Removal of Emerging Contaminants and Estrogenic Activity from Wastewater Treatment Plant Effluent with UV/Chlorine and UV/H2O2 Advanced Oxidation Treatment at Pilot Scale


Abstract

1. Introduction
2. Materials and Methods
2.1. Experimental Concept
- Experiment 1: Variation of UV energy consumption (0.0, 0.4, 0.7, and 1.0 kWh/m3) at 0 and 3 mg/L oxidant concentrations (FAC or H2O2).
- Experiment 2: Variation of oxidant concentration (1–6 mg/L FAC or H2O2) at 0.4 kWh/m3 UV energy consumption.
2.2. Chemicals and Reagents
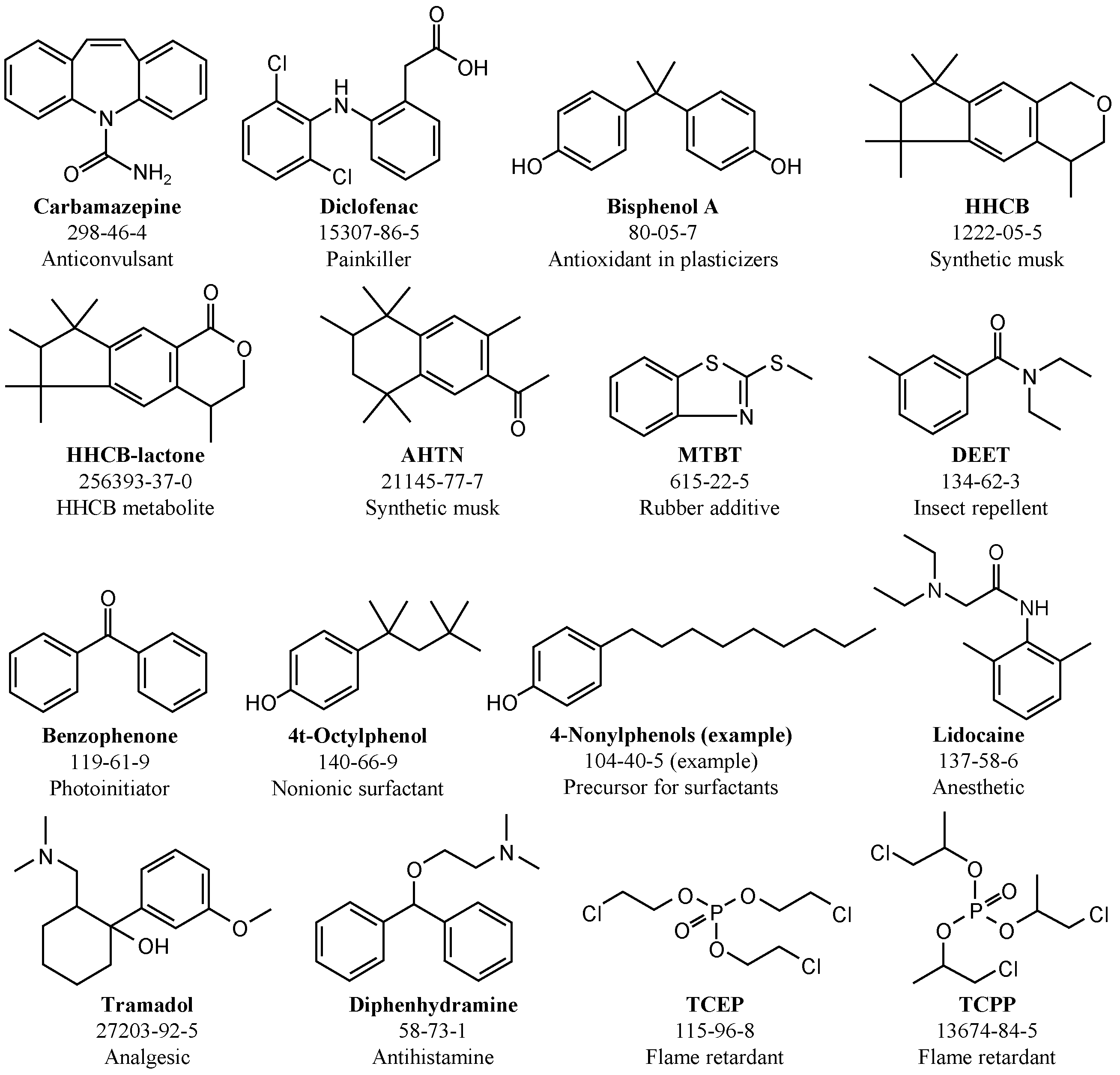
2.3. Wastewater Treatment Plant Effluent (WWTE) and Emerging Contaminants (ECs)
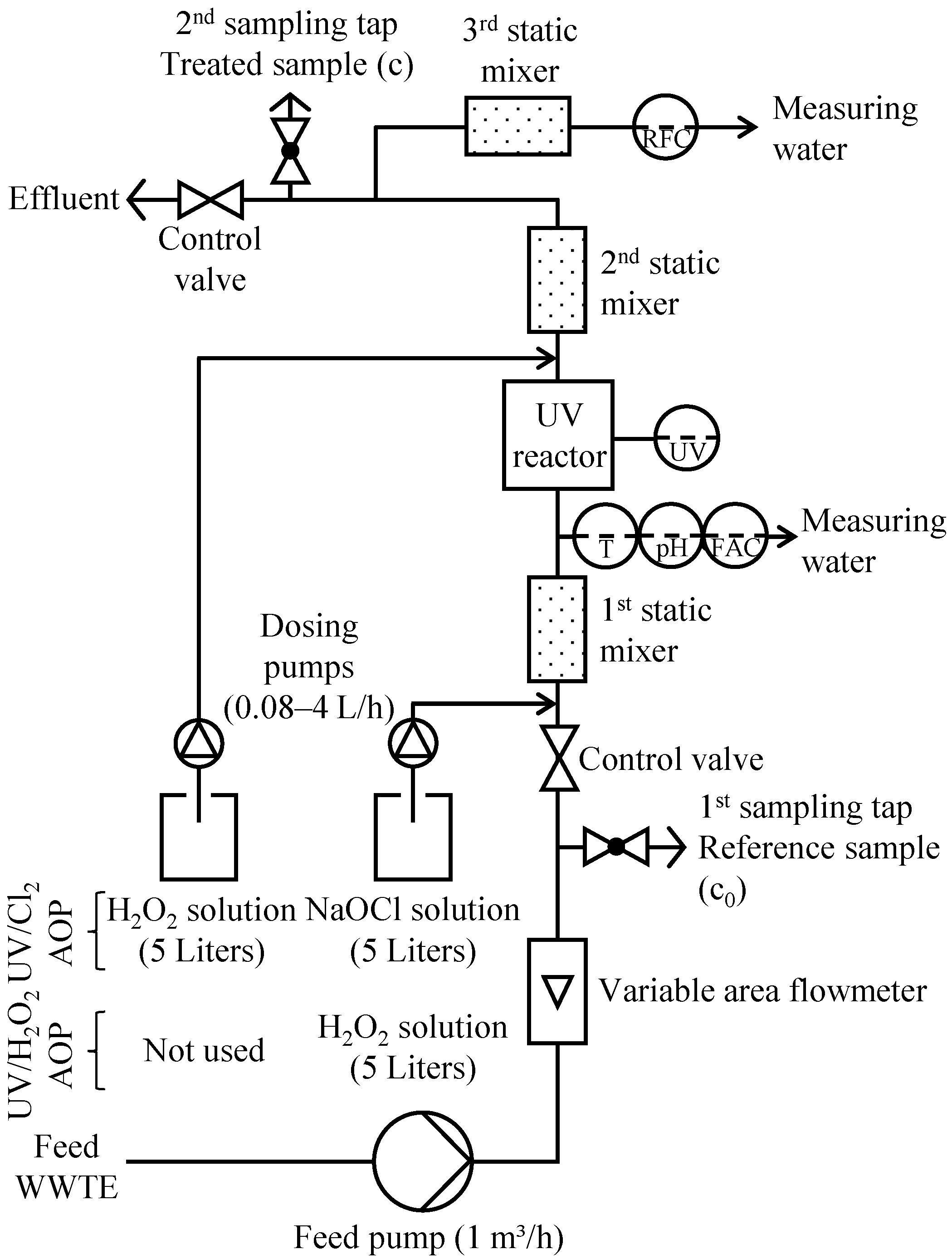
2.4. UV Pilot Plant
2.5. Experimental Procedure
2.5.1. Variation of UV Energy Consumption at 0 and 3 mg/L Oxidant Concentrations (Experiment 1)
2.5.2. Variation of Oxidant Concentration at 0.4 kWh/m3 UV Energy Consumption (Experiment 2)
2.6. Analytical Methods
2.6.1. Free Cl2, Combined Cl2, Total Cl2
2.6.2. Chlorite (ClO2−), Chlorate (ClO3−), Perchlorate (ClO4−)
2.6.3. Emerging Contaminants (ECs)
2.6.4. Total Estrogenic Activity (TEA)
2.6.5. Adsorbable Organohalogens (AOX)
2.6.6. Number of Measurements
3. Results and Discussion
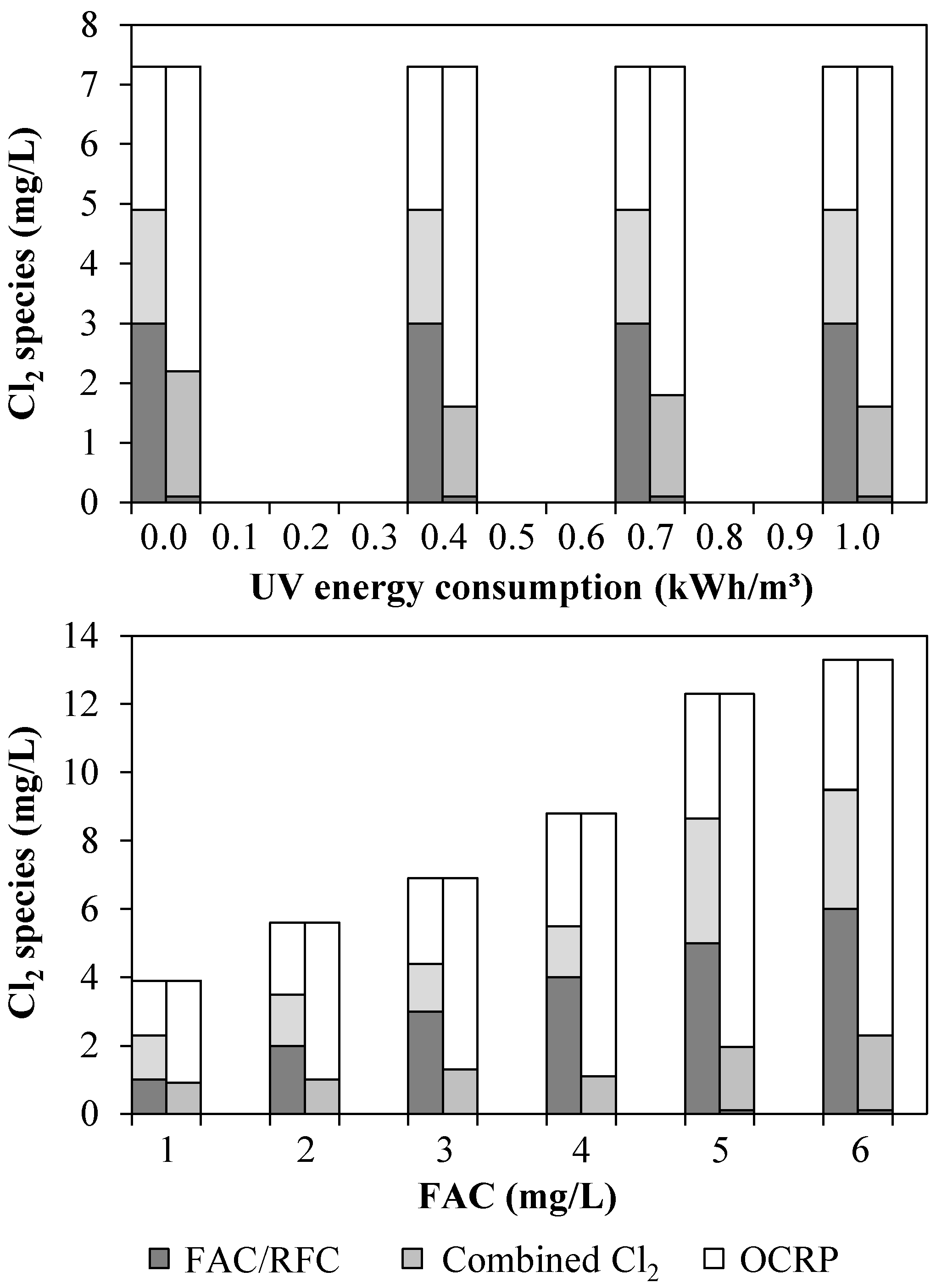
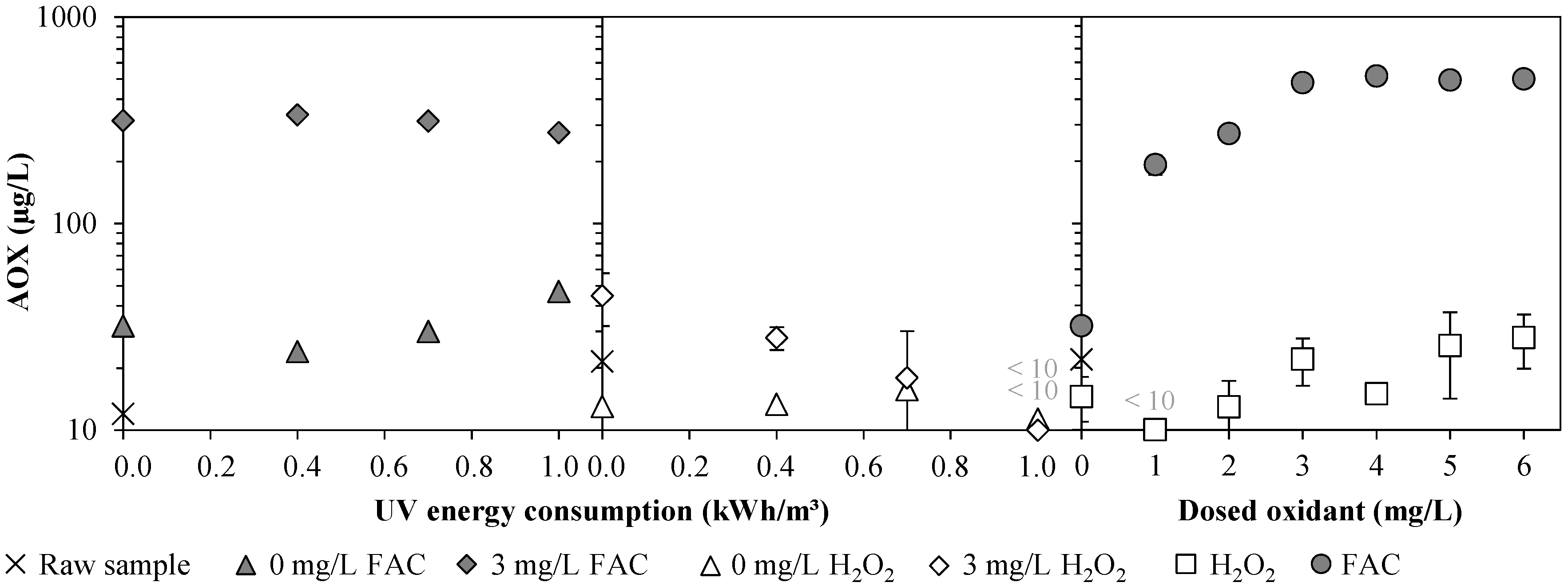
