1. Introduction
Parkinson’s disease (PD) is an age-dependent and gradually progressive neurodegenerative disorder of great societal importance, ranking second only to Alzheimer’s disease (AD) in terms of its frequency in occurrence. PD is distinguished by a progressive degeneration of dopaminergic neurons within the nigrostriatal system of the brain, which leads to impairments primarily of motor function—resting tremor, hypokinesia, muscle rigidity, and postural instability—accompanied by nonmotor symptoms such as cognitive impairment and autonomic dysfunction [
1,
2,
3]. It is assumed that the etiology of PD is associated with genomic, epigenetic, and environmental factors leading to conformational changes and deposits in the cytoplasm of neurons of protein aggregates (Lewy bodies) due to abnormalities in the ubiquitin–proteasome system, impaired differentiation of dopaminergic neurons, and dysregulation of function and oxidative stress, which ultimately causes neuronal death [
4].
Recent studies have indicated the direct involvement of activated microglia in the processes of degeneration of dopaminergic neurons in the substantia nigra, specifically through the release of proinflammatory agents. The results revealed that the inflammation of microglia, triggered by bacterial lipopolysaccharide (LPS), causes excessive production of IL-1β, IL-6, IL-8, and TNF, along with the activation of inducible nitric oxide synthase (iNOS) [
5]. This leads to elevated levels of reactive oxygen species (ROS) and nitric oxide (NO), which have detrimental effects on the normal functioning and essential activity of neurons [
6,
7].
Rotenone, an isoflavonoid, occurs naturally in various parts of Leguminosa plants and is utilized as a widely effective insecticide and pesticide. In experimental models of Parkinson’s disease (PD), both in in vitro and in animals, rotenone, a neurotoxic compound, is commonly employed as a trigger to induce neurodegenerative changes. Rotenone can impede the flow of electrons in the mitochondrial respiratory chain by inhibiting the electron transfer and interrupting the movement of electrons from the iron–sulfur cluster in complex I to ubiquinone. Due to this process, an excess of electrons accumulates in the mitochondrial matrix, specifically as NADPH. This surplus of electrons results in the conversion of oxygen into ROS, which can cause harm to mitochondrial components and DNA. In addition to its impact on mitochondrial respiration, rotenone also obstructs the formation of microtubules from tubulin. Consequently, these effects lead to neuronal dysfunction and eventual cell death [
8,
9].
1,4-Naphthoquinones (1,4-NQs) play a special role in animals, including marine organisms, and plants as their metabolites. There is a broad spectrum of cytoprotective properties exhibited by compounds, which originate from natural sources as well as through synthetic modifications. It has been established that these compounds possess a diverse range of biological activities, including antifungal, antibacterial, antioxidant, cardioprotective, hepatoprotective, anti-ischemic, and various other properties. There is an increasing number of scientific papers exploring the protective role of 1,4-naphthoquinones in disorders characterized by the degeneration of neurons such as AD and PD [
10,
11,
12,
13,
14,
15,
16,
17,
18,
19].
In recent studies, we successfully synthesized a mini library of 5,8-dihydroxy-1,4-naphthoquinone derivatives (consisting of 44 compounds) derived from the natural pigments found in echinoderms. We evaluated the cytotoxic and neuroprotective effects of these compounds against mouse neuronal Neuro-2a cells in two in vitro models of PD, induced by paraquat (PQ) and 6-hydroxydopamine (6-OHDA) neurotoxins. Some compounds exhibiting pronounced neuroprotective properties [
20,
21,
22] were selected for future examinations. According to the published data, the most active studied 1,4-NQ derivatives at low concentrations were found to effectively defend cell biomembranes, normalize the cell cycle, suppress oxidative stress, normalize the mitochondrial function altered by neurotoxins, and eliminate induced inflammation.
In this study, we examined the protective effects of the two most potent 1,4-NQ derivatives, namely U-443 and U-573, demonstrating in preliminary in vitro experiments a significant ability to preserve the viability of neuronal cells in the presence of PQ, 6-OHDA, and rotenone, in both cellular and animal models of parkinsonism.
For this purpose, we used two cell cultures, Neuro-2a mouse neuroblastoma cells and RAW 264.7 mouse macrophage cells, mimicking brain neurons and microglia, respectively. Rotenone was used as a PD inducer and LPS was used to induce microglial inflammation. We evaluated the potential of the two selected 1,4-NQs to reduce the generation of ROS and NO in cells subjected to neurotoxin and LPS treatment. Additionally, we examined their impact on mitochondrial membrane potential (MMP), their influence on the production of proinflammatory cytokines such as TNF and IL-1β, their ability to inhibit the activity of cyclooxygenase 2 (COX-2) in macrophages, and their antioxidant activity in mouse brain homogenate. Furthermore, the effect of the two 1,4-naphthoquinones on spontaneous locomotion in an in vivo murine rotenone-induced early stage of the PD model was investigated.
3. Discussion
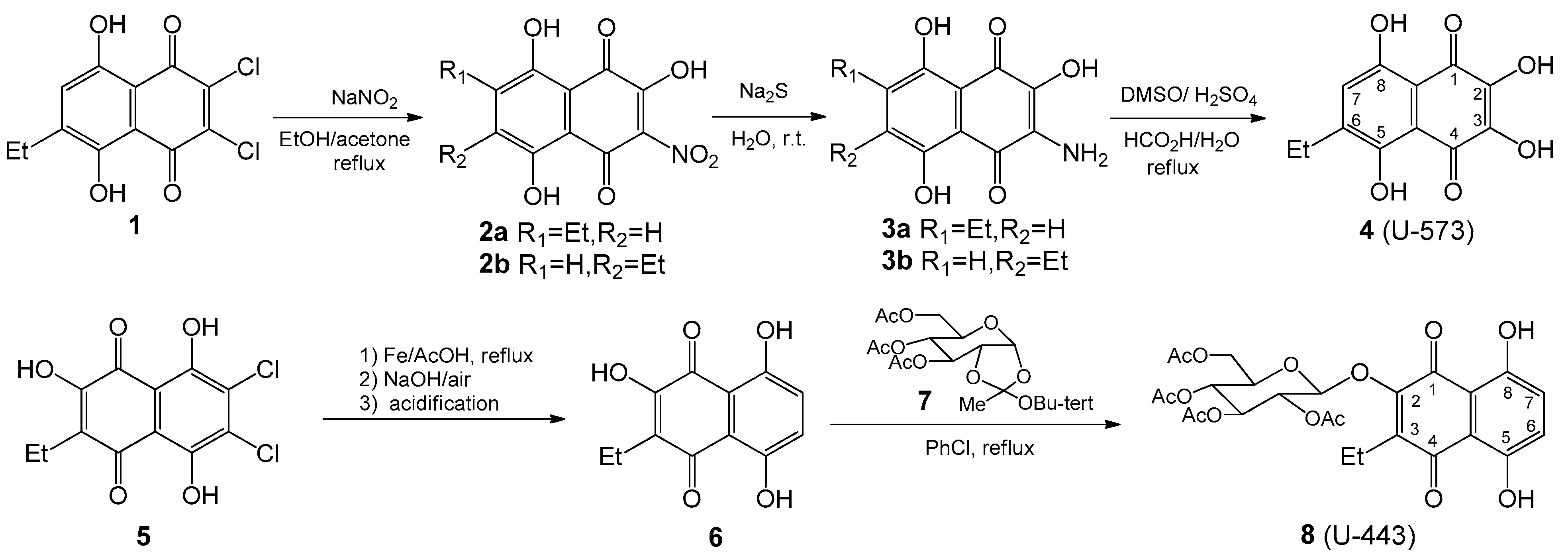
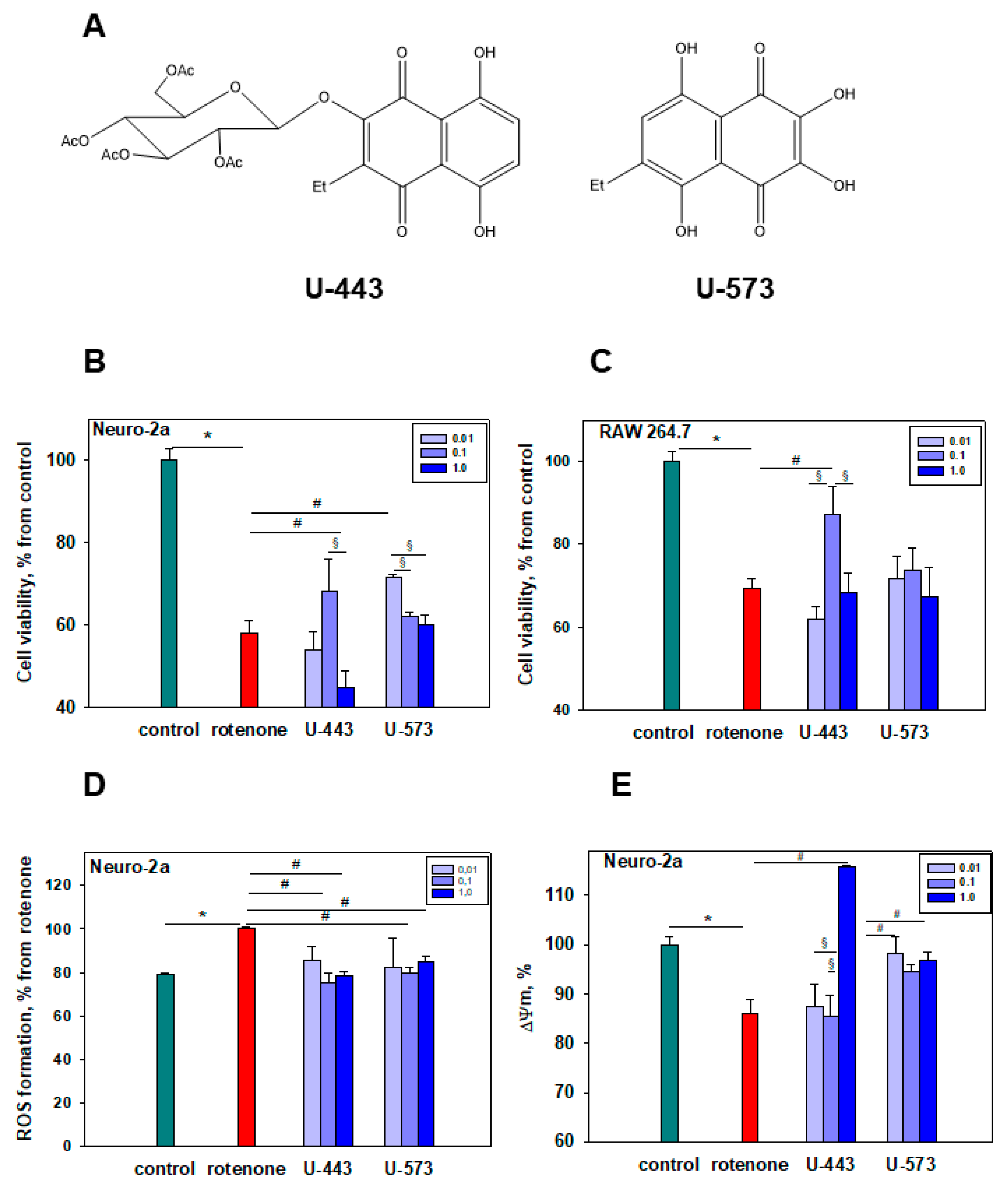
In this work, we selected two 1,4-naphthoquinones, synthesized sea urchin pigment
U-573 and synthetic acetylglucoside
U-443 of ophiuroid naphthazarine pigment
6, which are the most effective in a preliminary screening of cytoprotective activity in vitro model of neurotoxicity induced by rotenone. Ethylspinazarin
U-573 is a 6-deoxy analog of 6-ethyl-2,3,5,7,8-pentahydroxy-1,4-naphthoquinone
U-138 (echinochrome from sea urchin
Scaphechinus mirabilis) multifaceted medicine [
28]. Ethylspinazarin and echinochrome are structurally related quinones and both have excellent antioxidant properties [
28,
29]. Echinochrome
U-138 and ethylspinazarin
U-573 have two acidic β–hydroxyl groups at the 2 and 3 positions of the ethylene bond conjugated with quinone carbonyl [
30].
