
Molecular Docking and Dynamics Simulations Reveal the Antidiabetic Potential of a Novel Fucoxanthin Derivative from Chnoospora minima

 ,
,  , , , and
, , , and
Abstract
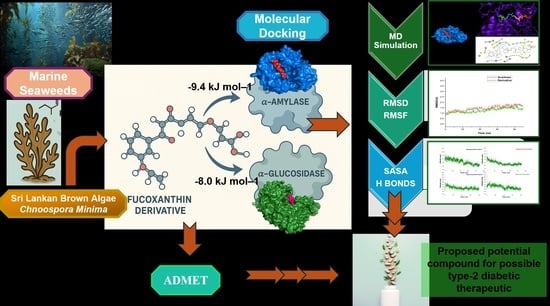
1. Introduction
2. Results
2.1. Phytochemicals
2.1.1. Total Phenolic Content (TPC)
2.1.2. Total Flavonoid Content (TFC)
2.2. Antidiabetic Activities
2.2.1. α-Amylase Activity
2.2.2. α-Glucosidase Activity
2.3. Fractionation, and Compound Isolation
2.4. Pharmacokinetic Profiles and Drug-likeness Characteristics of the Optimized Ligand
2.5. Protein Structure Validation
2.6. Molecular Docking
2.7. Molecular Dynamics (MD) Simulation
3. Discussion
4. Materials and Methods
4.1. Collection and Preparation of Algae Samples
4.2. De-polysaccharide Crude Methanol Extraction and Solvent-Solvent Partition
4.3. In Vitro Quantification of Phytochemicals
4.4. In Vitro Antidiabetic Activities
4.4.1. α-Amylase Inhibitory Activity
4.4.2. α-Glucosidase Inhibitory Activity
4.5. Bioassay-Guided Fractionation, Compound Isolation, and Structural Elucidation
4.6. Ligand Preparation and Structure Optimization
4.7. Pharmacokinetics and Drug-likeness Prediction
4.8. Protein Structures Retrieval and Optimization
4.9. Molecular Docking Study
4.10. Molecular Dynamics (MD) Simulation
4.11. Data Analysis and Visualization
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FD | Fucoxanthin Derivative |
| T2DM | Type 2 Diabetes Mellitus |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| SASA | Solvent Accessible Surface Area |
| TPSA | Topological Polar Surface Area |
| HIA | Human Intestinal Absorption |
| BBB | Blood–Brain Barrier |
| PPB | Plasma Protein Binding |
| HPLC | High Performance Liquid Chromatography |
| NMR | Nuclear Magnetic Resonance |
| H NMR | Hydrogen Nuclear Magnetic Resonance |
| 1H-1H COSY | 1H-1H Correlation Spectroscopy |
| HMQC | Hetero Nuclear Multiple Bond Correlation Spectroscopy |
| HMBC | 1H-13C Heteronuclear Multiple Bond Correlation Spectroscopy |
| C NMR | Carbon Nuclear Magnetic Resonance |
References
- Oyewusi, H.A.; Wu, Y.-S.; Safi, S.Z.; Wahab, R.A.; Hatta, M.H.M.; Batumalaie, K. Molecular dynamics simulations reveal the inhibitory mechanism of Withanolide A against α-glucosidase and α-amylase. J. Biomol. Struct. Dyn. 2023, 41, 6203–6218. [Google Scholar] [CrossRef]
- Dahiru, M.M.; Musa, N.; Abaka, A.M.; Abubakar, M.A. Potential Antidiabetic Compounds from Anogeissus leiocarpus: Molecular Docking, Molecular Dynamic Simulation, and ADMET Studies. Borneo J. Pharm. 2023, 6, 249–277. [Google Scholar] [CrossRef]
- Goyal, S.; Rani, J.; Bhat, M.A.; Vanita, V. Genetics of diabetes. World J. Diabetes 2023, 14, 656–679. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Obesity management for the treatment of type 2 diabetes: Standards of medical care in diabetes—2020. Diabetes Care 2020, 43, S89–S97. [Google Scholar] [CrossRef] [PubMed]