3.1. Chlorine Species and Adsorbable Organohalogens (AOX)
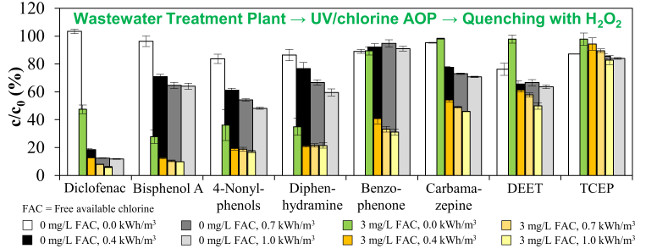
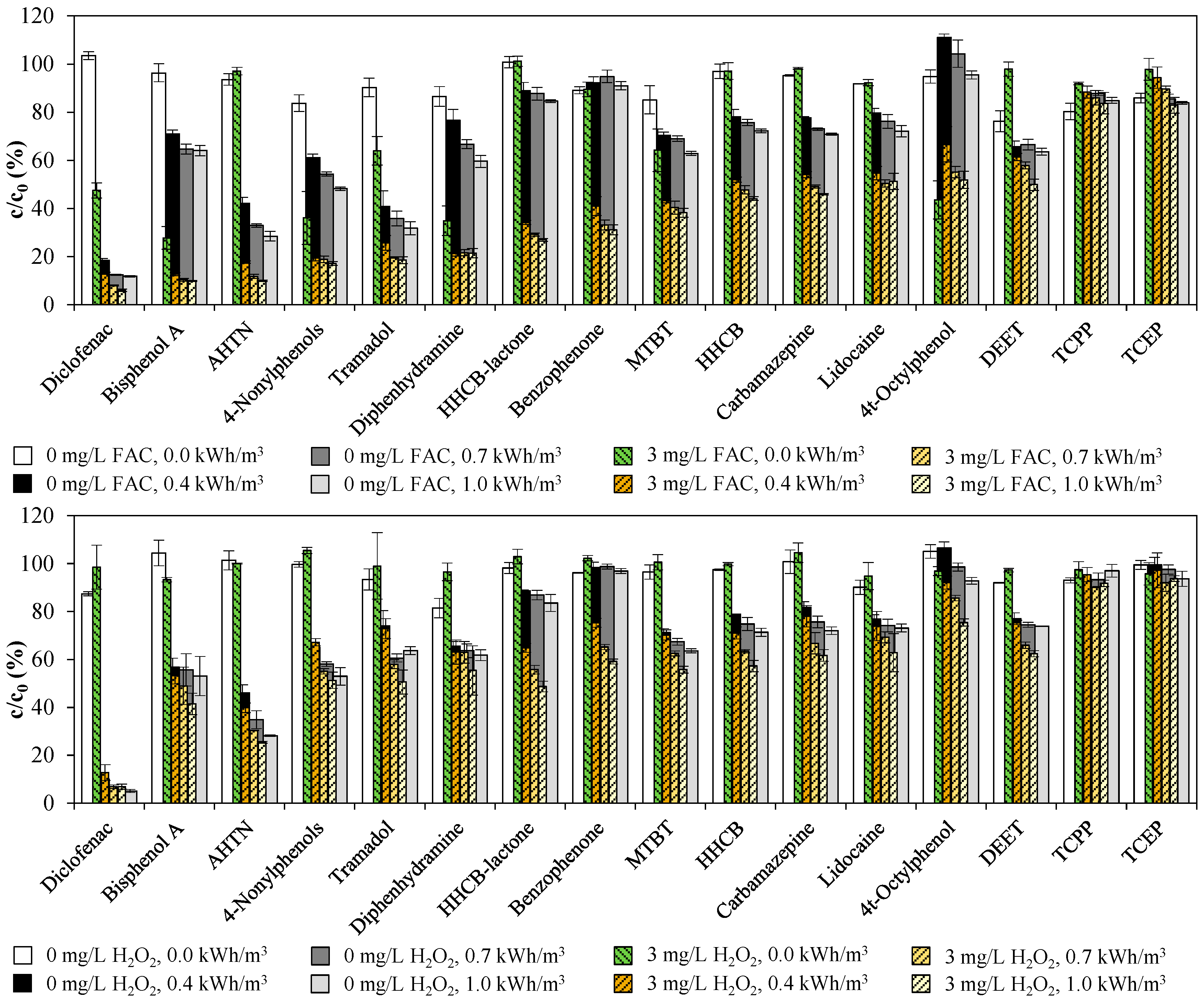
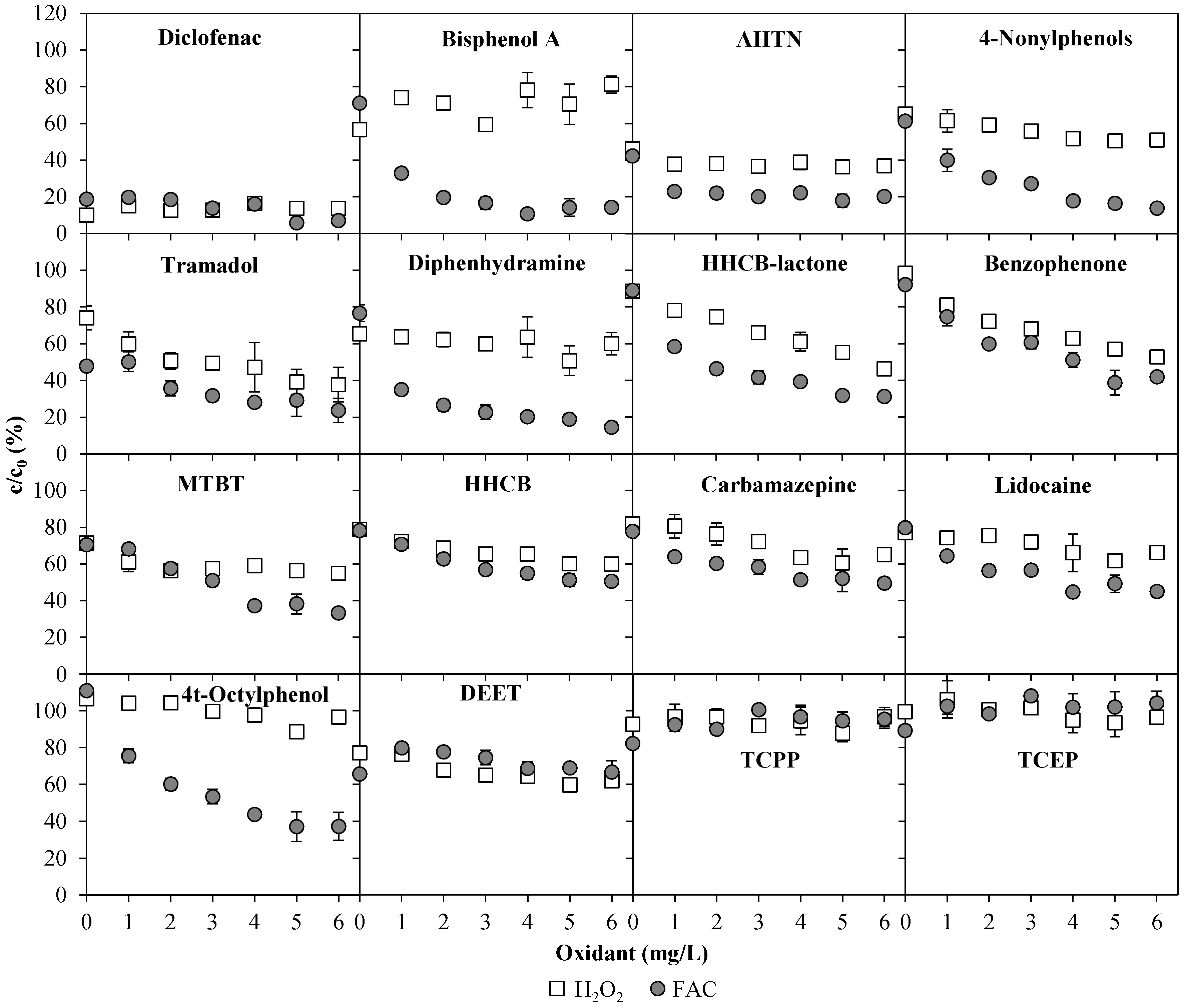
3.2. Emerging Contaminants
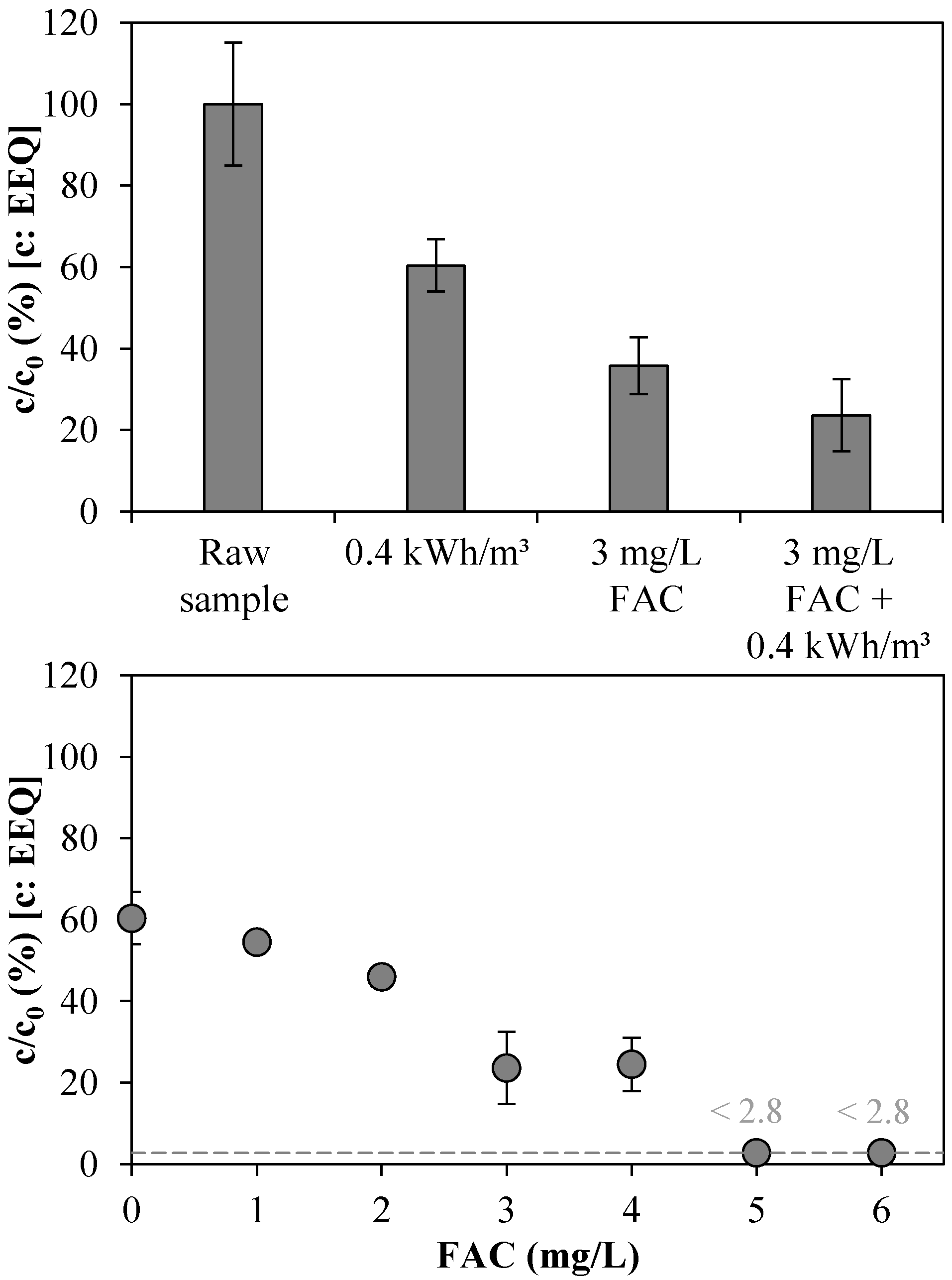
3.3. Total Estrogenic Activity in the UV/Chlorine AOP Experiments
4. Conclusions
Reference
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Kolpin, D.W.; Furlong, E.T.; Meyer, M.T.; Thurman, E.M.; Zaugg, S.D.; Barber, L.B.; Buxton, H.T. Pharmaceuticals, Hormones, and Other Organic Wastewater Contaminants in U.S. Streams, 1999−2000: A National Reconnaissance. Environ. Sci. Technol. 2002, 36, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Reemtsma, T.; Weiss, S.; Mueller, J.; Petrovic, M.; González, S.; Barcelo, D.; Ventura, F.; Knepper, T.P. Polar Pollutants Entry into the Water Cycle by Municipal Wastewater: A European Perspective. Environ. Sci. Technol. 2006, 40, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.; Lapworth, D.; Crane, E.; Hart, A. Review of risk from potential emerging contaminants in UK groundwater. Sci. Total Environ. 2012, 416, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.; Carvalho, R.; António, D.C.; Comero, S.; Locoro, G.; Tavazzi, S.; Paracchini, B.; Ghiani, M.; Lettieri, T.; Blaha, L.; et al. EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res. 2013, 47, 6475–6487. [Google Scholar] [CrossRef] [PubMed]
