
The Madangamines: Synthetic Strategies Toward Architecturally Complex Alkaloids




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
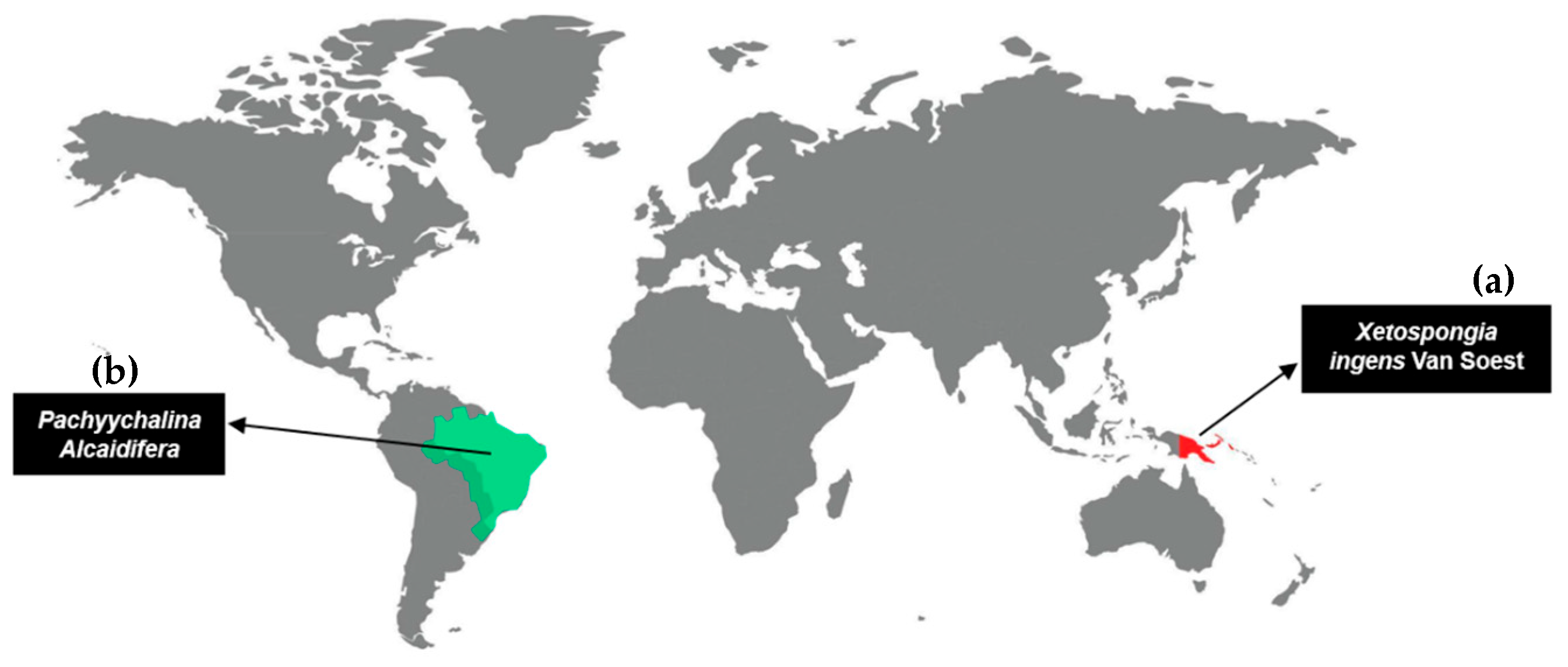
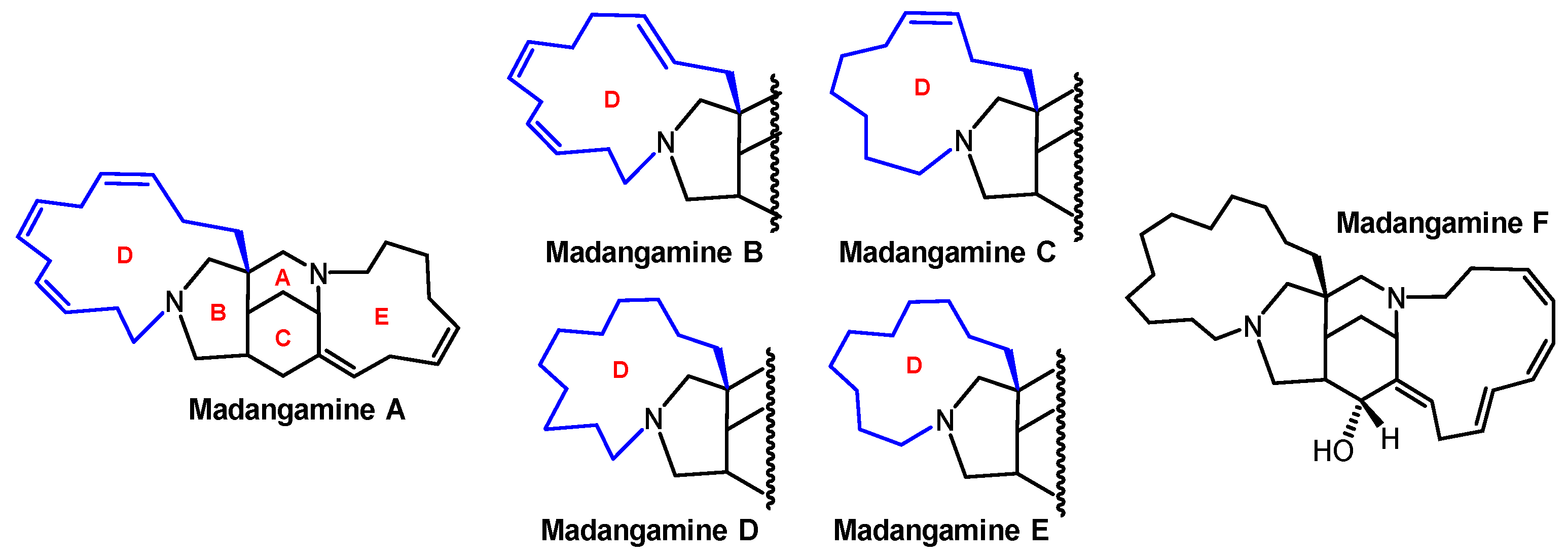
1. Introduction
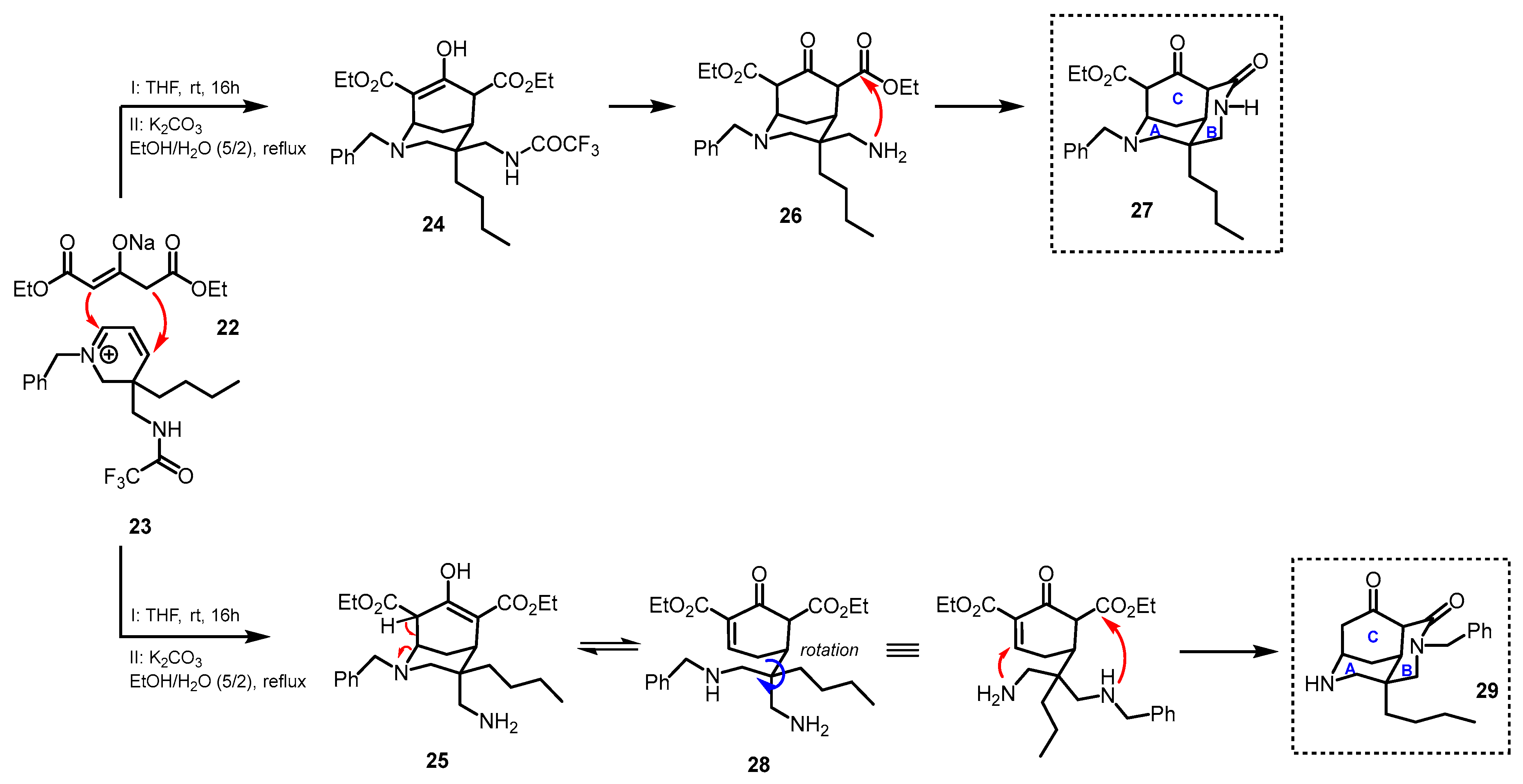
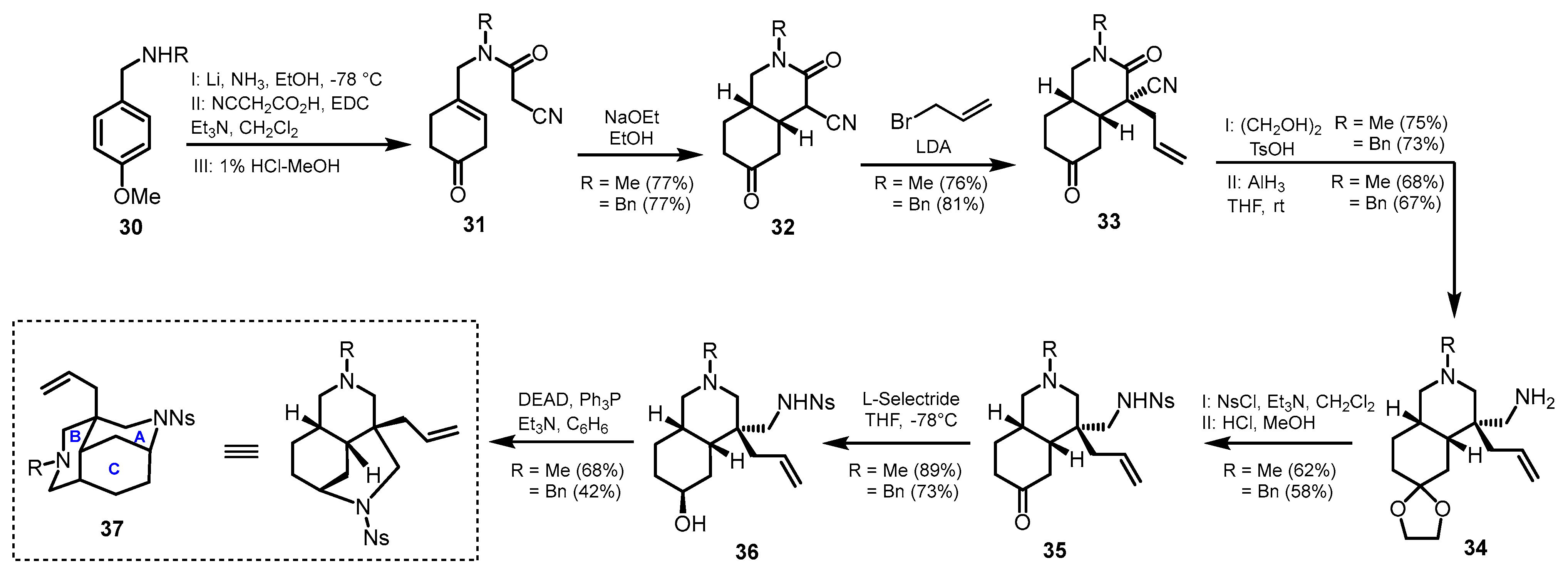
2. Synthetic Approaches to the Tricyclic Skeleton of Madangamines
Diazatricyclic Fragment (ABC Rings)
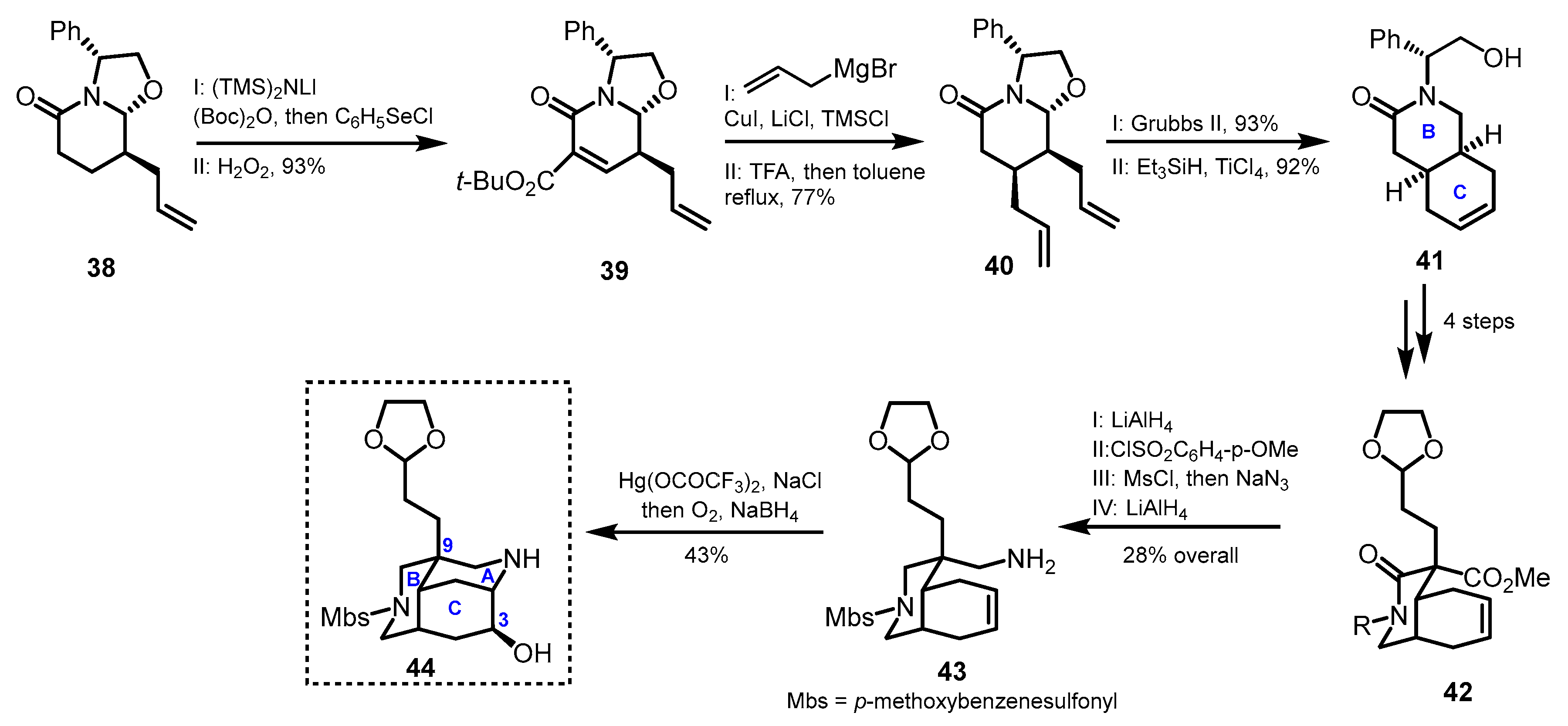
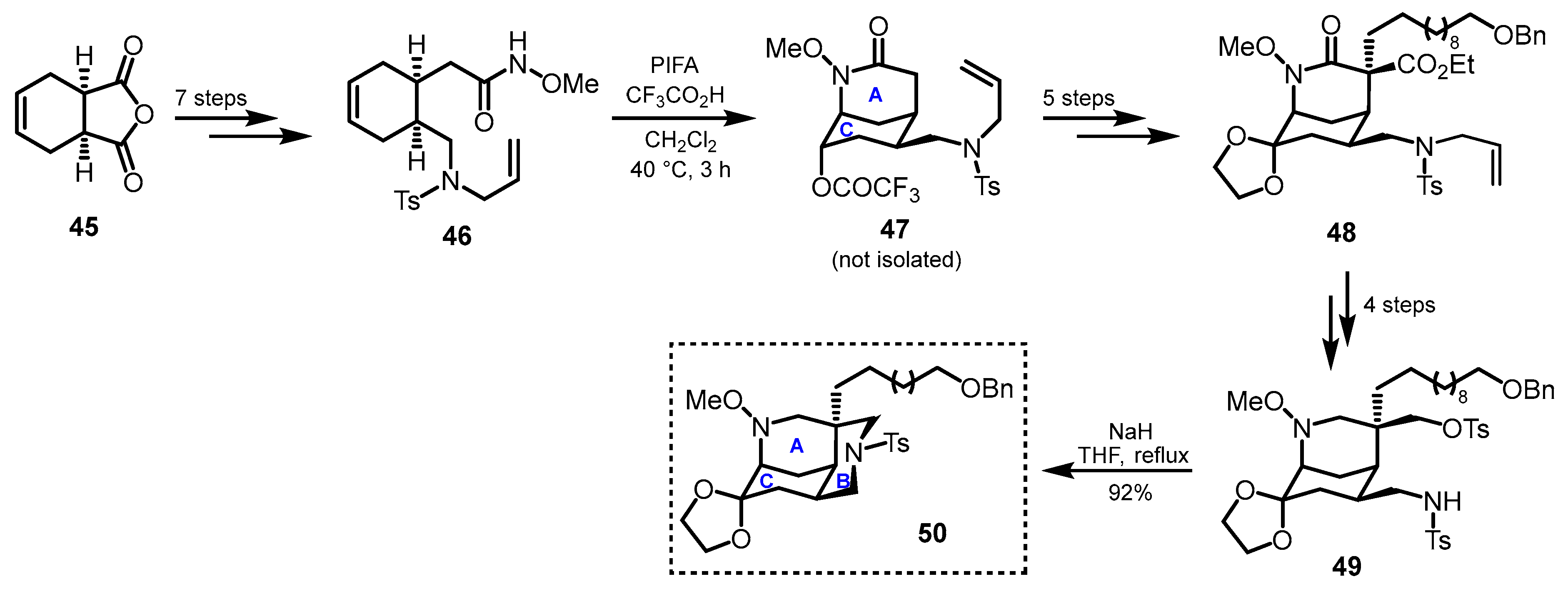
3. Synthetic Methods Enabling Macrocyclic Ring Construction
3.1. Azatricyclic Fragments (ACE Rings)
3.2. Model Studies on the Construction of Macrocyclic D-Ring
3.3. Tetracyclic Fragments (ABCD and ABCE Rings)
4. Total Syntheses of Madangamines
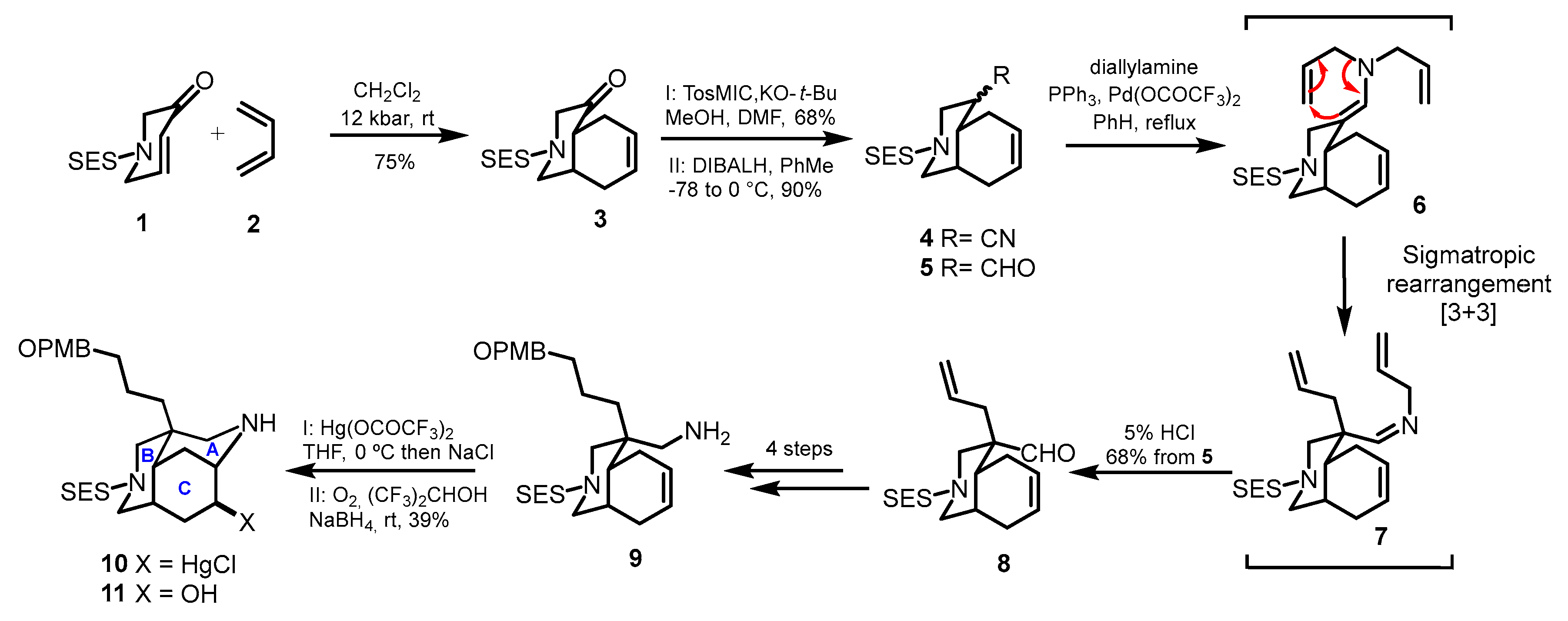
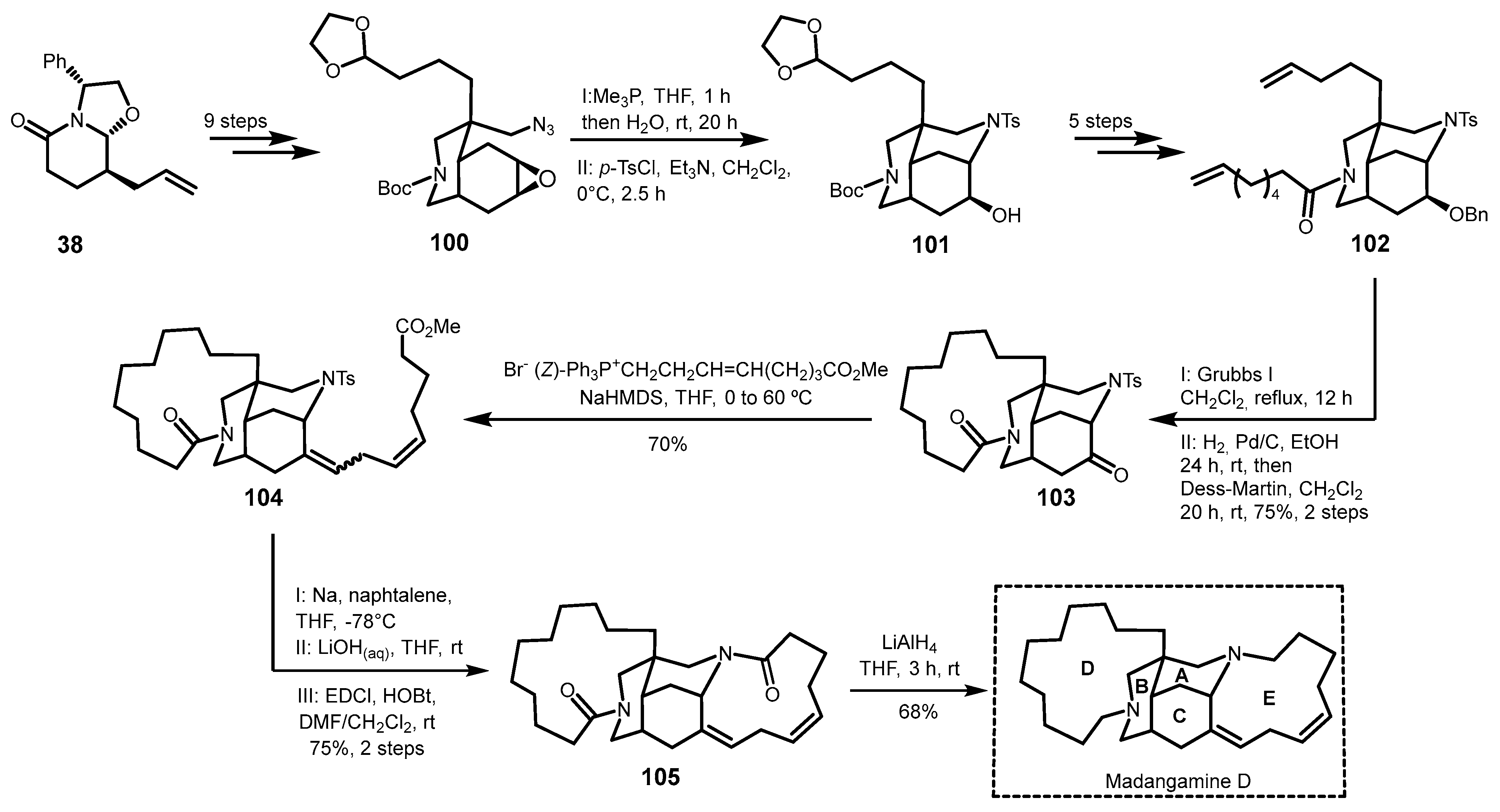
4.1. The First Total Synthesis: Synthesis of the Alkaloid Madandamine D
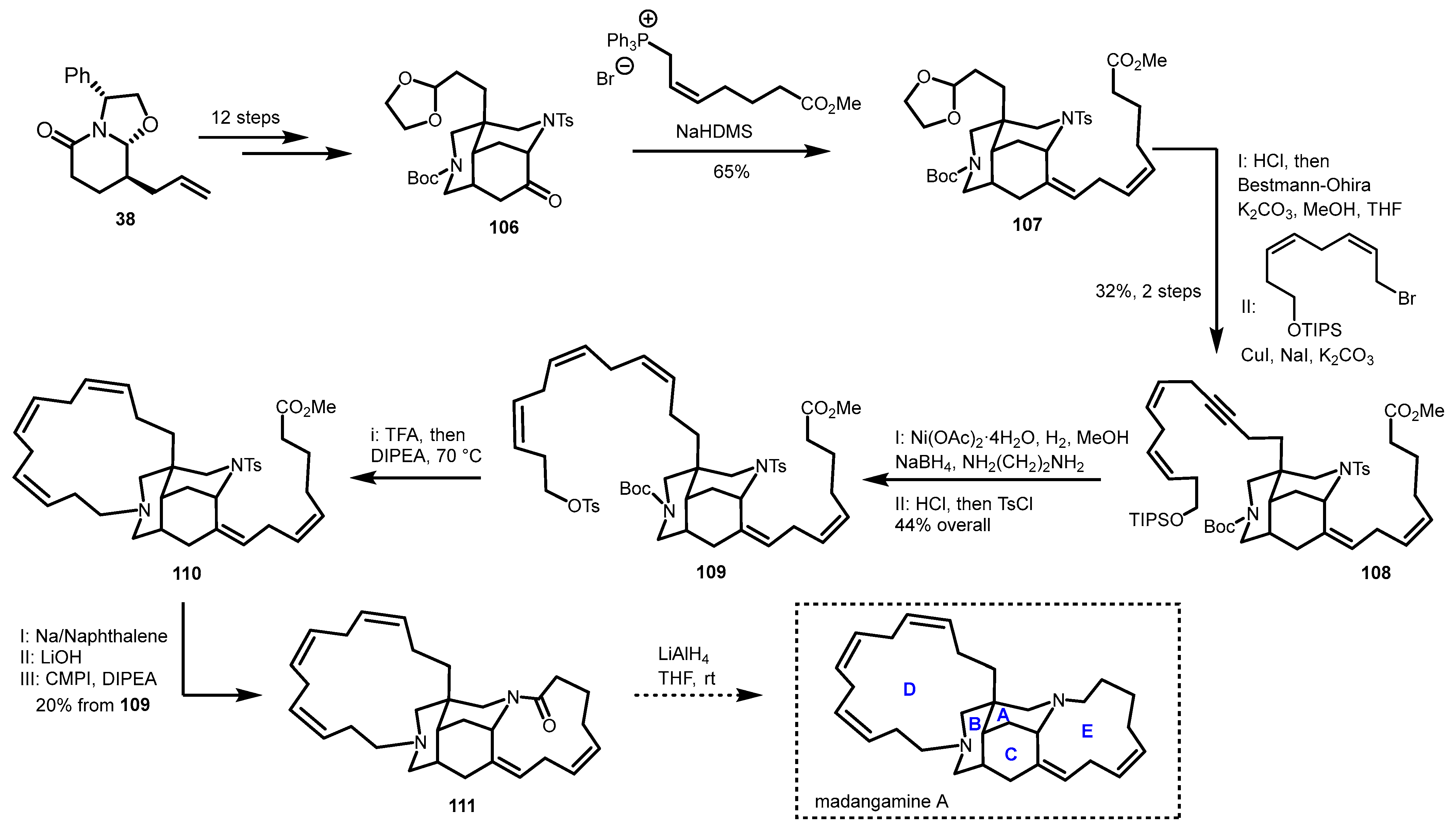
4.2. Formal Synthesis of Madangamine A
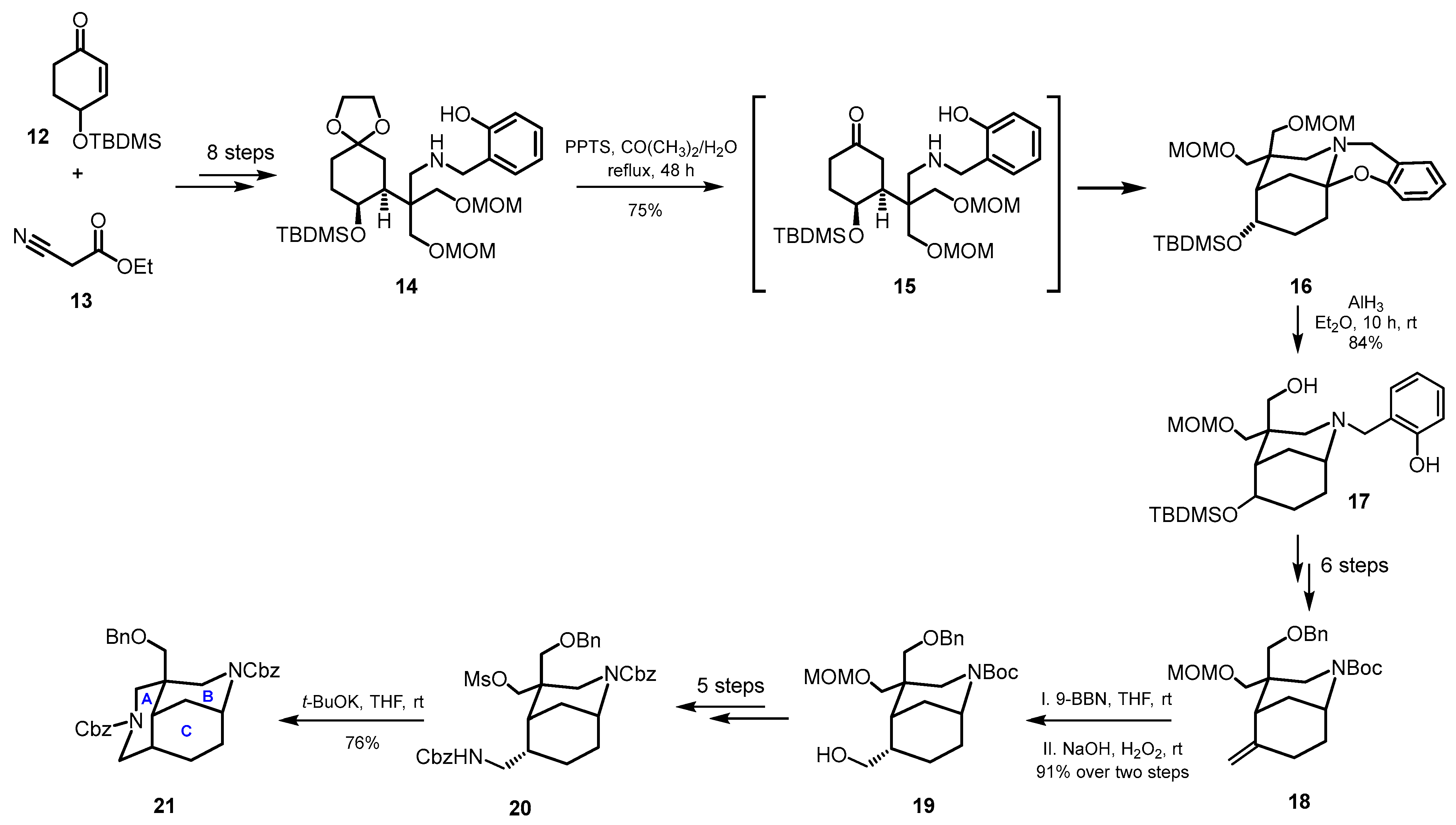