- Rannan-Eliya, R.P.; Wijemunige, N.; Perera, P.; Kapuge, Y.; Gunawardana, N.; Sigera, C.; Jayatissa, R.; Herath, H.M.M.; Gamage, A.; Weerawardena, N.; et al. Prevalence of diabetes and pre-diabetes in Sri Lanka: A new global hotspot-estimates from the Sri Lanka Health and Ageing Survey 2018/2019. BMJ Open Diabetes Res. Care 2023, 11, e003160. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee; ElSayed, N.A.; Aleppo, G.; Bannuru, R.R.; Bruemmer, D.; Collins, B.S.; Ekhlaspour, L.; Gaglia, J.L.; Hilliard, M.E.; Johnson, E.L.; et al. 2. Diagnosis and Classification of Diabetes: Standards of Care in Diabetes—2024. Diabetes Care 2024, 47, S20–S42. [Google Scholar] [CrossRef]
- Fatema, K.; Sharmin, A.A.; Sharna, J.F.; Haque, A.; Rahman, M.M.; Sarker, S.; Kazi, M.; Rahman, R.; Namakka, M.; Uzzaman, M.; et al. Antioxidant and Antidiabetic Effects of Flemingia macrophylla Leaf Extract and Fractions: In vitro, Molecular Docking, Dynamic Simulation, Pharmacokinetics, and Biological Activity Studies. Bioresources 2024, 19, 4960–4983. [Google Scholar] [CrossRef]
- Kawee-Ai, A.; Kim, A.T.; Kim, S.M. Inhibitory activities of microalgal fucoxanthin against α-amylase, α-glucosidase, and glucose oxidase in 3T3-L1 cells linked to type 2 diabetes. J. Oceanol. Limnol. 2019, 37, 928–937. [Google Scholar] [CrossRef]
- Gunathilaka, T.; Keertihirathna, L.R.; Peiris, D. Advanced Pharmacological Uses of Marine Algae as an Anti-Diabetic Therapy. Nat. Med. Plants 2022, 11, 79. [Google Scholar] [CrossRef]
- Jang, H.; Lee, J.; Park, Y.-K.; Lee, J.-Y. Exploring the health benefits and concerns of brown seaweed consumption: A comprehensive review of bioactive compounds in brown seaweed and its potential therapeutic effects. J. Agric. Food Res. 2024, 17, 101215. [Google Scholar] [CrossRef]
- Gunathilaka, T.L.; Samarakoon, K.; Ranasinghe, P.; Peiris, L.D.C. Antidiabetic Potential of Marine Brown Algae—A Mini Review. J. Diabetes Res. 2020, 2020, 1230218. [Google Scholar] [CrossRef] [PubMed]
- Gunathilaka, T.L.; Bandaranayake, U.; Boudjelal, M.; Ali, R.; Silva, R.M.; Samarakoon, K.W.; Ranasinghe, P.; Peiris, L.D.C. Chnoospora minima: A Robust Candidate for Hyperglycemia Management, Unveiling Potent Inhibitory Compounds and Their Therapeutic Potential. Mar. Biotechnol. 2024, 26, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Mohibbullah, M.; Haque, N.; Sohag, A.A.M.; Hossain, T.; Zahan, S.; Uddin, J.; Hannan, A.; Moon, I.S.; Choi, J.-S. A Systematic Review on Marine Algae-Derived Fucoxanthin: An Update of Pharmacological Insights. Mar. Drugs 2022, 20, 279. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yuan, J.-P.; Wu, C.-F.; Wang, J.-H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef]
- Abdulazeez, S. Molecular simulation studies on B-cell lymphoma/leukaemia 11A (BCL11A). Am. J. Transl. Res. 2019, 11, 3689–3697. [Google Scholar]