Unlike ethylspinazarin
U-573, the structure of the acetyl-
O-glucoside derivative
U-443 does not contain free β-hydroxyl groups. The compound
U-443, a glucoside derivative, is a simplified analog of the bioactive echinochrome acetylated tris-
O-glucoside
U-133. Previous research [
31] has demonstrated the therapeutic potential of
U-133 in preventing and/or slowing down neurodegeneration similar to Parkinson’s disease.
U-443 serves as a more readily available alternative with similar therapeutic properties.
A number of synthesized 1,4-naphthoquinones, including
U-573, have already demonstrated pronounced neuroprotective properties in paraquat- and 6-OHDA-induced models of Parkinson’s disease in vitro [
22]. In the present work, it was found that the synthetic 1,4-naphthoquinones,
U-443 and
U-573, are also capable of partially protecting neuronal cells from the cytotoxic effect of the neurotoxin rotenone. These 1,4-NQs have a modest impact on the level of some oxidative burst products and proinflammatory agents under rotenone and LPS influence. The fact that the studied 1,4-naphthoquinones similarly protect neuronal and macrophage cells from the damaging effects of both rotenone and LPS may indicate the similar molecular mechanisms of the protective action. Most likely, the inhibition of ROS production in these cells under rotenone and LPS actions is the key step that prevents the development of additional events such as MMP depolarization, mitochondrial dysfunction, proinflammatory cytokine release, DNA damage, apoptosis induction, and some other perturbations leading to cell death. Compounds
U-443 and
U-573 were used at concentrations less than EC
50 values, which did not suppress cell viability. These 1,4-NQs by themselves did not significantly affect the levels of ROS and NO in cell cultures, mitochondrial membrane potential, TNF production, and COX-2 activity (data not shown).
Evidently,
U-443 and
U-573 have the ability to hinder the production of reactive oxygen species in neuronal cells or macrophages, thereby regulating the activity of enzymes responsible for maintaining a balanced level of reactive oxygen species within cells. These compounds can stimulate various antioxidant enzymes, such as superoxide dismutase, catalase, and peroxiredoxins, to eliminate excessive reactive oxygen species. Additionally, they inhibit enzymes involved in the generation of ROS, including NADPH oxidase in phagocytic cells, xanthine oxidase, mitochondrial cytochrome C oxidase, and microsomal monooxygenases. These effects can be attributed to the activation of the Keap1/Nrf2/ARE signaling pathway by 1,4-naphthoquinones, which help regulate the cellular redox status [
18,
22]. At the same time, we showed that
U-443 and
U-573 partially block NO formation in RAW 264.7 cells. Perhaps this is due to the targeted inhibition of inducible iNO synthase, which is responsible for the synthesis of nitrogen monoxide in macrophages after stimulation.
In recent studies, it has been demonstrated that several 1,4-naphthoquinone derivatives synthesized by us effectively inhibit the activity of purinergic P2X7 receptors (P2X7R) in vitro. The inhibition results in marked blockade of the receptor macropore, a significant reduction in P2X7R-mediated production of ROS, NO, release of NLRP3 inflammasome-dependent proinflammatory cytokines, and pronounced protection of neuronal and macrophage cells from the ATP toxic impact. For one of the 1,4-NQ derivatives, compound
U-556, direct binding to this purinoceptor has been proven by surface plasmon resonance. In silico analysis showed the ability of some synthetic 1,4-NQs to specifically bind to an allosteric site located in the extracellular region of P2X7R and to almost completely eliminate receptor-mediated inflammation in mice [
23,
32,
33]. Thus, inhibition of P2X7R activity may also contribute to the compounds
U-443 and
U-573 protective potential.
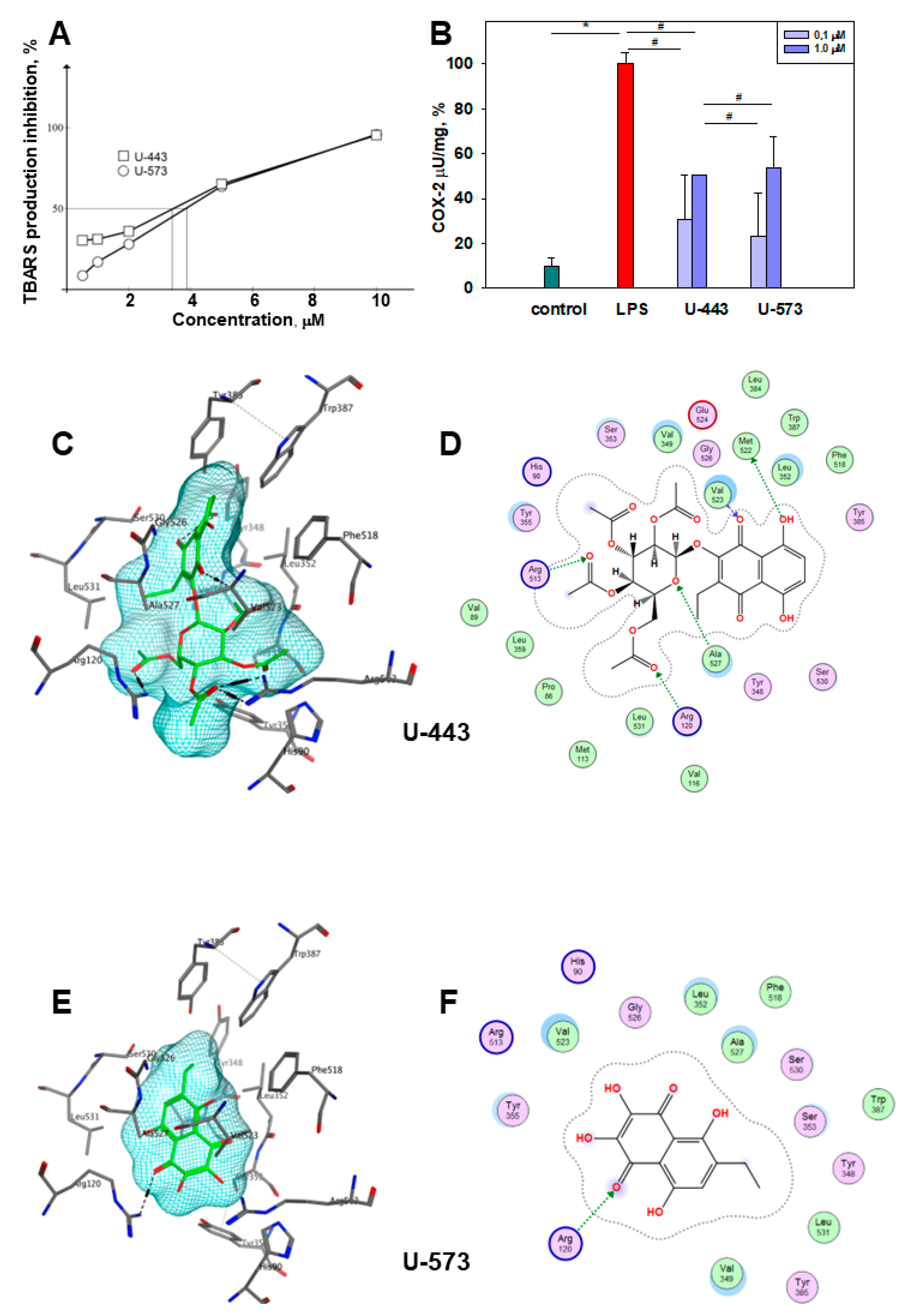
Several natural and synthetic 1,4-naphthoquinones have been documented to possess notable antioxidant activity [
15]. In the present study, compounds
U-443 and
U-537 demonstrated a pronounced inhibition of the induced peroxide radical production in mouse brain homogenate. Moreover, this capability may contribute to the reduction of reactive oxygen species formation and the attenuation of neuroinflammation in mouse brain tissue, which are commonly observed in the initiation and advancement of Parkinson’s disease.