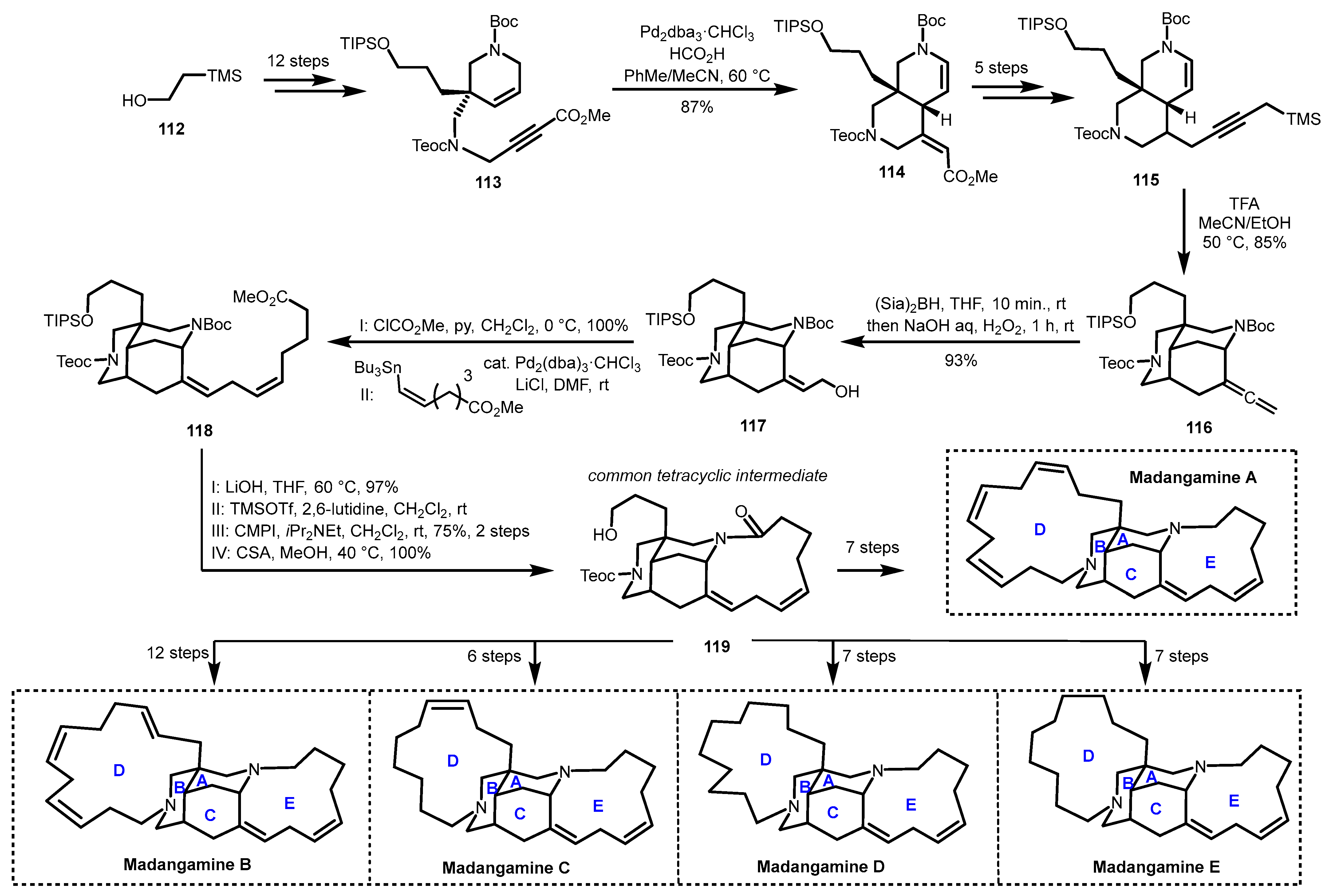
4.3. Divergent Strategy: Total Synthesis of Madangamines A, B, C, D, and E
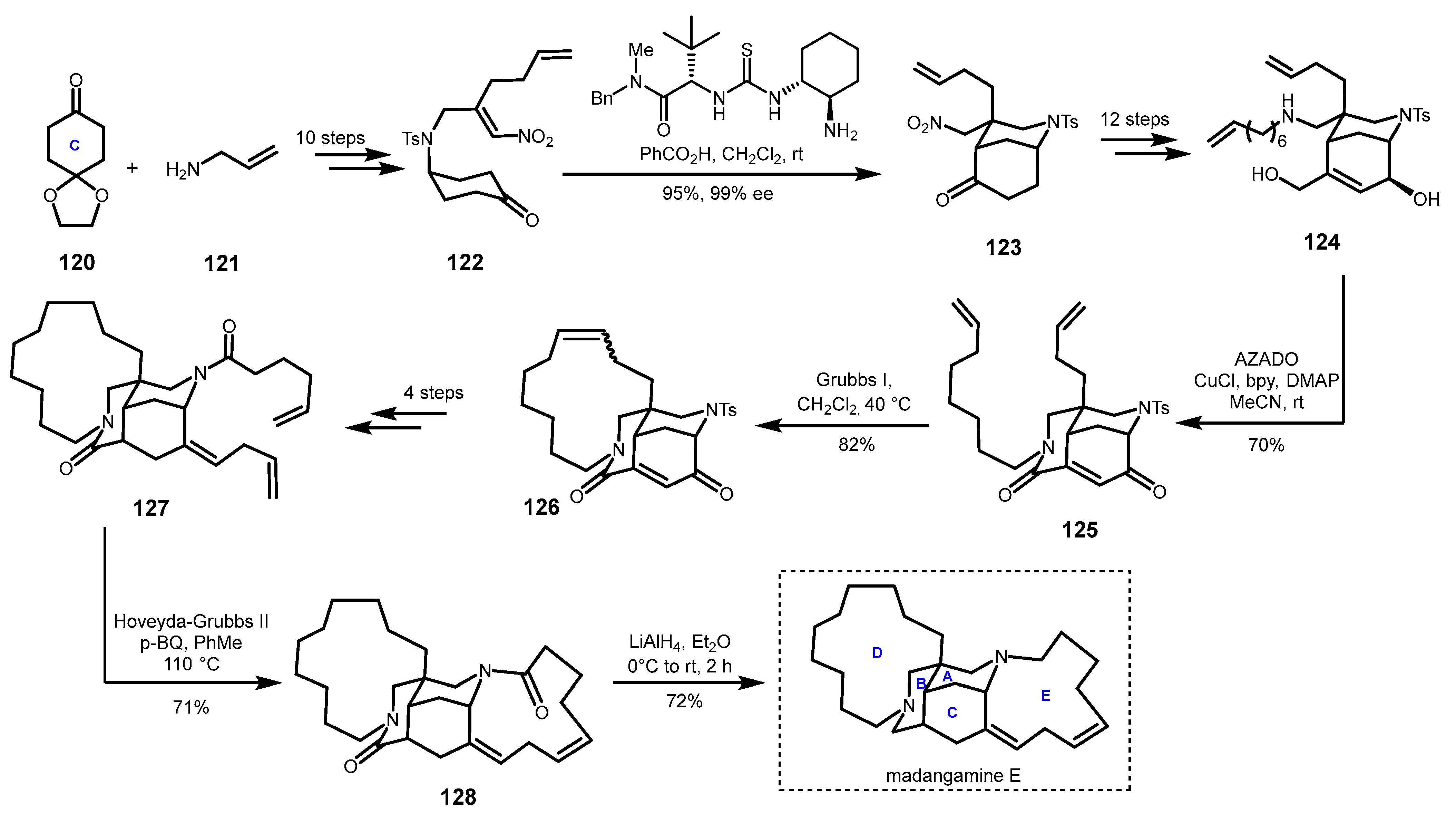
4.4. Enantioselective Total Synthesis of Madangamine E
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Cimino, G.; Mattia, C.; Mazzarella, L.; Puliti, R.; Scognamiglio, G.; Spinella, A.; Trivellone, E. Unprecedented alkaloid skeleton from the mediterranean sponge reniera sarai—X-ray structure of an acetate derivative of sarain-A. Tetrahedron 1989, 45, 3863–3872. [Google Scholar] [CrossRef]
- Kong, F.M.; Andersen, R.J.; Allen, T.M. Ingenamine, A novel pentacyclic alkaloid from the marine sponge Xestospongia ingens. Tetrahedron Lett. 1994, 35, 1643–1646. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Manzamine A, A novel antitumor alkaloid from a sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Crews, P.; Cheng, X.C.; Adamczeski, M.; Rodríguez, J.; Jaspars, M.; Schmitz, F.J.; Traeger, S.C.; Pordesimo, E.O. 1,2,3,4-tetrahydro-8-hydroxymanzamines, alkaloids from 2 different Haplosclerid sponges. Tetrahedron 1994, 50, 13567–13574. [Google Scholar] [CrossRef]