- Jin, Y.; Arroo, R. The protective effects of flavonoids and carotenoids against diabetic complications—A review of in vivo evidence. Front. Nutr. 2023, 10, 1020950. [Google Scholar] [CrossRef]
- Aryal, D.; Joshi, S.; Thapa, N.K.; Chaudhary, P.; Basaula, S.; Joshi, U.; Bhandari, D.; Rogers, H.M.; Bhattarai, S.; Sharma, K.R.; et al. Dietary phenolic compounds as promising therapeutic agents for diabetes and its complications: A comprehensive review. Food Sci. Nutr. 2024, 12, 3025–3045. [Google Scholar] [CrossRef]
- Widyawati, T.; Yusoff, N.A.; Bello, I.; Asmawi, M.Z.; Ahmad, M. Bioactivity-Guided Fractionation and Identification of Antidiabetic Compound of Syzygium polyanthum (Wight.)’s Leaf Extract in Streptozotocin-Induced Diabetic Rat Model. Molecules 2022, 27, 6814. [Google Scholar] [CrossRef]
- Kumarasinghe, H.; Gunathilaka, M. A systematic review of fucoxanthin as a promising bioactive compound in drug development. Phytochem. Lett. 2024, 61, 52–65. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Mbah, J.A.; Lifongo, L.L.; Owono, L.C.O.; Megnassan, E.; Mbaze, L.M.; Judson, P.N.; Sippl, W.; Efange, S.M. Assessing the pharmacokinetic profile of the CamMedNP natural products database: An in silico approach. Org. Med. Chem. Lett. 2013, 3, 10. [Google Scholar] [CrossRef]
- Wang, C.; Kim, J.H.; Kim, S.W. Synthetic biology and metabolic engineering for marine carotenoids: New opportunities and future prospects. Mar. Drugs 2014, 12, 4810–4832. [Google Scholar] [CrossRef] [PubMed]
- Shikov, A.N.; Flisyuk, E.V.; Obluchinskaya, E.D.; Pozharitskaya, O.N. Pharmacokinetics of marine-derived drugs. Mar. Drugs 2020, 18, 557. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef] [PubMed]
- Sippel, K.H.; Quiocho, F.A. Ion-dipole interactions and their functions in proteins. Protein Sci. 2015, 24, 1040–1046. [Google Scholar] [CrossRef]
- Alafnan, A.; Chettupalli, A.K.; Unnisa, A.; Hussain, T.; Anwar, S.; Alkhojali, W.M.; Khalifa, N.E.; Osman, M.E.D.; Younes, K.M.; Abouzied, A.S.; et al. In silico elucidation of plausible anti-obesity activity by Withaferin-A compound targeting alpha-amylase. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 3150–3158. [Google Scholar]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and molecular docking studies of fucosterol and fucoxanthin, BACE1 inhibitors from brown algae Undaria pinnatifida and Ecklonia stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef]
- Khenifi, M.L.; Serseg, T.; Migas, P.; Krauze-Baranowska, M.; Özdemir, S.; Bensouici, C.; Alghonaim, M.I.; Al-Khafaji, K.; Alsalamah, S.A.; Boudjeniba, M.; et al. HPLC-DAD-MS Characterization, Antioxidant Activity, α-amylase Inhibition, Molecular Docking, and ADMET of Flavonoids from Fenugreek Seeds. Molecules 2023, 28, 7798. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Xing, Z.; Zhu, B.; Hou, R.; Zhang, J.; Li, T.; Zhang, Z.; Wang, H.; Li, Z. Study on the binding behavior and functional properties of soybean protein isolate and β-carotene. Front. Nutr. 2022, 9, 984490. [Google Scholar] [CrossRef]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. [Google Scholar] [CrossRef]