In the investigated concentration range, it was noted that the cytoprotective effects of
U-443 and
U-537 compounds do not depend on the dosage. It is now understood that many quinones display dual effects on biological systems and demonstrate nonlinear, U-shaped dose–response curves. The action of quinones, dependent on interaction duration and dose, can result in either toxicity or cytoprotection. At lower doses, quinones with cytoprotective properties activate detoxification enzymes through electrophilic responses. Conversely, at higher doses, less selective and more reactive quinones exhibit cytotoxic properties by disrupting cell functions through the generation of semiquinone radicals and superoxide, ultimately leading to the formation of highly toxic hydroxyl radicals causing cell death [
34].
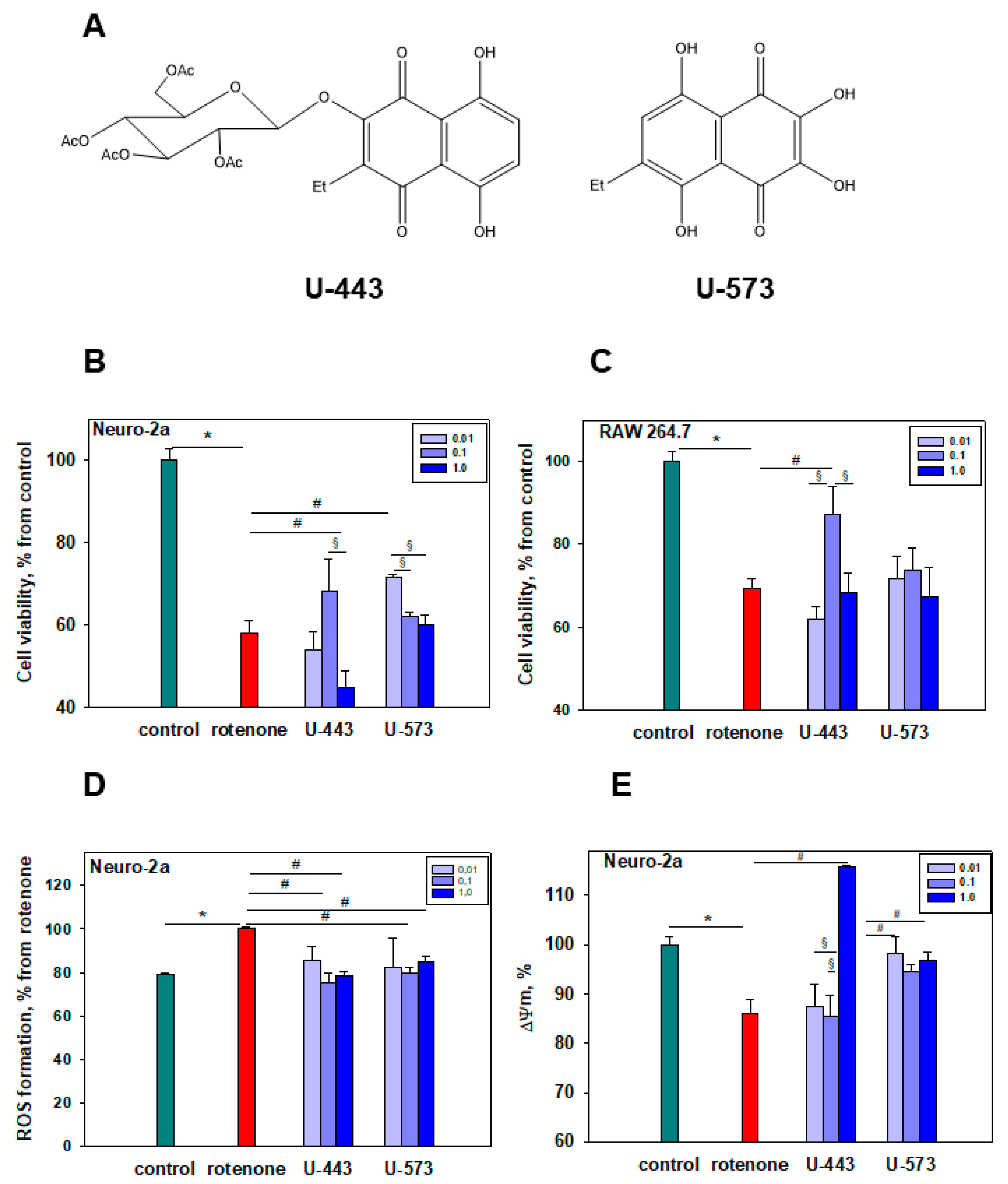
We found that, in some cases,
U-443 at the highest studied concentration of 1 μM exhibited the opposite cytoprotective effect. This concentration is close to its cytotoxicity EC
50 value. Upon closer inspection, this impacts the manifestation of its cytotoxic activity (
Figure 1B) and results in slight hyperpolarization, indicating the change in MMP (
Figure 1E) and increase in ROS and NO production (
Figure 2A,B) at this concentration when co-cultivated with the neurotoxin rotenone. This may be explained by the weak cytotoxic impact of compound
U-443 since a concentration of 1 μM is quite near to the onset of its cytotoxic activity.
Currently, it is known that some 1,4-NQs can exhibit cytotoxic properties due to the induction of semiquinone radicals and superoxide formation in exposed cells followed by affected redox signaling, alkylation, DNA damage, cell cycle blockade, and subsequent apoptosis. Some naphthoquinones have been shown to be cytotoxic to neuronal cells. Thus, natural 8-hydroxy-2-methoxy-1,4-naphthoquinone and 5-hydroxy-2-methoxy-1,4- naphthoquinone from plant
Juglans sinensis and series of synthetic 1,4-NQs derivatives exhibited significant cytotoxicity against human SH-SY5Y and mouse Neuro-2a neuroblastoma cells [
21,
35]. Exposure to some 1,2-NQs и 1,4-NQs provokes human neuroblastoma cell lines SK-N-SH apoptosis in a ROS-dependent pathway, including DNA damage, mitochondrial dysfunction, and resultant caspase 3 and 9 activation together with GSH decrease, proteasome inactivation and NQO1 upregulation [
36]. Treatment of mouse hippocampal neuronal HT22 cells with 2,3-dimethoxy-1,4-naphthoquinone increased cytosolic and mitochondrial ROS and apoptosis, involving CHOP/GADD153 upregulation, JNK and p38 MAPK activation, Bak/Bax activation, mitochondrial membrane potential loss, caspase-9 and caspase-3 activation, PARP cleavage, nucleosomal DNA fragmentation, and intracellular GSH reduction [
37].
In our previous investigations, we showed that the antiproliferative and cytotoxic activity of a series of 1,4-NQ derivatives against mouse neuronal cells largely related to their hydrophobicity/polarity, depending among other things on the acetylation of the sugar moiety of 1,4-NQ glycosides [
21]. The observed difference in cytotoxic activity between compounds
U-443 and
U-537 can be explained by differences in the nature of the substituents of the naphthoquinone core. Thus, unlike polyhydroxy-1,4-NQ
U-537, compound
U-443 contains a bulky lipophilic acetylglycoside fragment. Previously, we showed [
21] that the introduction of an acetylglycoside fragment leads to a cytotoxic effect increasing in comparison with the initial hydroxyquinone. Deacetylation of such glycosides leads to a several-fold cytotoxicity decrease.
Many quinones exhibit ambivalent and opposite effects on biological systems. Depending on the dose used, cellular targets, and exposure time, the effect of quinones on cells leads to toxicity or cytoprotection. In contrast to the cytotoxic activity of naphthoquinones at high concentrations, at low doses, stable and selective quinones often exhibit cytoprotective properties resulting from electrophilic counterattacks, leading to the detoxification enzymes’ appearance through the Keap1/Nrf2/ARE pathway, and scavenging action [
34,
38].
It has been established that a number of natural and synthetic naphthoquinones, including 1,4-naphthoquinone derivatives, are able to protect various types of cells from adverse effects. For example, 2-carbomethoxy-2,3-epoxy-3-prenyl-1,4-naphthoquinone (CMEP-NQ) has been found to effectively save human acute leukemia Jurkat T cells from some apoptotic cell death inducers, such as microtubule-damaging and DNA-damaging agents. The cytoprotection of CMEP-NQ was provided by the elevation of anti-apoptotic chaperone regulator BAG3 and cell differentiation protein MCL-1 levels, which leads to the mitochondrial apoptosis pathway inhibition, as well as by blocking mitochondrial damage caused by intracellular ROS production [
39]. Several novel short-chain 2,3-disubstituted naphthoquinone derivatives were found to protect human hepatic carcinoma HepG2 cells and rodent retinal precursor RGC5 cells against loss of viability and cell death in the presence of rotenone by protecting and restoring mitochondrial dysfunction caused by this toxin [
40]. Echinochrome (
U-138) was described recently to protect the primary culture of rat pulmonary fibroblasts from oxidative stress caused by hydrogen peroxide H
2O
2, probably due to the increase in mitochondrial biogenesis, rise of chaperone Hsp70 activity, and stimulation of hypoxia-inducible factor HIF-1 expression, which together significantly enhance the resistance of cells to damage [
41]. Ubiquinone short-chain synthetic analog, idebenone, was also shown to effectively protect human retinal pigment epithelia ARPE-19 cells exposed to H
2O
2 inducing oxidative damage. Idebenone cytoprotective action was accompanied by Keap1/Nrf2/ARE signaling pathway activation, reduction of the Bax/Bcl-2 ratio, recovery of the mitochondrial membrane potential to physiological levels, preservation of the ROS production, and minimization of caspase-3 activity [
42]. Plumbagin obtained from the roots of different medicinal plants was demonstrated to effectively protect neuronal cells and chondrocytes exposed to H
2O
2, which was accompanied by an enhancement in antioxidant enzyme activities and the expression of Nrf2; it also prevents dexamethasone-induced apoptosis in osteoblasts by restoring mitochondrial potential and has a pronounced neuroprotective effect on neuronal human SH-SY5Y cells, hepatoprotective potential leading to liver regeneration, as well as cardioprotective activity on rat H9c2 cardiomyocytes. In addition, the powerful anti-inflammatory activity of plumbagin has been studied in sufficient detail. According to numerous investigations performed on various immune cells, including murine lymphocytes, RAW264.7 macrophages, bone marrow-derived macrophages, microglia, and some others, this natural naphthoquinone exhibited its protective anti-inflammatory properties by normalizing an imbalance of ROS/NO, antioxidant molecules CAT, GPx and SOD, and proinflammatory cytokines TNF, IL-1β, IL-6, and IL-12, as well as an increase in Nf-kB oxidation. Raised expression of iNOS, granulocyte-colony stimulating factors G-CSF, monocyte chemoattractant protein MCP1, and MCP5 was also reverted by plumbagin treatment [
43]. Thus, protective properties are inherent in many naphthoquinones, which they exhibit on various types of cells.
