






Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes


Abstract
1. Introduction
2. Materials and Methods
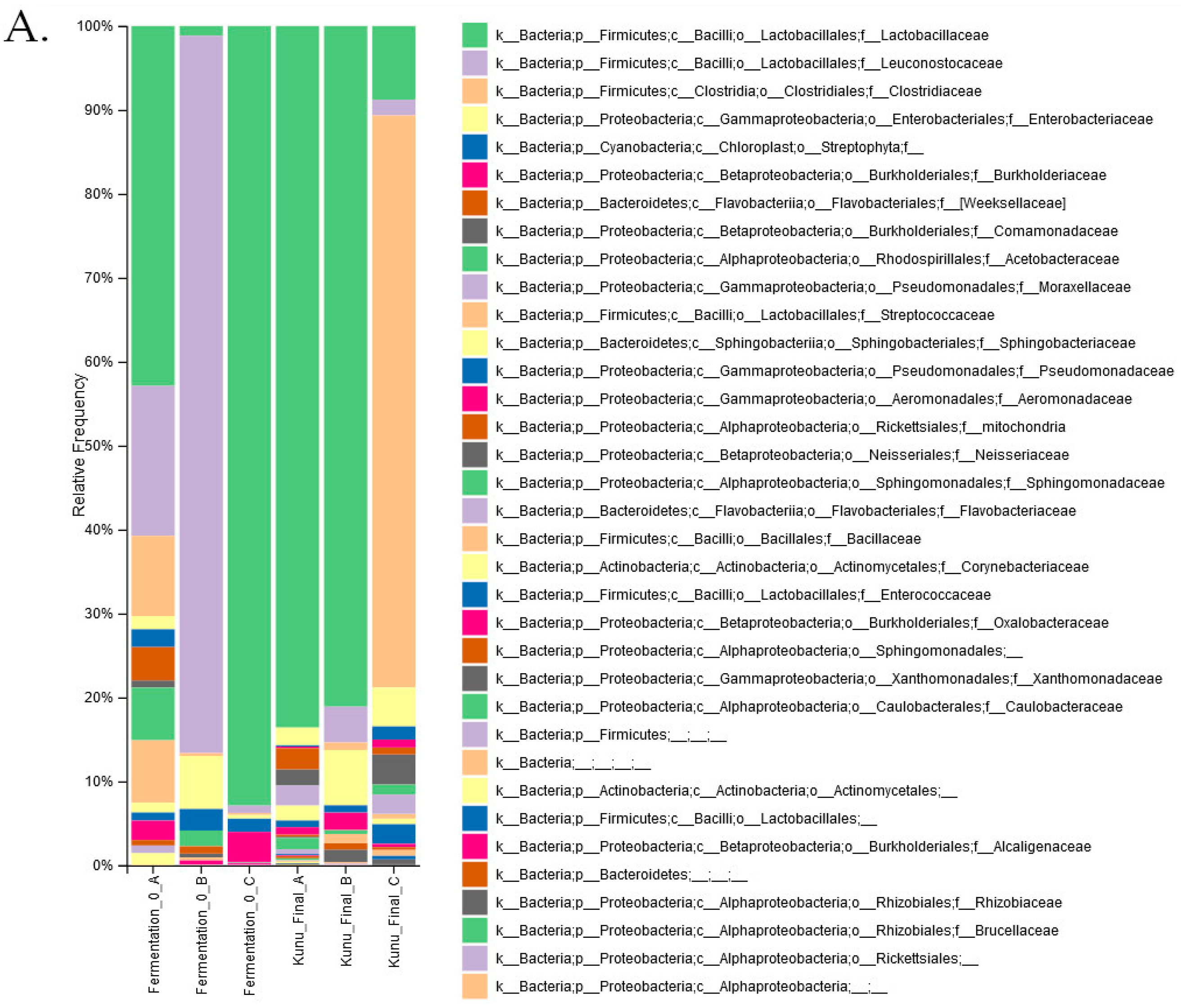
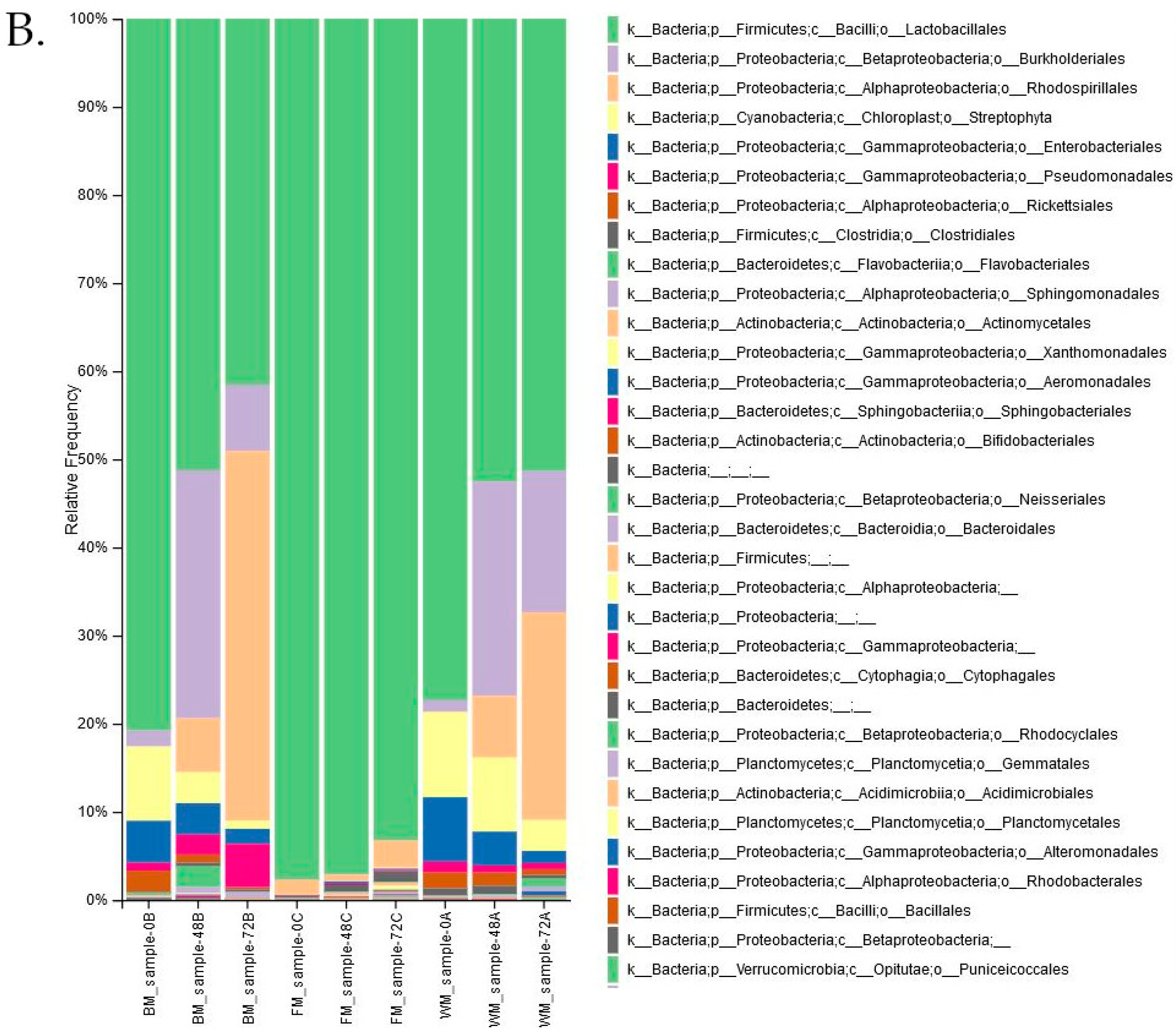
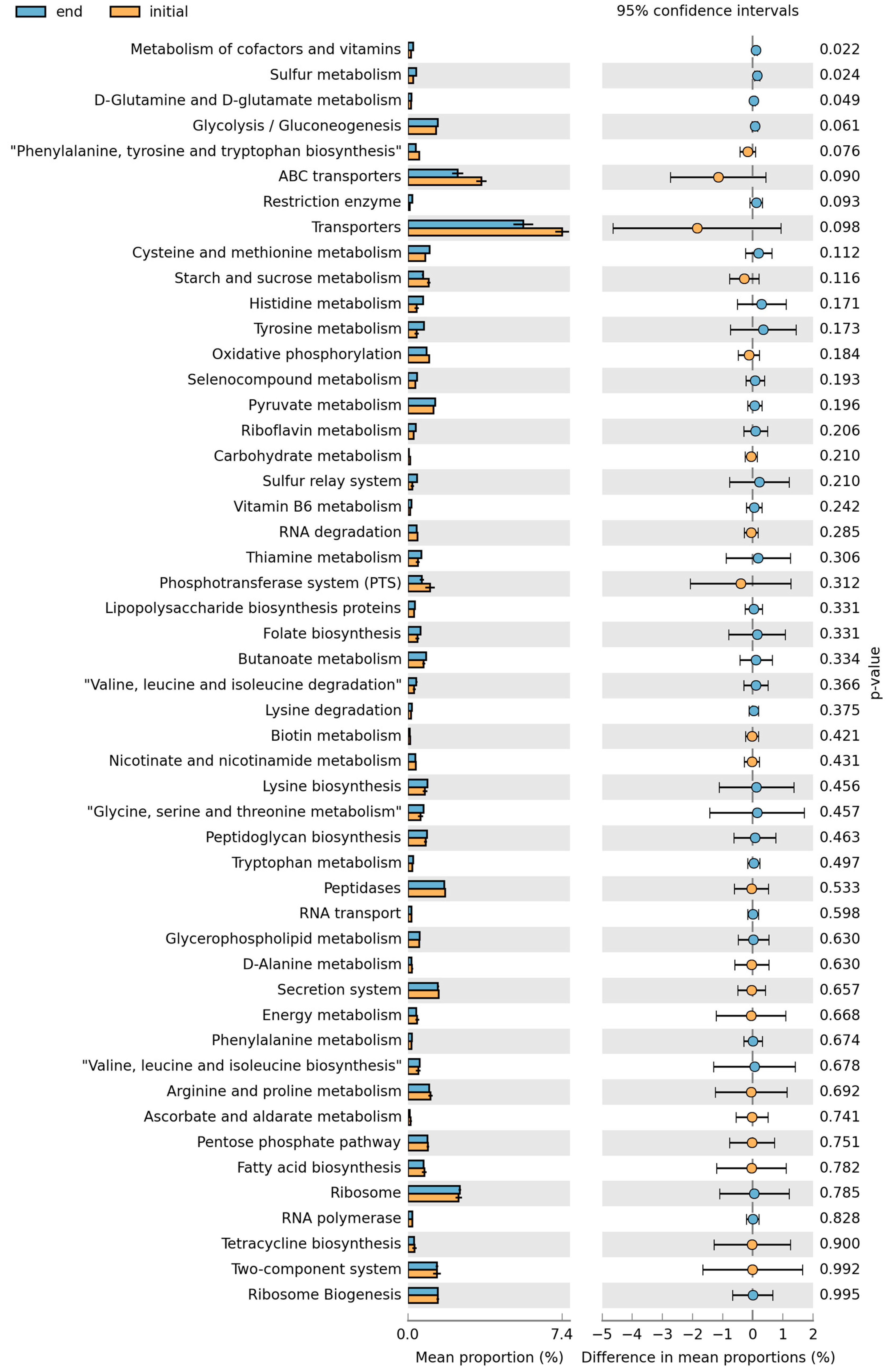
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sivamaruthi, B.; Kesika, P.; Chaiyasut, C. Thai Fermented Foods as a Versatile Source of Bioactive Microorganisms—A Comprehensive Review. Sci. Pharm. 2018, 86, 37. [Google Scholar] [CrossRef] [PubMed]
- Chaves-López, C.; Serio, A.; Grande-Tovar, C.D.; Cuervo-Mulet, R.; Delgado-Ospina, J.; Paparella, A. Traditional Fermented Foods and Beverages from a Microbiological and Nutritional Perspective: The Colombian Heritage: Colombian Fermented Foods and Beverages. Compr. Rev. Food Sci. Food Saf. 2014, 13, 1031–1048. [Google Scholar] [CrossRef]
- Li, S.; Tayie, F.A.K.; Young, M.F.; Rocheford, T.; White, W.S. Retention of Provitamin A Carotenoids in High β-Carotene Maize (Zea Mays) During Traditional African Household Processing. J. Agric. Food Chem. 2007, 55, 10744–10750. [Google Scholar] [CrossRef] [PubMed]
- Blandino, A.; Al-Aseeri, M.E.; Pandiella, S.S.; Cantero, D.; Webb, C. Cereal-Based Fermented Foods and Beverages. Food Res. Int. 2003, 36, 527–543. [Google Scholar] [CrossRef]
- Messia, M.C.; Reale, A.; Maiuro, L.; Candigliota, T.; Sorrentino, E.; Marconi, E. Effects of Pre-Fermented Wheat Bran on Dough and Bread Characteristics. J. Cereal Sci. 2016, 69, 138–144. [Google Scholar] [CrossRef]
- Pranoto, Y.; Anggrahini, S.; Efendi, Z. Effect of Natural and Lactobacillus Plantarum Fermentation on In-Vitro Protein and Starch Digestibilities of Sorghum Flour. Food Biosci. 2013, 2, 46–52. [Google Scholar] [CrossRef]