- Launay, M.A.; Dittmer, U.; Steinmetz, H. Organic micropollutants discharged by combined sewer overflows—Characterisation of pollutant sources and stormwater-related processes. Water Res. 2016, 104, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Ternes, T.A. Occurrence of drugs in German sewage treatment plants and rivers. Water Res. 1998, 32, 3245–3260. [Google Scholar] [CrossRef]
- Auriol, M.; Filali-Meknassi, Y.; Tyagi, R.D.; Adams, C.D.; Surampalli, R.Y. Endocrine disrupting compounds removal from wastewater, a new challenge. Process. Biochem. 2006, 41, 525–539. [Google Scholar] [CrossRef]
- Bolong, N.; Ismail, A.F.; Salim, M.R.; Matsuura, T. A review of the effects of emerging contaminants in wastewater and options for their removal. Desalination 2009, 239, 229–246. [Google Scholar] [CrossRef]
- Reungoat, J.; Escher, B.I.; Macova, M.; Argaud, F.X.; Gernjak, W.; Keller, J. Ozonation and biological activated carbon filtration of wastewater treatment plant effluents. Water Res. 2012, 46, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Gerrity, D.; Gamage, S.; Holady, J.C.; Mawhinney, D.B.; Quiñones, O.; Trenholm, R.A.; Snyder, S.A. Pilot-scale evaluation