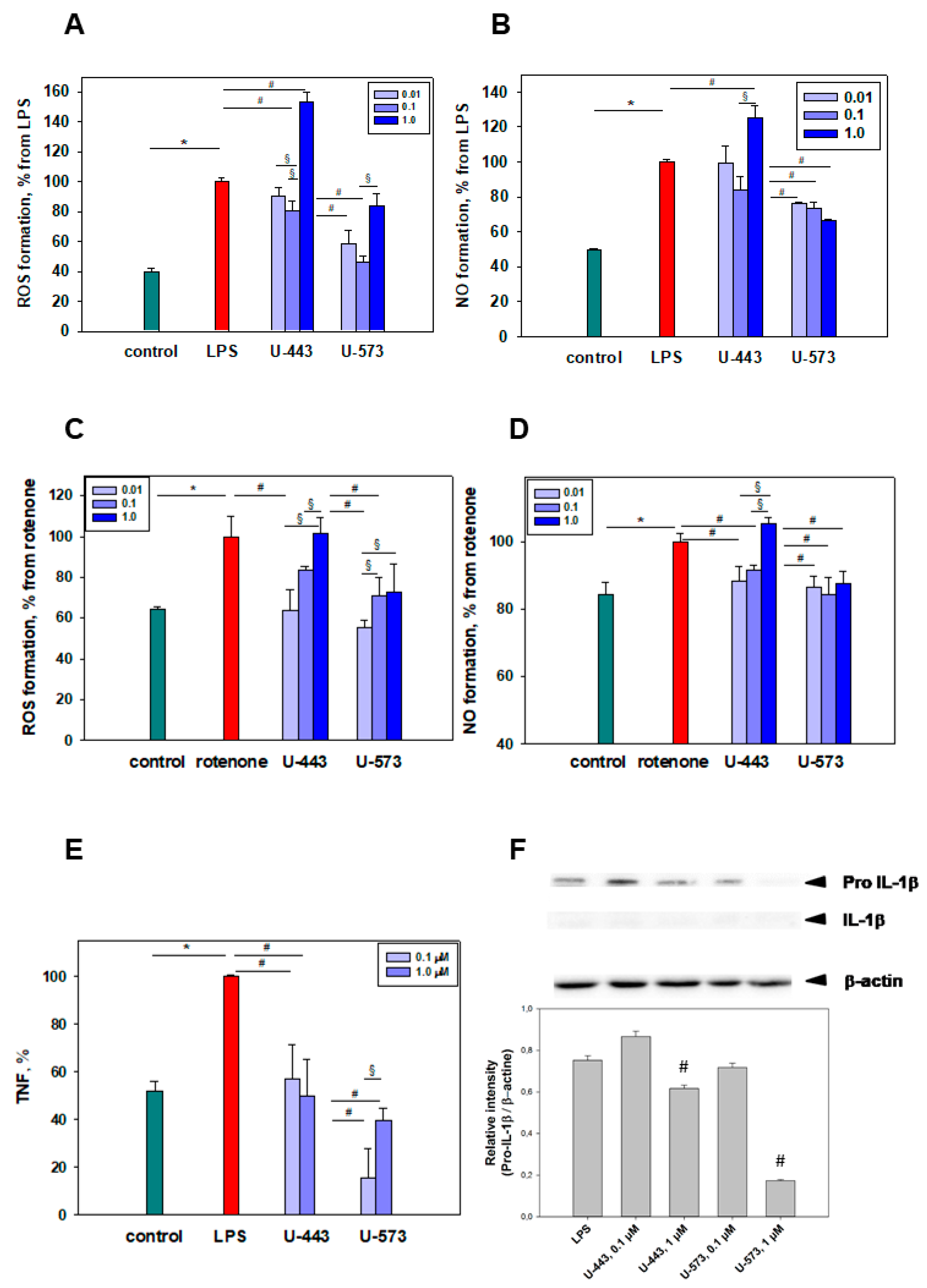
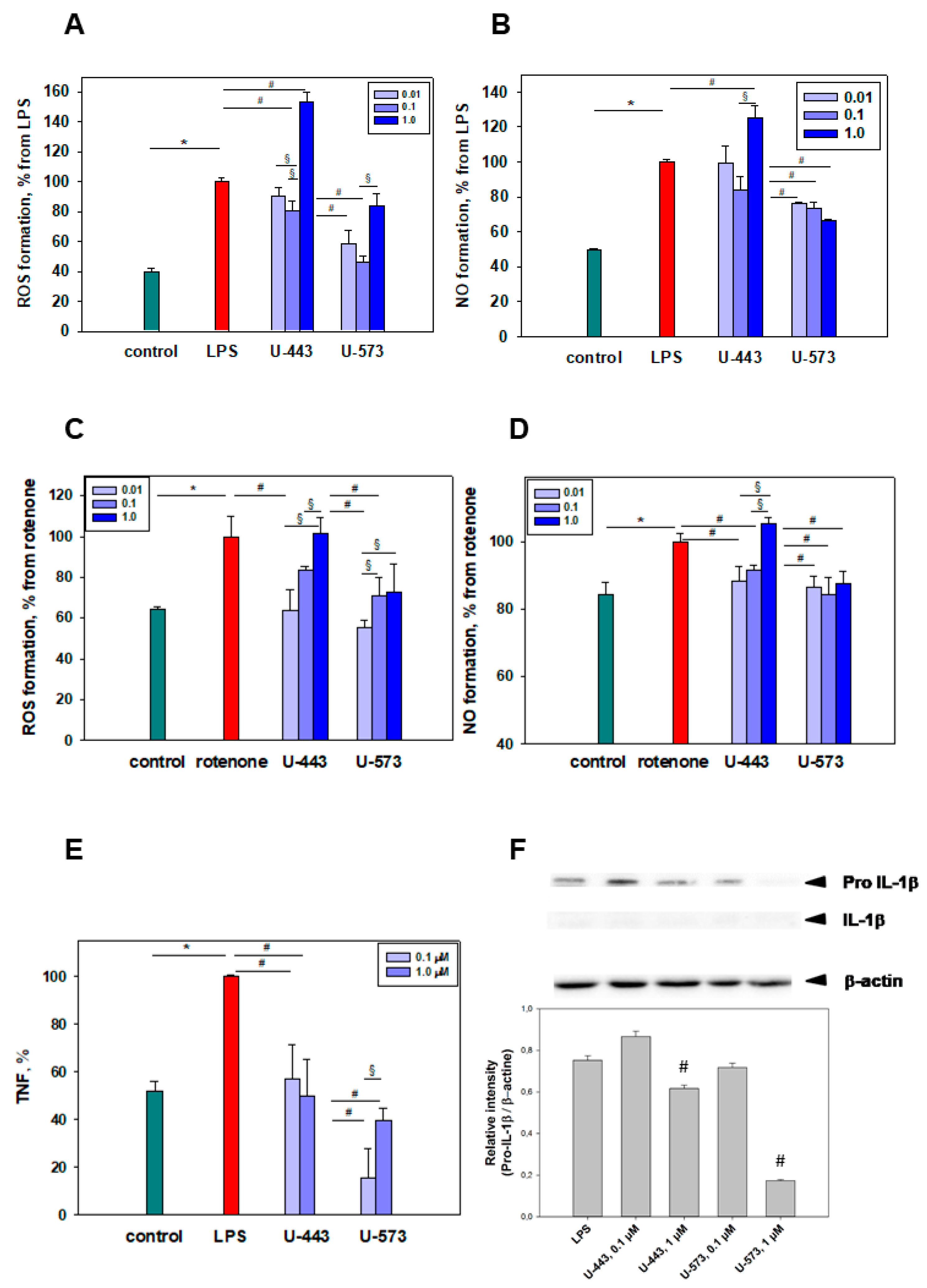
The moderate inhibition of proinflammatory cytokine TNF and pro-IL-1β production by 1,4-naphthoquinone derivatives, following exposure to LPS found in the present study, may take place either at the level of the TLR4 receptor or in the downstream signaling pathway. Recently, it has been discovered that 1,4-naphthoquinones effectively block the interleukin-1 receptor-associated kinase (IRAK1) in LPS-stimulated human THP-1 macrophages. IRAK1 kinase plays a crucial role as a signaling intermediate in the IL1R/TLR downstream cascade, activating various transcription factors that contribute to the regulation of immune responses and inflammation. The interaction between certain 1,4-naphthoquinones and IRAK1 has been shown to inhibit the production of proinflammatory cytokines such as TNF, IL-1β, IL-6, IL-8, and IL-10 in macrophages [
44]. In our experiments, the minimal presence of IL-1β detected following cell stimulation with LPS is associated with the particular behavior of RAW 264.7 cells. These macrophages are known to release pro-IL-1β but not mature IL-1β due to the absence of the apoptotic speck-like protein required for the further processing of pro-IL-1β into its mature form [
45].
