | regions of increased T2 and FLAIR signal and of hypointense T1 signal in the globus pallidus bilaterally with mild diffusion restriction | globus pallidus, putamen, caudate nuclei, basal ganglia T2 hyperintensity | asymmetric ventricular dilatation, partial agenesis of the posterior part of the corpus callosum, basal ganglia, a slight increase of T2 WM signal intensity, germinal cyst in the thalamo-caudate notch | asymmetric cavitation of globus pallidus, bilateral T2-WI hyperintensity, restricted diffusion | globus pallidus, caudate nuclei T2 hyperintensity |
| References | Our case | [27] | [4] | [4] | [4] | [28] | [29] | [30] | [31] | [32] | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muntean, C.; Tripon, F.; Bogliș, A.; Bănescu, C. Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review. Int. J. Environ. Res. Public Health 2022, 19, 2088. https://doi.org/10.3390/ijerph19042088
Muntean C, Tripon F, Bogliș A, Bănescu C. Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review. International Journal of Environmental Research and Public Health. 2022; 19(4):2088. https://doi.org/10.3390/ijerph19042088
Chicago/Turabian StyleMuntean, Carmen, Florin Tripon, Alina Bogliș, and Claudia Bănescu. 2022. "Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review" International Journal of Environmental Research and Public Health 19, no. 4: 2088. https://doi.org/10.3390/ijerph19042088
APA StyleMuntean, C., Tripon, F., Bogliș, A., & Bănescu, C. (2022). Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review. International Journal of Environmental Research and Public Health, 19(4), 2088. https://doi.org/10.3390/ijerph19042088