- Tangyu, M.; Muller, J.; Bolten, C.J.; Wittmann, C. Fermentation of Plant-Based Milk Alternatives for Improved Flavour and Nutritional Value. Appl. Microbiol. Biotechnol. 2019, 103, 9263–9275. [Google Scholar] [CrossRef]
- Tamene, A.; Baye, K.; Kariluoto, S.; Edelmann, M.; Bationo, F.; Leconte, N.; Humblot, C. Lactobacillus Plantarum P2R3FA Isolated from Traditional Cereal-Based Fermented Food Increase Folate Status in Deficient Rats. Nutrients 2019, 11, 2819. [Google Scholar] [CrossRef]
- Steinkraus, K.H. Classification of Fermented Foods: Worldwide Review of Household Fermentation Techniques. Food Control 1997, 8, 311–317. [Google Scholar] [CrossRef]
- Poutanen, K.S.; Kårlund, A.O.; Gómez-Gallego, C.; Johansson, D.P.; Scheers, N.M.; Marklinder, I.M.; Eriksen, A.K.; Silventoinen, P.C.; Nordlund, E.; Sozer, N.; et al. Grains—A Major Source of Sustainable Protein for Health. Nutr. Rev. 2022, 80, 1648–1663. [Google Scholar] [CrossRef]
- Gupta, R.K.; Gangoliya, S.S.; Singh, N.K. Reduction of Phytic Acid and Enhancement of Bioavailable Micronutrients in Food Grains. J. Food Sci. Technol. 2015, 52, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Jood, S. Effect of Germination and Probiotic Fermentation on PH, Titratable Acidity, Dietary Fibre, β-Glucan and Vitamin Content of Sorghum Based Food Mixtures. J. Nutr. Food Sci. 2012, 02, 1–4. [Google Scholar] [CrossRef]
- Obafemi, Y.D.; Oranusi, S.U.; Ajanaku, K.O.; Akinduti, P.A.; Leech, J.; Cotter, P.D. African Fermented Foods: Overview, Emerging Benefits, and Novel Approaches to Microbiome Profiling. NPJ Sci. Food 2022, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Zain, M.E. Impact of Mycotoxins on Humans and Animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef]