- Kobayashi, J.; Watanabe, D.; Kawasaki, N.; Tsuda, M. Nakadomarin A, a novel hexacyclic manzamine-related alkaloid from Amphimedon sponge. J. Org. Chem. 1997, 62, 9236–9239. [Google Scholar] [CrossRef]
- Kong, F.M.; Andersen, R.J.; Allen, T.M. Madangamine-A, A novel cytotoxic alkaloid from the marine sponge Xestospongia ingens. J. Nat. Prod. 1994, 116, 6007–6008. [Google Scholar] [CrossRef]
- Kong, F.M.; Graziani, E.I.; Andersen, R.J. Madangamines B-E, pentacyclic alkaloids from the marine sponge Xestospongia ingens. J. Nat. Prod. 1998, 61, 267–271. [Google Scholar] [CrossRef]
- de Oliveira, J.; Nascimento, A.M.; Kossuga, M.H.; Cavalcanti, B.C.; Pessoa, C.O.; Moraes, M.O.; Macedo, M.L.; Ferreira, A.G.; Hajdu, E.; Pinheiro, U.S.; et al. Cytotoxic alkylpiperidine alkaloids from the Brazilian marine sponge Pachychalina alcaloidifera. J. Nat. Prod. 2007, 70, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kawano, S.; Suto, T.; Sato, T.; Chida, N.; Simizu, S. Identification of madangamine A as a novel lysosomotropic agent to inhibit autophagy. Bioorg. Med. Chem. 2021, 34, 116041. [Google Scholar] [CrossRef]
- Suto, T.; Yanagita, Y.; Nagashima, Y.; Takikawa, S.; Kurosu, Y.; Matsuo, N.; Sato, T.; Chida, N. Unified total synthesis of madangamines A, C, and E. J. Am. Chem. Soc. 2017, 139, 2952–2955. [Google Scholar] [CrossRef]
- Suto, T.; Yanagita, Y.; Nagashima, Y.; Takikawa, S.; Kurosu, Y.; Matsuo, N.; Miura, K.; Simizu, S.; Sato, T.; Chida, N. Unified total synthesis of madangamine Alkaloids. Bull. Chem. Soc. Jpn. 2019, 92, 545–571. [Google Scholar] [CrossRef]
- Shiomi, S.; Shennan, B.D.A.; Yamazaki, K.; de Arriba, A.L.F.; Vasu, D.; Hamlin, T.A.; Dixon, D.J. A new organocatalytic desymmetrization reaction enables the enantioselective total synthesis of madangamine E. J. Am. Chem. Soc. 2022, 144, 1407–1415. [Google Scholar] [CrossRef]