- Idris, M.O.; Yekeen, A.A.; Alakanse, O.S.; Durojaye, O.A. Computer-aided screening for potential TMPRSS2 inhibitors: A combination of pharmacophore modeling, molecular docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2020, 39, 5638–5656. [Google Scholar] [CrossRef]
- Khan, A.; Gui, J.; Ahmad, W.; Haq, I.; Shahid, M.; Khan, A.A.; Shah, A.; Khan, A.; Ali, L.; Anwar, Z.; et al. The SARS-CoV-2 B.1.618 variant slightly alters the spike RBD-ACE2 binding affinity and is an antibody escaping variant: A computational structural perspective. RSC Adv. 2021, 11, 30132–30147. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Sharifi-Rad, J.; Seca, A.M.L.; Pinto, D.C.G.A.; Michalak, I.; Trincone, A.; Mishra, A.P.; Nigam, M.; Zam, W.; Martins, N. Current trends on seaweeds: Looking at chemical composition, phytopharmacology, and cosmetic applications. Molecules 2019, 24, 4182. [Google Scholar] [CrossRef] [PubMed]
- Jeyaseelan, E.C.; Jeyaseelan, T.C.; Thavaranjit, A.C. Antibacterial Activity of Some Selected Algae Present in the Costal Lines of Jaffna Peninsula. Int. J. Pharm. Biol. Arch. 2012, 3, 352–356. [Google Scholar]
- Lakmal, H.C.; Samarakoon, K.W.; Lee, W.; Lee, J.-H.; Abeytunga, D.; Lee, H.-S.; Jeon, Y.-J. Anticancer and antioxidant effects of selected Sri Lankan marine algae. J. Natl. Sci. Found. Sri Lanka 2014, 42, 315. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-Ravent, R.M. Analysis of Total Phenols and Other Oxidation Substrates and Antioxidants by Means of Folin-Ciocalteu Reagent. Methods Enzymol. 1999, 299, 152–178. [Google Scholar] [CrossRef]
- Kokilam, G.; Vasuki, S.; Sajitha, N. Biochemical composition, alginic acid yield and antioxidant activity of brown seaweeds from mandapam region, gulf of mannar. J. Appl. Pharm. Sci. 2013, 3, 99–104. [Google Scholar]
- Gunathilaka, T.L.; Samarakoon, K.W.; Ranasinghe, P.; Peiris, L.C.D. In-Vitro Antioxidant, Hypoglycemic Activity, and Identification of Bioactive Compounds in Phenol-Rich Extract from the Marine Red Algae Gracilaria edulis (Gmelin) Silva. Molecules 2019, 24, 3708. [Google Scholar] [CrossRef]
- Zhou, B.; Huang, N.; Zeng, W.; Zhang, H.; Chen, G.; Liang, Z. Development of a strategy for the screening of α-glucosidase-producing microorganisms. J. Microbiol. 2020, 58, 163–172. [Google Scholar] [CrossRef]
- Jawarkar, R.D.; Sharma, P.; Jain, N.; Gandhi, A.; Mukerjee, N.; Al-Mutairi, A.A.; Zaki, M.E.A.; Al-Hussain, S.A.; Samad, A.; Masand, V.H.; et al. QSAR, Molecular Docking, MD Simulation and MMGBSA Calculations Approaches to Recognize Concealed Pharmacophoric Features Requisite for the Optimization of ALK Tyrosine Kinase Inhibitors as Anticancer Leads. Molecules 2022, 27, 4951. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract/Fraction | TPC (mg GAE/g) | TFC (mg QE/g) |