- Wafula, E.N.; Muhonja, C.N.; Kuja, J.O.; Owaga, E.E.; Makonde, H.M.; Mathara, J.M.; Kimani, V.W. Lactic Acid Bacteria from African Fermented Cereal-Based Products: Potential Biological Control Agents for Mycotoxins in Kenya. J. Toxicol. 2022, 2022, 2397767. [Google Scholar] [CrossRef] [PubMed]
- Meijer, N.; Kleter, G.; de Nijs, M.; Rau, M.-L.; Derkx, R.; van der Fels-Klerx, H.J. The Aflatoxin Situation in Africa: Systematic Literature Review. Compr. Rev. Food Sci. Food Saf. 2021, 20, 2286–2304. [Google Scholar] [CrossRef]
- Leeuwendaal, N.K.; Stanton, C.; O’Toole, P.W.; Beresford, T.P. Fermented Foods, Health and the Gut Microbiome. Nutrients 2022, 14, 1527. [Google Scholar] [CrossRef]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligné, B.; Gänzle, M.; Kort, R.; Pasin, G.; Pihlanto, A.; et al. Health Benefits of Fermented Foods: Microbiota and Beyond. Curr. Opin. Biotechnol. 2017, 44, 94–102. [Google Scholar] [CrossRef]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef]
- Drewes, J.L.; Housseau, F.; Sears, C.L. Sporadic Colorectal Cancer: Microbial Contributors to Disease Prevention, Development and Therapy. Br. J. Cancer 2016, 115, 273–280. [Google Scholar] [CrossRef]
- Gold, A.; Zhu, J. Not Just a Gut Feeling: A Deep Exploration of Functional Bacterial Metabolites That Can Modulate Host Health. Gut Microbes 2022, 14, 2125734. [Google Scholar] [CrossRef] [PubMed]