of ozone and biological activated carbon for trace organic contaminant mitigation and disinfection. Water Res. 2011, 45, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.A.; Adham, S.; Redding, A.M.; Cannon, F.S.; DeCarolis, J.; Oppenheimer, J.; Wert, E.C.; Yoon, Y. Role of membranes and activated carbon in the removal of endocrine disruptors and pharmaceuticals. Desalination 2007, 202, 156–181. [Google Scholar] [CrossRef]
- Dolar, D.; Gros, M.; Rodriguez-Mozaz, S.; Moreno, J.; Comas, J.; Rodriguez-Roda, I.; Barceló, D. Removal of emerging contaminants from municipal wastewater with an integrated membrane system, MBR-RO. J. Hazard. Mater. 2012, 239–240, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Esplugas, S.; Bila, D.M.; Krause, L.G.T.; Dezotti, M. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents. J. Hazard. Mater. 2007, 149, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, M.; Gracia-Lor, E.; Bijlsma, L.; Morales, E.; Pastor, L.; Hernández, F. Removal of emerging contaminants in sewage water subjected to advanced oxidation with ozone. J. Hazard. Mater. 2013, 260, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Sichel, C.; Garcia, C.; Andre, K. Feasibility studies: UV/chlorine advanced oxidation treatment for the removal of emerging contaminants. Water Res. 2011, 45, 6371–6380. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, J.; Fu, W.; Shang, C.; Li, Y.; Chen, Y.; Gan, W.; Fang, J. PPCP degradation by UV/chlorine treatment and its impact on DBP formation potential in real waters. Water Res. 2016, 98, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fang, J.; Shang, C. Kinetics and pathways of ibuprofen degradation by the UV/chlorine advanced oxidation process. Water Res. 2016, 90, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-L.; Wu, Q.-Y.; Huang, N.; Wang, T.; Hu, H.-Y. Synergistic effect between UV and chlorine (UV/chlorine) on the degradation of carbamazepine: Influence factors and radical species. Water Res. 2016, 98, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, B.; Liungai, Z.; Hu, H.-Y.; Chen, C.; Qiao, J.; Lu, Y. The removal of estrogenic activity with UV/chlorine technology and identification of novel estrogenic disinfection by-products. J. Hazard. Mater. 2016, 307, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.X.; Margerum, D.W. Kinetics of Reversible Chlorine Hydrolysis: Temperature Dependence and General-Acid/Base-Assisted Mechanisms. Inorg. Chem. 1994, 33, 1050–1055. [Google Scholar] [CrossRef]
- Morris, J.C. The Acid Ionization Constant of HOCl from 5 to 35°. J. Phys. Chem. 1966, 70, 3798–3805. [Google Scholar] [CrossRef]