|---|---|---|
| Crude methanol extract | 57.01 ± 6.12 a | 0.79 ± 0.04 d |
| Hexane fraction | 2.96 ± 0.41 d | 0.21 ± 0.06 e |
| Chloroform fraction | 36.42 ± 2.74 b | 3.31 ± 0.04 b |
| Ethyl acetate fraction | 58.11 ± 4.28 a | 5.24 ± 1.01 a |
| Aqueous fraction | 19.90 ± 2.11 c | 1.05 ± 0.07 c |
| Extract/Fraction | α- Amylase (µg/mL) | α- Glucosidase (µg/mL) |
|---|---|---|
| Ethyl acetate fraction | 30.56 ± 0.56 d | 14.78 ± 0.26 d |
| Chloroform fraction | 5.34 ± 0.32 e | 6.02 ± 0.18 e |
| Aqueous fraction | 92.12 ± 1.20 b | 36.92 ± 1.06 c |
| Hexane fraction | 149.31 ± 0.94 a | 83.92 ± 0.54 a |
| Crude methanol extract | 45.63 ± 0.04 d | 58.88 ± 2.01 b |
| Acarbose | 72.41 ± 0.24 c | 1.02 ± 0.07 f |
| Position | 13C δ (ppm) | 1H δ (ppm), Integration, Multiplicity, J (Hz) | Position′ | 13C δ (ppm) | 1H δ (ppm), Integration, Multiplicity, J (Hz) |
|---|---|---|---|---|---|
| C1 | 35.14 | - | C1′ | 35.76 | - |
| C2 | 47.07 | 1.33 (1H, dd, J = 12.3) 1.47 (1H, dd, J = 13.8) | C2′ | 45.42 | 1.39 (1H, t, J = 12.63) 1.97 (1H, m) |
| C3 | 64.31 | 3.79 (1H, m) | C3′ | 67.97 | 5.36 (1H, tt J = 4.29, 11.38) |
| C4 | 41.66 | 1.76 (1H, dd, J = 9.18, 13.8) 2.30 (1H, ddd, J = 1.44, 4.74, 9.18) | C4′ | 45.22 | 1.49 (1H, t, J = 12.92) 2.26 (1H, ddd, J = 2.04, 4.2, 12.84) |
| C5 | 66.12 | - | C5′ | 72.67 | - |
| C6 | 67.06 | - | C6′ | 117.51 | - |
| C7 | 40.8 | 3.63, 2.58 (2H, d, J = 18.3) | C7′ | 103.36 | 6.03 (1H, s) |
| C8 | 197.83 | - | C8′ | 202.33 | - |
| C9 | 134.52 | - | C9′ | 132.46 | 6.61 (1H, dd, J = 11.64, 14.16) |
| C10 | 139.06 | 7.13 (1H, d, J = 10.92) | C10′ | 128.51 | 6.11 (1H, d, J = 11.52) |
| C11 | 123.36 | 6.55 (1H, dd, J = 12.05, 15) | C11′ | 125.66 | 6.73 (1H, dd, J = 11.94, 14.6) |
| C12 | 144.99 | 6.65 (1H, d, J = 14.64) | C12′ | 137.09 | 6.25 (1H, d, J = 11.7) |
| C13 | 135.41 | - | C13′ | 138.05 | 6.65 (1H, dd, J = 11.94, 14.64) |
| C14 | 136.6 | 6.39 (1H, dd, J = 11.58) | C14′ | 132.15 | 6.39 (1H, d, J = 11.58) |
| C15 | 129.4 | 6.61 (1H, dd, J = 11.64, 14.16) | C15′ | 132.13 | 6.55 (1H, dd, J = 12.05, 15) |
| C16 | 25.03 | 1.02 (3H, s) | C16′ | 32.07 | 1.05 (3H, s) |
| C17 | 28.11 | 0.94 (3H, s) | C17′ | 29.18 | 1.36 (3H, s) |
| C18 | 21.14 | 1.20 (3H, s) | C18′ | 31.27 | 1.33 (3H, s) |
| C19 | 11.8 | 1.92 (3H, s) | C19′ | 13.99 | 1.79 (3H, s) |
| C20 | 12.67 | 1.97 (3H, s) | C20′ | 12.95 | 1.97 (3H, s) |
| C21′ | 170.38 | 2.02 (3H, s) | |||
| C22′ | 21.4 | - |
| Class | Property | Value |
|---|---|---|
| Physicochemical Property | Molecular Weight | 674.42 |
| nHA | 7 | |
| nHD | 2 | |
| nRot | 13 | |
| TPSA | 113.43 | |
| logS | −5.261 | |
| logP | 4.068 | |
| Drug likeness | Lipinski Rule | 1 violation |
| Vebers rule | 1 violation | |
| Absorption | Caco-2 Permeability | −4.947 |
| HIA | Yes | |