- Lebeer, S.; Vanderleyden, J.; De Keersmaecker, S.C.J. Genes and Molecules of Lactobacilli Supporting Probiotic Action. Microbiol. Mol. Biol. Rev. 2008, 72, 728–764. [Google Scholar] [CrossRef] [PubMed]
- Leech, J.; Cabrera-Rubio, R.; Walsh, A.M.; Macori, G.; Walsh, C.J.; Barton, W.; Finnegan, L.; Crispie, F.; O’Sullivan, O.; Claesson, M.J.; et al. Fermented-Food Metagenomics Reveals Substrate-Associated Differences in Taxonomy and Health-Associated and Antibiotic Resistance Determinants. mSystems 2020, 5, e00522-20. [Google Scholar] [CrossRef] [PubMed]
- Deka, P.; Mehetre, G.T.; Lalnunmawii, E.; Upadhyaya, K.; Singh, G.; Hashem, A.; Al-Arjani, A.-B.F.; Fathi Abd_Allah, E.; Singh, B.P. Metagenomic Analysis of Bacterial Diversity in Traditional Fermented Foods Reveals Food-Specific Dominance of Specific Bacterial Taxa. Fermentation 2021, 7, 167. [Google Scholar] [CrossRef]
- Anyogu, A.; Olukorede, A.; Anumudu, C.; Onyeaka, H.; Areo, E.; Adewale, O.; Odimba, J.N.; Nwaiwu, O. Microorganisms and Food Safety Risks Associated with Indigenous Fermented Foods from Africa. Food Control 2021, 129, 108227. [Google Scholar] [CrossRef]
- Soro-Yao, A.A.; Brou, K.; Amani, G.; Thonart, P.; Djè, K.M. The Use of Lactic Acid Bacteria Starter Cultures during the Processing of Fermented Cereal-Based Foods in West Africa: A Review. Trop. Life Sci. Res. 2014, 25, 81–100. [Google Scholar]
- Chileshe, J.; van den Heuvel, J.; Handema, R.; Zwaan, B.J.; Talsma, E.F.; Schoustra, S. Nutritional Composition and Microbial Communities of Two Non-Alcoholic Traditional Fermented Beverages from Zambia: A Study of Mabisi and Munkoyo. Nutrients 2020, 12, 1628. [Google Scholar] [CrossRef]