- Deborde, M.; von Gunten, U. Reactions of chlorine with inorganic and organic compounds during water treatment—Kinetics and mechanisms: A critical review. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Subhani, M.S. Radiation chemistry and photochemistry of oxychlorine ions. Part 2—Photodecomposition of aqueous solutions of hypochlorite ions. J. Chem. Soc. Faraday Trans. 1 1972, 68, 958–969. [Google Scholar] [CrossRef]
- Feng, Y.; Smith, D.W.; Bolton, J.R. Photolysis of aqueous free chlorine species (HOCl and OCl−) with 254 nm ultraviolet light. J. Environ. Eng. Sci. 2007, 6, 277–284. [Google Scholar] [CrossRef]
- Jin, J.; El-Din, M.G.; Bolton, J.R. Assessment of the UV/chlorine process as an advanced oxidation process. Water Res. 2011, 45, 1890–1896. [Google Scholar] [CrossRef] [PubMed]
- Held, A.M.; Halko, D.J.; Hurst, J.K. Mechanisms of chlorine oxidation of hydrogen peroxide. J. Am. Chem. Soc. 1978, 100, 5732–5740. [Google Scholar] [CrossRef]
- Graedel, T.E.; Goldberg, K.I. Kinetic studies of raindrop chemistry: 1. Inorganic and organic processes. J. Geophys. Res. 1983, 88, 10865. [Google Scholar] [CrossRef]
- American Chemical Society. Database of SciFinder. Available online: https://scifinder.cas.org (accessed on 28 April 2016).