| Pgp-inhibitor | 0.894 | |
| Distribution | PPB | 78.242 |
| BBB | 0 | |
| Metabolism | CYP1A2 inhibitor | Excellent |
| CYP1A2 substrate | Excellent | |
| CYP2C19 inhibitor | Poor | |
| CYP2C19 substrate | Excellent | |
| CYP2C9 inhibitor | Poor | |
| CYP2C9 substrate | Excellent | |
| CYP2D6 inhibitor | Excellent | |
| CYP2D6 substrate | Excellent | |
| CYP3A4 inhibitor | Medium | |
| CYP3A4 substrate | Yes | |
| Excretion | CLplasma | 7.864 |
| T1/2 | 0.744 | |
| Toxicity | Toxicity Class | 3 |
| AMES Mutagenicity | 0.708 | |
| Skin Sensitization | Yes | |
| Respiratory | No | |
| Eye Irritation | No |
| Protein Structure | Total No. of Residues | Favored Region | Allowed Region | Disallowed Region |
|---|---|---|---|---|
| α-amylase | 460 | 449 (97.61%) | 11 (2.39%) | 0 (0%) |
| α-glucosidase | 803 | 771 (96.02%) | 30 (3.74%) | 2 (0.25%) |
| Protein | Ligand | Binding Affinity (Kcal/mol) | No. of H-Bonds | H-Bonds Forming Residues (Bond Distance in A0) | No. of Non-Bonded Contacts |
|---|---|---|---|---|---|
| α-Amylase | Fucoxanthin Derivative | −9.4 | 3 | SER108 (2.80), HSD305 (3.19), GLY306 (3.28) | 59 |
| Acarbose | −8.5 | 5 | GLU282 (2.80), ASP402 (2.89), ASP402 (3.09), GLY403 (3.13), ARG421 (3.08) | 55 | |
| α-Glucosidase | Fucoxanthin Derivative | −8.0 | 1 | VAL718 (2.84) | 67 |
| Acarbose | −7.4 | 5 | ASP91 (2.91), ALA93 (2.83), PRO94 (2.71), GLN118 (3.12), GLN118 (3.01) | 61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigera, S.; Theekshana, K.D.; Dinanja, S.G.; Eranga, P.; Karunathilake, N.; Abeywardhana, S.; Weerasinghe, L.; Senapathi, T.; Peiris, D.C. Molecular Docking and Dynamics Simulations Reveal the Antidiabetic Potential of a Novel Fucoxanthin Derivative from Chnoospora minima. Mar. Drugs 2025, 23, 471. https://doi.org/10.3390/md23120471
Sigera S, Theekshana KD, Dinanja SG, Eranga P, Karunathilake N, Abeywardhana S, Weerasinghe L, Senapathi T, Peiris DC. Molecular Docking and Dynamics Simulations Reveal the Antidiabetic Potential of a Novel Fucoxanthin Derivative from Chnoospora minima. Marine Drugs. 2025; 23(12):471. https://doi.org/10.3390/md23120471
Chicago/Turabian StyleSigera, Sachini, Kavindu D. Theekshana, Sathmi G. Dinanja, Pasindu Eranga, Nayanatharie Karunathilake, Shamali Abeywardhana, Laksiri Weerasinghe, Tharindu Senapathi, and Dinithi C. Peiris. 2025. "Molecular Docking and Dynamics Simulations Reveal the Antidiabetic Potential of a Novel Fucoxanthin Derivative from Chnoospora minima" Marine Drugs 23, no. 12: 471. https://doi.org/10.3390/md23120471
APA StyleSigera, S., Theekshana, K. D., Dinanja, S. G., Eranga, P., Karunathilake, N., Abeywardhana, S., Weerasinghe, L., Senapathi, T., & Peiris, D. C. (2025). Molecular Docking and Dynamics Simulations Reveal the Antidiabetic Potential of a Novel Fucoxanthin Derivative from Chnoospora minima. Marine Drugs, 23(12), 471. https://doi.org/10.3390/md23120471