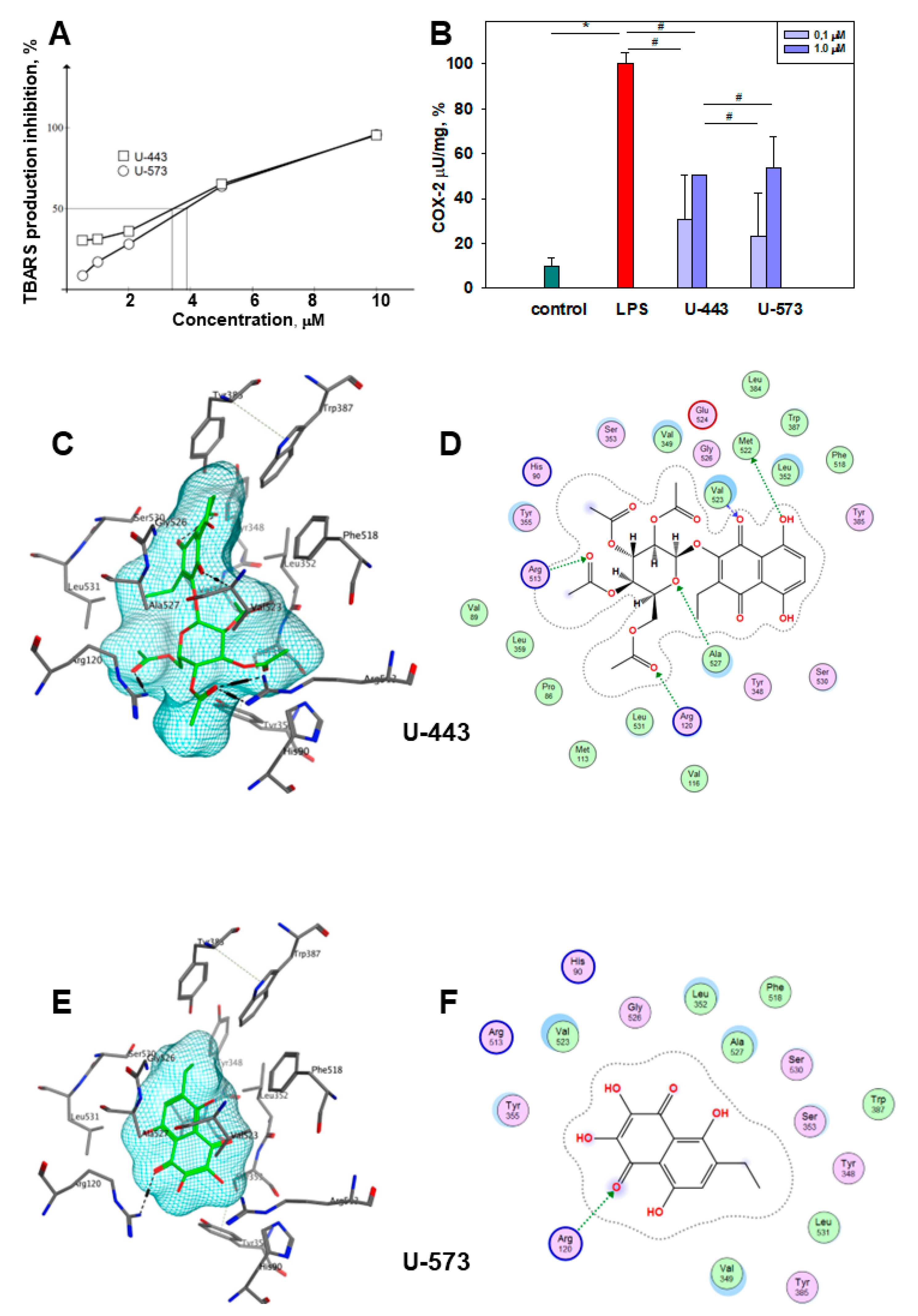
LPS-induced COX-2 activation can be inhibited in several ways. First, effective 1,4-NQs can directly interact with cyclooxygenase-2 and selectively inhibit the activity of the enzyme due to the binding of the catalytic center responsible for the activity of cyclooxygenase to amino acids, especially Arg 120, similar to the known inhibitor of this enzyme, SC-558 [
46]. Binding to Arg 120 plays a pivot role in the interaction of COX-2 with the substrate and with inhibitors [
47,
48]. Such activity has been previously noted for a number of 1,4-naphthoquinone derivatives [
49,
50,
51]. Our data on modeling (docking) of compounds
U-443 and
U-573 clearly indicate the direct interaction of these 1,4-NQs with the active center of COX-2, which may be the reason for the manifestation of their properties inhibiting enzymatic activity. Second, suppression of the production of a number of cytokines noted in our study can also directly affect COX-2 and be a direct cause of a decrease in COX-2 activity.
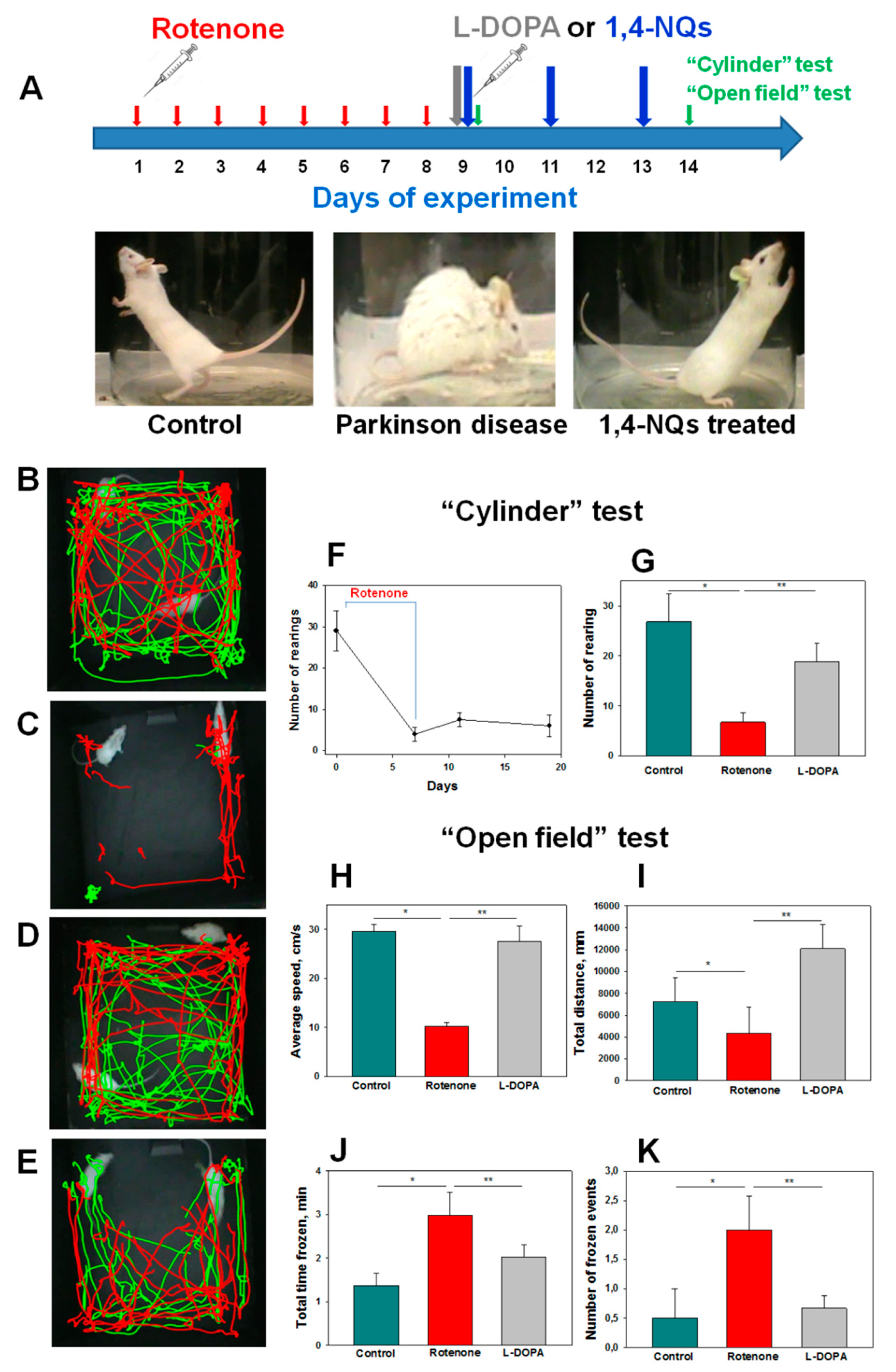
Rotenone with high efficiency blocks the transfer of electrons in complex I of the mitochondrial chain. Under the action of rotenone throughout the brain, a general suppression of Complex I occurred, as a result of which, in the group of animals receiving rotenone, signs of PD appeared, such as the motor deficit, selective nigrostriatal dopaminergic degeneration, and formation of ubiquitin- and synuclein-positive nigral inclusions. The toxic effect on cells caused by rotenone-induced oxidative stress can initiate neurodegeneration processes in Parkinson’s disease [
52]. The process of mitochondrial respiration and dopamine metabolism leads to the formation of reactive oxygen species. In Complex I, near the rotenone binding site, there is a site through which an electron leakage passes, which promotes the formation of ROS; disruption of the functioning of Complex I leads to an increase in ROS [
53]. In addition to oxidative stress in neurons, rotenone promotes activation of the microglia surrounding neurons. In the early and progressive stages of PD, astrocytes and microglia are activated in various ways. This, in turn, contributes to an increase in the number of dead neurons, which leads to an increase in the level of superoxide anions, free radicals, IL-1β, TNF, PGE2, NO, and other substances. The combination of these processes initiates dopaminergic neurodegeneration [
54].