- Diaba, F.; Pujol-Grau, C.; Martínez-Laporta, A.; Fernández, I.; Bonjoch, J. Synthesis of the tetracyclic ABCD ring systems of madangamines D-F. Org. Lett. 2015, 17, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Are, C.; Pérez, M.; Ballette, R.; Proto, S.; Caso, F.; Yayik, N.; Bosch, J.; Amat, M. Access to enantiopure advanced intermediates en route to madangamines. Chem. Eur. J. 2019, 25, 15929–15933. [Google Scholar] [CrossRef]
- Matzanke, N.; Gregg, R.J.; Weinreb, S.M.; Parvez, M. A concise approach to the tricyclic core of the cytotoxic marine alkaloid madangamine A. J. Org. Chem. 1997, 62, 1920–1921. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kusanagi, T.; Kibayashi, C. Synthesis of the diazatricyclic core of the marine alkaloids madangamines. Tetrahedron Lett. 2004, 45, 6509–6512. [Google Scholar] [CrossRef]
- Tong, H.M.; Martin, M.T.; Chiaroni, A.; Bénéchie, M.; Marazano, C. An approach to the tricyclic core of madangamines based upon a biogenetic scheme. Org. Lett. 2005, 7, 2437–2440. [Google Scholar] [CrossRef]