- Parker, M.; Zobrist, S.; Donahue, C.; Edick, C.; Mansen, K.; Nadjari, M.H.Z.; Heerikhuisen, M.; Sybesma, W.; Molenaar, D.; Diallo, A.M.; et al. Naturally Fermented Milk From Northern Senegal: Bacterial Community Composition and Probiotic Enrichment With Lactobacillus Rhamnosus. Front. Microbiol. 2018, 9, 2218. [Google Scholar] [CrossRef]
- Ezekiel, C.N.; Ayeni, K.I.; Ezeokoli, O.T.; Sulyok, M.; van Wyk, D.A.B.; Oyedele, O.A.; Akinyemi, O.M.; Chibuzor-Onyema, I.E.; Adeleke, R.A.; Nwangburuka, C.C.; et al. High-Throughput Sequence Analyses of Bacterial Communities and Multi-Mycotoxin Profiling During Processing of Different Formulations of Kunu, a Traditional Fermented Beverage. Front. Microbiol. 2019, 9, 3282. [Google Scholar] [CrossRef]
- Schmidhuber, J.; Tubiello, F.N. Global Food Security under Climate Change. Proc. Natl. Acad. Sci. USA 2007, 104, 19703–19708. [Google Scholar] [CrossRef]
- Diaz, M.; Kellingray, L.; Akinyemi, N.; Adefiranye, O.O.; Olaonipekun, A.B.; Bayili, G.R.; Ibezim, J.; du Plessis, A.S.; Houngbédji, M.; Kamya, D.; et al. Comparison of the Microbial Composition of African Fermented Foods Using Amplicon Sequencing. Sci. Rep. 2019, 9, 13863. [Google Scholar] [CrossRef] [PubMed]
- Chibuzor-Onyema, I.E.; Ezeokoli, O.T.; Sulyok, M.; Notununu, I.; Petchkongkaew, A.; Elliott, C.T.; Adeleke, R.A.; Krska, R.; Ezekiel, C.N. Metataxonomic Analysis of Bacterial Communities and Mycotoxin Reduction during Processing of Three Millet Varieties into Ogi, a Fermented Cereal Beverage. Food Res. Int. 2021, 143, 110241. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- PuTTY: A Free SSH and Telnet Client. Available online: https://www.chiark.greenend.org.uk/~sgtatham/putty (accessed on 16 October 2022).