- International Organization for Standardization. Water Quality—Determination of Dissolved Anions by Liquid Chromatography of Ions—Part 1: Determination of Bromide, Chloride, Fluoride, Nitrate, Nitrite, Phosphate and Sulfate; ISO 10304-1:2007; International Organization for Standardization: Geneva, Switzerland, 2007. [Google Scholar]
- Soto, A.M.; Sonnenschein, C.; Chung, K.L.; Fernandez, M.F.; Olea, N.; Serrano, F.O. The E-SCREEN Assay as a Tool to Identify Estrogens: An Update on Estrogenic Environmental Pollutants. Environ. Health Perspect. 1995, 103, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Körner, W.; Hanf, V.; Schuller, W.; Kempter, C.; Metzger, J.; Hagenmaier, H. Development of a sensitive E-screen assay for quantitative analysis of estrogenic activity in municipal sewage plant effluents. Sci. Total Environ. 1999, 225, 33–48. [Google Scholar] [CrossRef]
- Schultis, T. Erfassung der Estrogenen Wirksamkeit von Umweltproben und Reinsubstanzen durch Biologische Testsysteme—Entwicklung und Vergleich von In Vitro-Assays. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2005. [Google Scholar]
- International Organization for Standardization. Water Quality—Determination of Adsorbable Organically Bound Halogens (AOX); ISO 9562:2004; International Organization for Standardization: Geneva, Switzerland, 2007. [Google Scholar]
- Placak, O.R.; Ruchhoft, C.C. Studies of Sewage Purification: XVII. The Utilization of Organic Substrates by Activated Sludge. Public Health Rep. 1947, 62, 697–716. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Drinking Water and Health, Volume 7: Disinfectants and Disinfectant By-Products; National Academies Press: Washington, DC, USA, 1987. [Google Scholar]
- Krasner, S.W.; Westerhoff, P.; Chen, B.; Rittmann, B.E.; Amy, G. Occurrence of disinfection byproducts in United States wastewater treatment plant effluents. Environ. Sci. Technol. 2009, 43, 8320–8325. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.; Huang, J.; Templeton, M.R.; Graham, N. Occurrence and control of nitrogenous disinfection by-products in drinking water—A review. Water Res. 2011, 45, 4341–4354. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.C.; Wong, M.H.; Liang, Y. Amino Acids as Precursors of Trihalomethane and Haloacetic Acid Formation During Chlorination. Arch. Environ. Contam. Toxicol. 2009, 56, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Pehlivanoglu-Mantas, E.; Sedlak, D.L. Measurement of dissolved organic nitrogen forms in wastewater effluents: Concentrations, size distribution and NDMA formation potential. Water Res. 2008, 42, 3890–3898. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; von Gunten, U. Oxidative transformation of micropollutants during municipal wastewater treatment: Comparison of kinetic aspects of selective (chlorine, chlorine dioxide, ferrate VI, and ozone) and non-selective oxidants (hydroxyl radical). Water Res. 2010, 44, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Encinas, S.; Bosca, F.; Miranda, M.A. Photochemistry of 2,6-Dichlorodiphenylamine and 1-Chlorocarbazole, the Photoactive Chromophores of Diclofenac, Meclofenamic Acid and Their Major Photoproducts. Photochem. Photobiol. 1998, 68, 640–645. [Google Scholar] [CrossRef]
- Zhou, S.; Xia, Y.; Li, T.; Yao, T.; Shi, Z.; Zhu, S.; Gao, N. Degradation of carbamazepine by UV/chlorine advanced oxidation process and formation of disinfection by-products. Environ. Sci. Pollut. Res. Int. 2016, 23, 16448–16455. [Google Scholar] [CrossRef] [PubMed]
- Soufan, M.; Deborde, M.; Legube, B. Aqueous chlorination of diclofenac: Kinetic study and transformation products identification. Water Res. 2012, 46, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Gallard, H.; Leclercq, A.; Croué, J.-P. Chlorination of bisphenol A: Kinetics and by-products formation. Chemosphere 2004, 56, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Deborde, M.; Rabouan, S.; Gallard, H.; Legube, B. Aqueous Chlorination Kinetics of Some Endocrine Disruptors. Environ. Sci. Technol. 2004, 38, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.J.; Linden, K.G. Chlorine photolysis and subsequent OH radical production during UV treatment of chlorinated water. Water Res. 2007, 41, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Cédat, B.; de Brauer, C.; Métivier, H.; Dumont, N.; Tutundjan, R. Are UV photolysis and UV/H2O2 process efficient to treat estrogens in waters? Chemical and biological assessment at pilot scale. Water Res. 2016, 100, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Rosario-Ortiz, F.L.; Wert, E.C.; Snyder, S.A. Evaluation of UV/H2O2 treatment for the oxidation of pharmaceuticals in wastewater. Water Res. 2010, 44, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-Y.; Hu, H.-Y.; Zhao, X.; Sun, Y.-X. Effect of Chlorination on the Estrogenic/Antiestrogenic Activities of Biologically Treated Wastewater. Environ. Sci. Technol. 2009, 43, 4940–4945. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.G.; Borglin, S.E.; Green, F.B.; Grayson, A.; Wozei, E.; Stringfellow, W.T. Biologically directed environmental monitoring, fate, and transport of estrogenic endocrine disrupting compounds in water: A review. Chemosphere 2006, 65, 1265–1280. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Kamata, M.; Akatsuka, Y.; Takeda, M.; Ohno, K.; Kamei, T.; Magara, Y. Effects of chlorine on the decrease of estrogenic chemicals. Water Res. 2004, 38, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, E.J.; Linden, K.G. Degradation of Endocrine Disrupting Chemicals Bisphenol A, Ethinyl Estradiol, and Estradiol during UV Photolysis and Advanced Oxidation Processes. Environ. Sci. Technol. 2004, 38, 5476–5483. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, E.J.; Chen, P.J.; Kullman, S.; Linden, K.G. Destruction of estrogenic activity in water using UV advanced oxidation. Sci. Total Environ. 2007, 377, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bolton, J.R.; Andrews, S.A.; Hofmann, R. Formation of disinfection by-products in the ultraviolet/chlorine advanced oxidation process. Sci. Total Environ. 2015, 518–519, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Deng, S.; Li, D.; Ren, L.; Shan, D.; Wang, B.; Huang, J.; Wang, Y.; Yu, G. Sorption behavior and mechanism of organophosphate flame retardants on activated carbons. Chem. Eng. J. 2018, 332, 286–292. [Google Scholar] [CrossRef]
- Lee, W.; Westerhoff, P. Formation of organic chloramines during water disinfection: Chlorination versus chloramination. Water Res. 2009, 43, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Blatchley, E.R.; Wang, X.; Ren, P. UV/chlorine process for ammonia removal and disinfection by-product reduction: Comparison with chlorination. Water Res. 2015, 68, 804–811. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Variation of UV Energy Consumption between 0 and 1 kWh/m3 (Experiment 1) | Variation of Oxidant Concentration at 0.4 kWh/m3 (Experiment 2) | |||
|---|---|---|---|---|---|
| 0 and 3 mg/L FAC | 0 and 3 mg/L H2O2 | 1–4 mg/L FAC | 5–6 mg/L FAC | 1–6 mg/L H2O2 | |
| Temperature (°C) | 14.9 | 18.9 | 14.6 | 14.8 | 19.5 |
| pH | 7.0 | 7.0 | 7.0 | 7.0 | 7.0 |
| COD (mg/L) | 17.8 ± 1.3 | 20.4 ± 1.8 | 23.6 ± 0.3 | 23.2 ± 0.1 | 21.3 ± 0.8 |
| DOC (mg/L) | 5.8 ± 0.9 | 5.9 ± 0.3 | 6.0 ± 1.1 | 6.9 ± 0.2 | 5.5 ± 0.1 |
| NH4+-N (mg/L) | <0.15 | <0.11 | <0.1 | <0.1 | <0.1 |
| EEQ (ng/L) | 1.83 ± 0.28 | n. m. | 3.92 ± 0.31 | 1.77 ± 0.25 | n. m. |
| AOX (µg/L) | 12 * | 25 ± 1 | 21 ± 5 | 25 ± 5 | 22 ± 8 |
| ClO2− (mg/L) | n. m. | n. m. | n. m. | <0.20 | n. m. |
| ClO3− (mg/L) | n. m. | n. m. | n. m. | <0.06 | n. m. |
| ClO4− (mg/L) | n. m. | n. m. | n. m. | <0.13 | n. m. |
| Emerging Contaminant (µg/L) | Variation of UV Energy Consumption between 0 and 1 kWh/m3 (Experiment 1) | Variation of Oxidant Concentration at 0.4 kWh/m3 (Experiment 2) | |||
|---|---|---|---|---|---|
| 0 and 3 mg/L FAC | 0 and 3 mg/L H2O2 | 1–4 mg/L FAC | 5–6 mg/L FAC | 1–6 mg/L H2O2 | |
| Carbamazepine | 0.47 ± 0.00 | 0.48 ± 0.02 | 0.75 ± 0.02 | 0.83 ± 0.02 | 0.43 ± 0.02 |
| Diclofenac | 1.16 ± 0.02 | 2.28 ± 0.09 | 2.15 ± 0.08 | 2.55 ± 0.18 | 1.83 ± 0.14 |
| Bisphenol A | 0.77 ± 0.00 | 0.61 ± 0.04 | 0.85 ± 0.02 | 0.57 ± 0.30 | 0.53 ± 0.12 |
| HHCB | 1.20 ± 0.01 | 1.14 ± 0.02 | 1.24 ± 0.03 | 1.19 ± 0.06 | 1.12 ± 0.02 |
| HHCB-lactone | 1.33 ± 0.02 | 1.21 ± 0.03 | 1.61 ± 0.05 | 1.56 ± 0.09 | 1.20 ± 0.08 |
| AHTN | 0.16 ± 0.00 | 0.14 ± 0.00 | 0.18 ± 0.01 | 0.18 ± 0.01 | 0.14 ± 0.01 |
| MTBT | 0.24 ± 0.00 | 0.29 ± 0.01 | 0.24 ± 0.01 | 0.22 ± 0.03 | 0.28 ± 0.01 |
| DEET | 0.08 ± 0.00 | 1.99 ± 0.01 | 0.05 ± 0.00 | 0.04 ± 0.01 | 0.28 ± 0.02 |
| Benzophenone | 0.14 ± 0.00 | 0.20 ± 0.00 | 0.12 ± 0.00 | 0.13 ± 0.02 | 0.21 ± 0.01 |
| 4t-Octylphenol | 0.04 ± 0.00 | 0.04 ± 0.00 | 0.04 ± 0.00 | 0.05 ± 0.01 | 0.03 ± 0.00 |
| 4-Nonylphenols | 1.93 ± 0.03 | 1.67 ± 0.06 | 1.65 ± 0.13 | 1.55 ± 0.24 | 2.01 ± 0.12 |
| Lidocaine | 0.12 ± 0.01 | 0.22 ± 0.00 | 0.27 ± 0.00 | 0.23 ± 0.01 | 0.17 ± 0.01 |
| Tramadol | 0.11 ± 0.01 | 0.17 ± 0.01 | 0.16 ± 0.01 | 0.09 ± 0.02 | 0.21 ± 0.02 |
| Diphenhydramine | 0.25 ± 0.01 | 0.25 ± 0.01 | 0.22 ± 0.01 | 0.27 ± 0.02 | 0.24 ± 0.01 |
| TCEP | 1.06 ± 0.02 | 0.69 ± 0.02 | 1.76 ± 0.03 | 0.35 ± 0.04 | 1.27 ± 0.16 |
| TCPP | 0.91 ± 0.05 | 1.56 ± 0.05 | 1.42 ± 0.02 | 1.66 ± 0.22 | 1.17 ± 0.09 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rott, E.; Kuch, B.; Lange, C.; Richter, P.; Kugele, A.; Minke, R. Removal of Emerging Contaminants and Estrogenic Activity from Wastewater Treatment Plant Effluent with UV/Chlorine and UV/H2O2 Advanced Oxidation Treatment at Pilot Scale. Int. J. Environ. Res. Public Health 2018, 15, 935. https://doi.org/10.3390/ijerph15050935
Rott E, Kuch B, Lange C, Richter P, Kugele A, Minke R. Removal of Emerging Contaminants and Estrogenic Activity from Wastewater Treatment Plant Effluent with UV/Chlorine and UV/H2O2 Advanced Oxidation Treatment at Pilot Scale. International Journal of Environmental Research and Public Health. 2018; 15(5):935. https://doi.org/10.3390/ijerph15050935
Chicago/Turabian StyleRott, Eduard, Bertram Kuch, Claudia Lange, Philipp Richter, Amélie Kugele, and Ralf Minke. 2018. "Removal of Emerging Contaminants and Estrogenic Activity from Wastewater Treatment Plant Effluent with UV/Chlorine and UV/H2O2 Advanced Oxidation Treatment at Pilot Scale" International Journal of Environmental Research and Public Health 15, no. 5: 935. https://doi.org/10.3390/ijerph15050935
APA StyleRott, E., Kuch, B., Lange, C., Richter, P., Kugele, A., & Minke, R. (2018). Removal of Emerging Contaminants and Estrogenic Activity from Wastewater Treatment Plant Effluent with UV/Chlorine and UV/H2O2 Advanced Oxidation Treatment at Pilot Scale. International Journal of Environmental Research and Public Health, 15(5), 935. https://doi.org/10.3390/ijerph15050935