- Quirante, J.; Paloma, L.; Diaba, F.; Vila, X.; Bonjoch, J. Synthesis of diazatricyclic core of madangamines from cis-perhydroisoquinolines. J. Org. Chem. 2008, 73, 768–771. [Google Scholar] [CrossRef]
- Amat, M.; Pérez, M.; Proto, S.; Gatti, T.; Bosch, J. First enantioselective synthesis of the diazatricyclic core of madangamine alkaloids. Chem. Eur. J. 2010, 16, 9438–9441. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Gerasimov, M.V.; DeJong, S.; Wardrop, D.J. Oxamidation of unsaturated o-alkyl hydroxamates: Synthesis of the madangamine diazatricylic (ABC rings) skeleton. Org. Lett. 2017, 19, 6570–6573. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, M.S.; Douglas, C.J. Synthesis of the madangamine alkaloid core by a C-C bond activation cascade. Org. Lett. 2019, 21, 6149–6154. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Inoue, J.; Yamazaki, N.; Aoyagi, S.; Kibayashi, C. Synthesis of the 11-membered ring of the marine alkaloids, madangamines. Tetrahedron Lett. 2006, 47, 3489–3492. [Google Scholar] [CrossRef]
- Proto, S.; Amat, M.; Pérez, M.; Ballette, R.; Romagnoli, F.; Mancinelli, A.; Bosch, J. Model studies on the synthesis of madangamine alkaloids. Assembly of the macrocyclic rings. Org. Lett. 2012, 14, 3916–3919. [Google Scholar] [CrossRef] [PubMed]
- Ballette, R.; Pérez, M.; Proto, S.; Amat, M.; Bosch, J. Total synthesis of (+) madangamine D. Angew. Chem.-Int. Ed. 2014, 53, 6202–6205. [Google Scholar] [CrossRef] [PubMed]
- Are, C.; Pérez, M.; Bosch, J.; Amat, M. Enantioselective formal synthesis of (+) madangamine A. Chem. Commun. 2019, 55, 7207–7210. [Google Scholar] [CrossRef] [PubMed]
- Yokoshima, S. Synthesis of 2-azabicyclo[3.3.1]nonanes: Lessons from synthetic studies of macrocyclic diamine alkaloids. Chem. Lett. 2021, 50, 96–102. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, L.; Hong, R. Madangamine alkaloids: Madness and tranquility. Tetrahedron Chem. 2022, 3, 100025. [Google Scholar] [CrossRef]
- Amat, M.; Pérez, M.; Ballette, R.; Proto, S.; Bosch, J. Chapter Three—The Alkaloids of the Madangamine Group. In The Alkaloids: Chemistry and Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 74, pp. 159–199. [Google Scholar]
- Wender, P.A.; Verma, V.A.; Paxton, T.J.; Pillow, T.H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos, V.; Maulen, C.; Parra, C.; Bradshaw, B. The Madangamines: Synthetic Strategies Toward Architecturally Complex Alkaloids. Mar. Drugs 2025, 23, 301. https://doi.org/10.3390/md23080301
Ríos V, Maulen C, Parra C, Bradshaw B. The Madangamines: Synthetic Strategies Toward Architecturally Complex Alkaloids. Marine Drugs. 2025; 23(8):301. https://doi.org/10.3390/md23080301
Chicago/Turabian StyleRíos, Valentina, Cristian Maulen, Claudio Parra, and Ben Bradshaw. 2025. "The Madangamines: Synthetic Strategies Toward Architecturally Complex Alkaloids" Marine Drugs 23, no. 8: 301. https://doi.org/10.3390/md23080301
APA StyleRíos, V., Maulen, C., Parra, C., & Bradshaw, B. (2025). The Madangamines: Synthetic Strategies Toward Architecturally Complex Alkaloids. Marine Drugs, 23(8), 301. https://doi.org/10.3390/md23080301