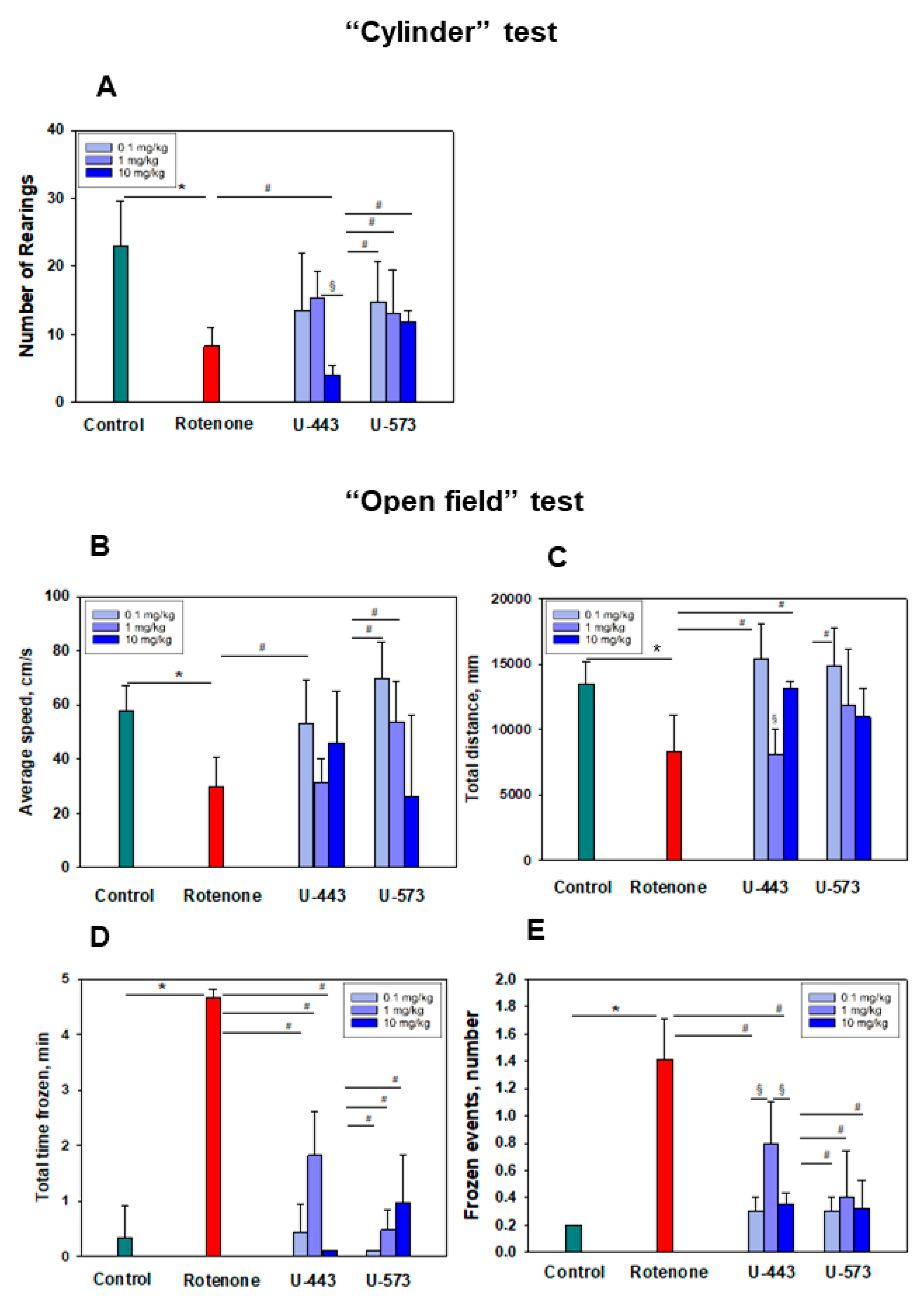
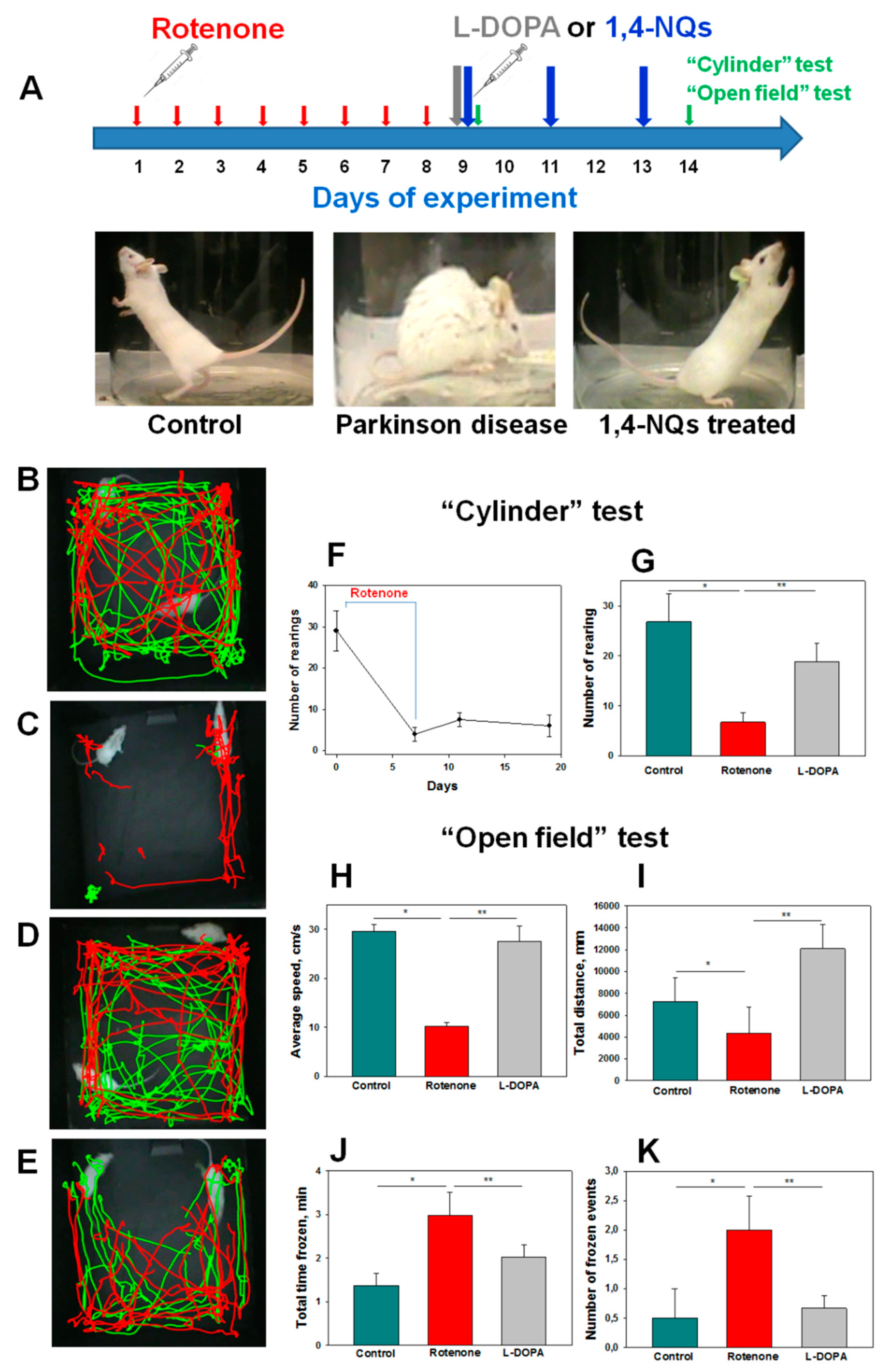
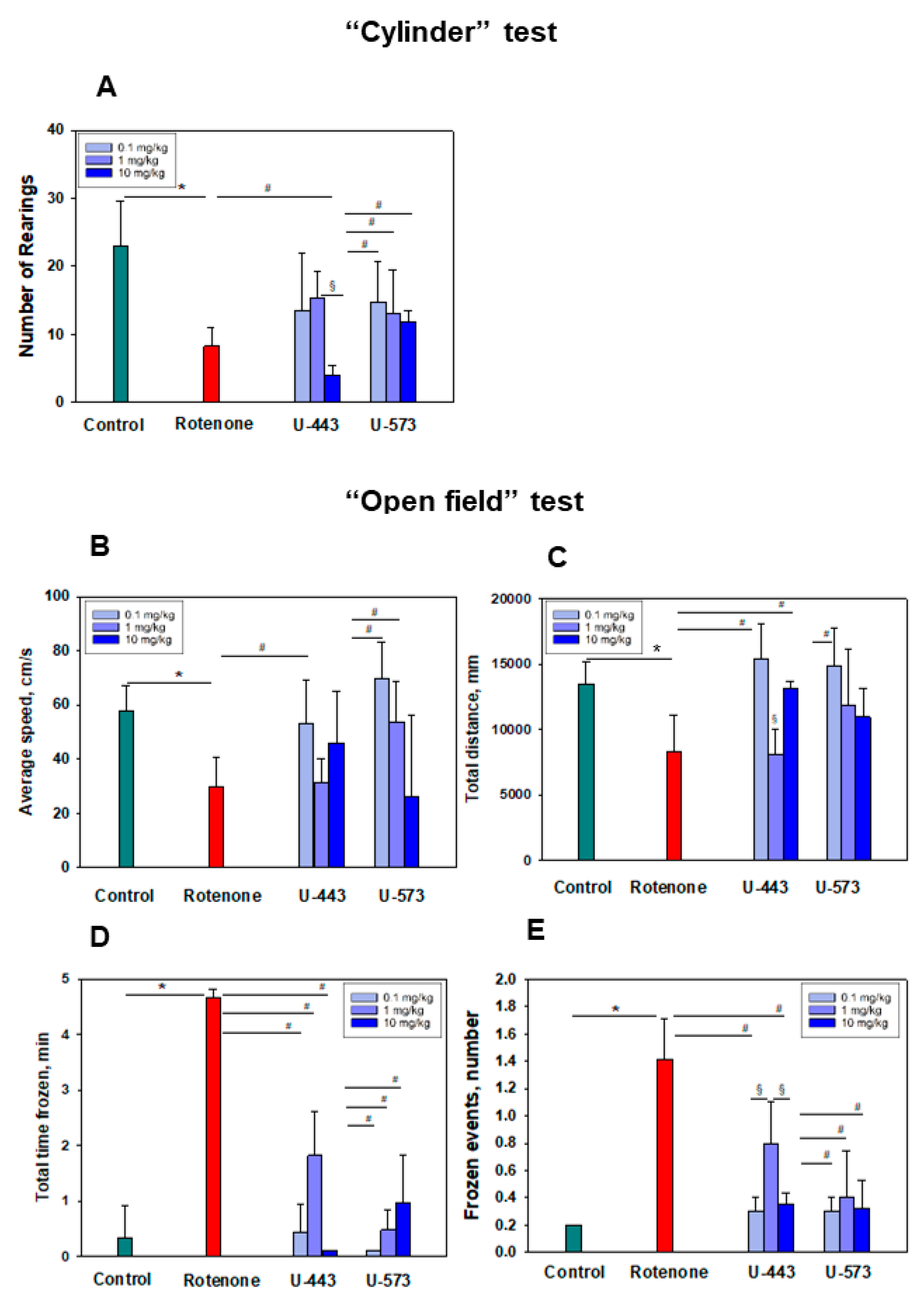
Both tested compounds showed comparable efficacy as antiparkinsonian agents in the “Cylinder” and “Open Field” tests. It is highly probable that during the early stage of Parkinson’s disease induction by rotenone in live organisms, the investigated compounds U-443 and U-573 exhibit antiparkinsonian properties through their capability to decrease the presence of reactive oxygen species and NO in neurons and microglia. These compounds also demonstrate the ability to inhibit the production of proinflammatory cytokines and suppress the activity of COX-2, resulting in cytoprotection, enhanced neuronal survival, and reduced inflammatory processes in microglia.