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]
- Brown, S.T.; Buitrago, P.; Hanna, E.; Sanielevici, S.; Scibek, R.; Nystrom, N.A. Bridges-2: A Platform for Rapidly-Evolving and Data Intensive Research. In Proceedings of the Practice and Experience in Advanced Research Computing, Boston, MA, USA, 17 July 2021; pp. 1–4. [Google Scholar]
- Foster, I. Globus Online: Accelerating and Democratizing Science through Cloud-Based Services. IEEE Internet Comput. 2011, 15, 70–73. [Google Scholar] [CrossRef]
- Downloading SRA Toolkit Ncbi/Sra-Tools Wiki. Available online: https://github.com/ncbi/sra-tools (accessed on 16 October 2022).
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 17 October 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) Format or: How I Learned to Stop Worrying and Love the Ome-Ome. GigaScience 2012, 1, 2047-217X. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Pswarayi, F.; Gänzle, M. African Cereal Fermentations: A Review on Fermentation Processes and Microbial Composition of Non-Alcoholic Fermented Cereal Foods and Beverages. Int. J. Food Microbiol. 2022, 378, 109815. [Google Scholar] [CrossRef]
- Zapaśnik, A.; Sokołowska, B.; Bryła, M. Role of Lactic Acid Bacteria in Food Preservation and Safety. Foods 2022, 11, 1283. [Google Scholar] [CrossRef]
- Owade, J.O.; Abong’, G.O.; Okoth, M.W.; Mwang’ombe, A.W.; Jobor, J.O. Comparative Profiling of Lactic Acid Bacteria Isolates in Optimized and Spontaneous Fermentation of Cowpea Leaves. Food Sci. Nutr. 2021, 9, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Lynch, S.V. The Gut Microbiome: Relationships with Disease and Opportunities for Therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef]
- Holzapfel, W.H.; Haberer, P.; Geisen, R.; Björkroth, J.; Schillinger, U. Taxonomy and Important Features of Probiotic Microorganisms in Food and Nutrition. Am. J. Clin. Nutr. 2001, 73, 365S–373S. [Google Scholar] [CrossRef] [PubMed]
- Sade, N.; Peleg, Z. Future Challenges for Global Food Security under Climate Change. Plant Sci. 2020, 295, 110467. [Google Scholar] [CrossRef]
- Wheeler, T.; von Braun, J. Climate Change Impacts on Global Food Security. Science 2013, 341, 508–513. [Google Scholar] [CrossRef]
- Lupton, F.G.H. Lost Crops of Africa: Volume 1. Exp. Agric. 1997, 33, 247–252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Lactobacillales Genus | Diaz et. al. [31] | Ezekiel et. al. [29] | Chibuzor-Onyema et. al. [32] |
|---|---|---|---|
| Lactobacillus | ***** | ** | ******** |
| Lactococcus | + | + | + |
| Leuconostoc | + | + | |
| Pediococcus | + | + | **** |
| Streptococcus | + | + | |
| Aerococcus | |||
| Alloiococcus | |||
| Carnobacterium | |||
| Dolosigranulum | |||
| Enterococcus | ** | ||
| Oenococcus | |||
| Tetragenococcus | |||
| Vagococcus | |||
| Weissella | + | + | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dania, M.I.; Faraji, B.; Wachira, J. Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. Int. J. Environ. Res. Public Health 2022, 19, 16621. https://doi.org/10.3390/ijerph192416621
Dania MI, Faraji B, Wachira J. Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. International Journal of Environmental Research and Public Health. 2022; 19(24):16621. https://doi.org/10.3390/ijerph192416621
Chicago/Turabian StyleDania, Margaret I., Bahram Faraji, and James Wachira. 2022. "Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes" International Journal of Environmental Research and Public Health 19, no. 24: 16621. https://doi.org/10.3390/ijerph192416621
APA StyleDania, M. I., Faraji, B., & Wachira, J. (2022). Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. International Journal of Environmental Research and Public Health, 19(24), 16621. https://doi.org/10.3390/ijerph192416621