We did not find a clear dose dependence of the antiparkinsonian activity of 1,4-naphthoquinones or their effects on the behavioral responses of animals with induced PD in the dose range of 0.1–10.0 mg/kg. Despite the fact that the studied naphthoquinones do not exhibit acute toxic properties in animals up to a dose of 100 mg/kg, the use of drugs at lower doses of 0.1–1.0 mg/kg seems to be preferable in future experiments.
In a previous study employing QSAR methodologies, it was observed that the neuroprotective activity of the investigated compounds relies on the hydrophobicity, polarity, charge, and shape of the 1,4-naphthoquinone molecule. These aforementioned properties suggest that these small molecules have the potential to effectively penetrate the blood–brain barrier and reach neurons and microglial cells, consequently offering neuroprotection in animal models of Parkinson’s disease induced by rotenone.
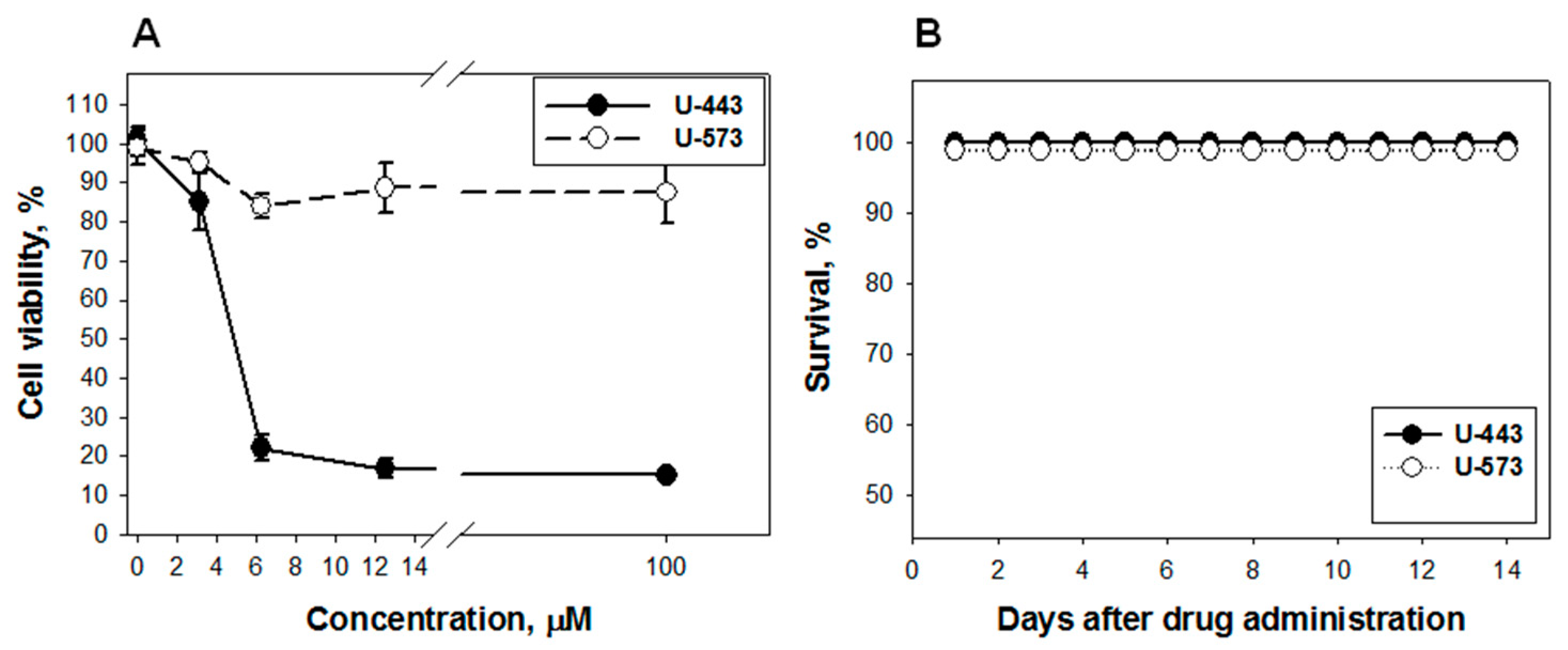
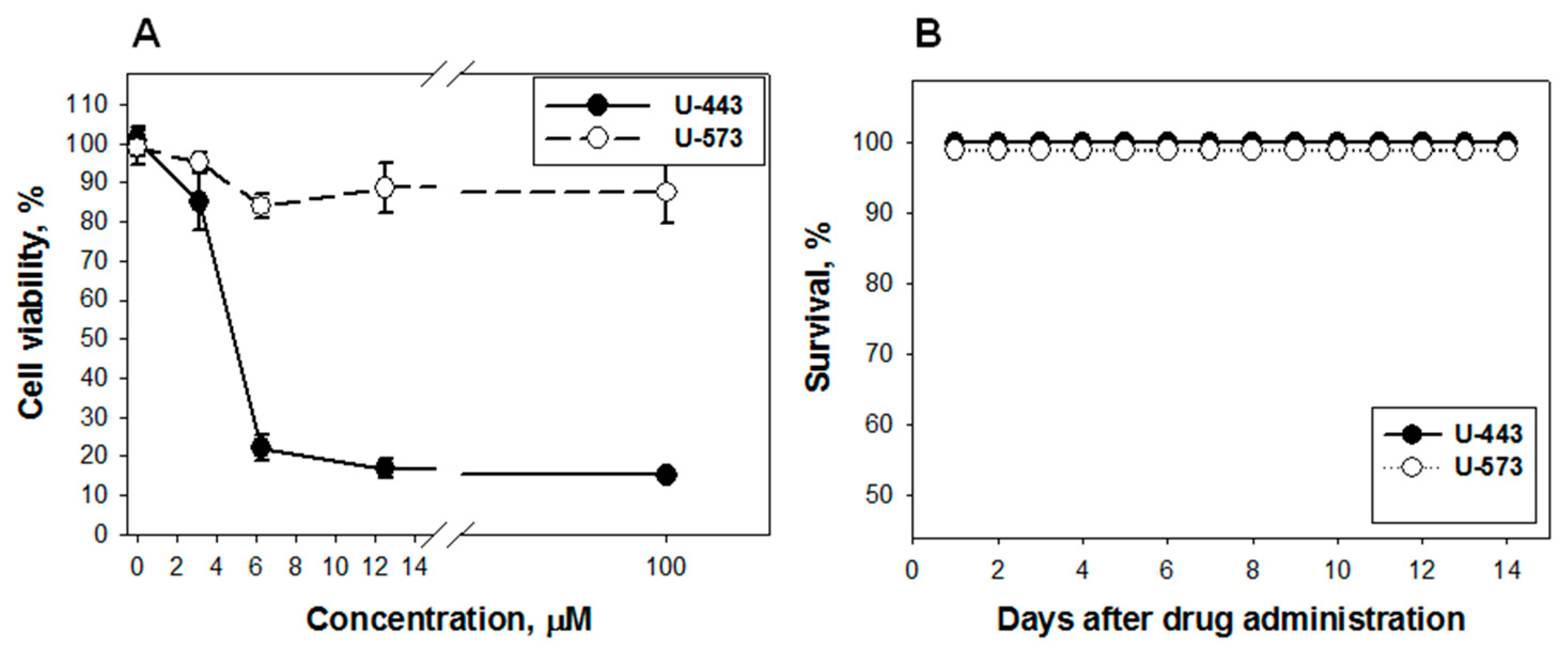
Detailed studies of the cytotoxicity of these 1,4-NQs in relation to Neuro-2a cells were carried out and published by us earlier [
21]. For compound
U-573, cytotoxicity was established (EC
50 > 100 µM), which indicates a very weak cytotoxic effect, while
U-443 was more cytotoxic (EC
50 = 4.46 µM), which excludes this substance from candidates for promising cytoprotectors. At the same time, compound
U-573 exhibits cytoprotective properties against Neuro-2a cells in the presence of various neurotoxins, such as paraquat, 6-OHDA, or rotenone, at concentrations much lower than cytotoxic ones, 0.1 and 0.01 µM [
22]. Compound
U-573 has very low toxicity in animals (LD
100 > 100 mg/kg), and in an in vivo study, in many cases, this compound at low doses showed statistically significant effects. This allows us to consider 1,4-naphthoquinone
U-573 as a promising small biomolecule for further investigation of its neuroprotective properties and ability to prevent and correct neurodegenerative disorders.
4. Materials and Methods
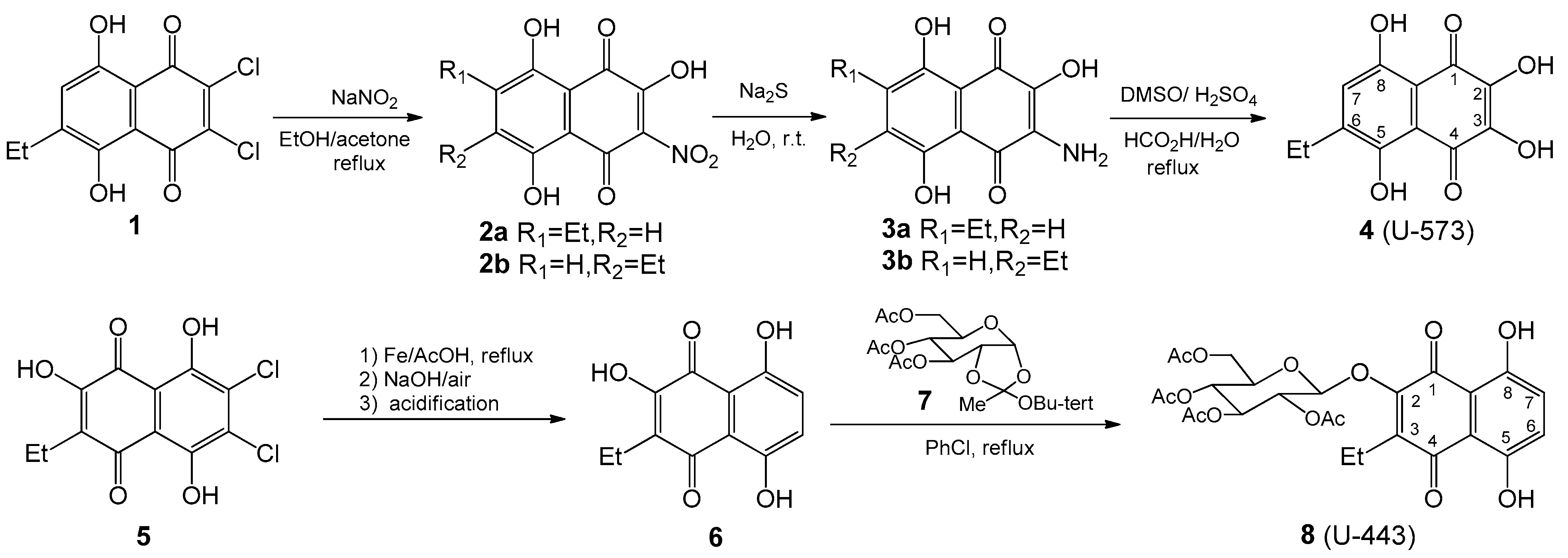
4.1. Synthesis of 1,4-Naphthoquinones General
6-Ethyl-2,3-dihydroxynaphthazarin
4 (
U-573) was synthesized from ethyldichloronaphtazarin as described in our works [
22,
24]; 2-Hydroxy-3-ethylnaphthazarin
6 was obtained by reductive dechlorination of appropriate dichlorohydroxyethylnaphthazarin
5 according to our previous paper [
26]; the synthesis of 3,4,6-tri-
O-acetyl-α-d- glucopyranose 1,2-(tert-butoxy orthoacetate)
7 was accomplished using Kochetkov’s method, as described in reference [
26]; 2-Hydroxy-3-ethylnaphthazarin
6 was condensed with 3,4,6-tri-
O-acetyl-α-d-glucopyranose 1,2-(tert-butoxyorthoacetate)
7 in chlorobenzene at reflux and led to acetylglucoside
8 (
U-443) as described in our previous works [
17,
25]. NMR spectra for
4 (
U-573) were recorded on a Bruker AVANCE-500 at frequencies 500 MHz for
1H spectra and 125 MHz for
13C in DMSO-d
6. NMR spectra for
8 (
U-443) were recorded on a Bruker AVANCE-700 at frequencies 700 MHz for
1H spectra and 176 MHz for
13C in CDCl
3 (
Figures S1 and S2 in Supplementary Materials). The purity of the synthesized compounds was assessed on LaChrom (Merck Hitachi, Tokyo, Japan) system (pump L-7100, UV/VIS detector L-7400, column oven L-7300, and integrator D-7500). Separations were carried out using Agilent Technologies column Zorbax Eclipse XDB-C18, 5 μm (150 mm × 4.6 mm) with a guard column Hypersil ODS (4.0 mm × 4 mm). The binary gradient employed phase A: water/glacial acetic acid (v/v, 100/1); phase B: acetonitrile/glacial acetic acid (v/v, 100/1) according to the following profile: 0–35 min, 5–35% B. The flow rate was 1 mL/min. The operating temperature was set at 30 °C, and the detection wavelength was set at 480 nm (
Figures S3 and S4).
1,4-NQs were prepared as a 10 mM stock solution in DMSO and then dissolved in ddH2O to the final concentrations before experiments.
4.2. Cell and Cultivation Conditions
Neuro-2a CCL-131TM mouse neuroblastoma cell line and RAW 264.7 TIB-71™ mouse macrophage cell line were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were cultured in DMEM medium (Biolot, St. Petersburg, Russia), which was supplemented with 10% fetal bovine serum (Biolot, St. Petersburg, Russia) and 1% penicillin/streptomycin (Biolot, St. Petersburg, Russia). The cultures were maintained in a CO2 incubator at a temperature of 37 °C with 5% CO2.
4.3. Cell Viability Assessment (MTS Test)
Neuro-2a or RAW 264.7 cells (1.0 × 104/200 µL) were seeded into 96-well plates and cultured at 37 °C in a 5% CO2 incubator for 24 h. Afterward, U-443 or U-573 compounds were added to the cell wells at concentrations of 0.01, 0.1, and 1.0 μM. The plates were then incubated for 1 h. Then, a solution of rotenone (Sigma-Aldrich, St. Louis, MO, USA) at a final concentration of 10.0 μM was added to the wells. This concentration of rotenone suppressed cell viability by around 30–40%. After 24 h cytoprotective effect of compounds was determined by the MTS assay. The absorbance in each well was detected at 490/630 nm using a PHERAstar FS plate reader (BMG Labtech, Ortenberg, Germany). The experiments were conducted in triplicate and the results were expressed as the percentage of inhibition relative to the untreated cells exposed to rotenone.
To study the cytotoxic activity of U-443 or U-573, these compounds were added at final concentrations of 0.01–100.0 μM to the wells of 96-well plates with Neuro-2a cells and the cells were incubated for an additional 24 h. Cell viability was then determined as described above.
4.4. Measurement of Mitochondrial Membrane Potential (MMP)
Neuro-2a cells (1.0 × 10
4 cells/well in a 96-well plate) were treated with different concentrations of compounds
U-443 or
U-573 (0.01–1.0 μM) for 1 h at 37 °C in a 5% CO
2 incubator. Following this, a solution of rotenone at a concentration of 10.0 μM was added, and the cells were further incubated for 1 h. Subsequently, a solution of tetramethylrhodamine methyl (TMRM) at a concentration of 500 nM (Sigma-Aldrich, St. Louis, MO, USA) was added to each well and incubated for 30 min at 37 °C. Fluorescence intensity was measured using a PHERAstar FS plate reader (BMG Labtech, Ortenberg, Germany) with excitation at 540 nm and emission at 590 nm. Data analysis was performed using MARS Data Analysis v. 3.01R2 (BMG Labtech, Ortenberg, Germany), and the results were presented as a percentage of the control [
55].
4.5. Analysis of ROS and NO Levels
Cells (1.0 × 104 cells/well) were treated with compounds U-443 or U-573 at concentrations ranging from 0.01 to 1.0 μM for 1 h. Subsequently, rotenone was added to each well at a final concentration of 10.0 μM, and the cells were incubated for an additional 1 h. To evaluate the production of ROS, a 20 μL solution of 2,7-dichlorodihydrofluorescein diacetate (H2DCF-DA, Molecular Probes, Eugene, OR, USA) at a final concentration of 10.0 μM was added to each well, and the plates were further incubated for 10 min at 37 °C in the absence of light.
To evaluate the nitric oxide levels in cells, a solution of the NO-sensitive probe DAF-FM (Molecular Probes, Eugene, OR, USA) was introduced to each well at a concentration of 5.0 μM. The plates were then incubated for 40 min at 37 °C in the absence of light. Fluorescence intensity was measured using a PHERAstar FS high-speed plate reader (BMG Labtech, Ortenberg, Germany) with excitation at 485 nm and emission at 518 nm. The obtained data were analyzed using MARS Data Analysis v. 3.01R2 (BMG Labtech, Ortenberg, Germany), and the results were expressed as a percentage of the control [
55].
4.6. TNF ELISA Assay
RAW 264.7 cells (2.0 × 104/200 µL) were plaсed in 96-well plates and allowed to adhere for 2 h at 37 °C in a 5% CO2 incubator. Following the adhesion period, cells were treated with compounds U-443 or U-573 at concentrations ranging from 0.1 to 1.0 µM for 1 h. Subsequently, LPS (Escherichia coli 055:B5, Sigma, St. Louis, MO, USA) was introduced to each well at a concentration of 1.0 μg/mL, and the cells were incubated for 24 h. Positive and negative controls were included where cells were incubated without LPS and compounds, and with LPS alone, respectively. Following that, the samples underwent centrifugation at 1000× g for 20 min, and the resulting supernatants were collected and stored on ice for future applications. The cells were gently washed with cold PBS and then resuspended in fresh lysis buffer (0.1 mL/well). Plates containing the samples were subsequently centrifuged at 1500× g for 10 min at 2–8 °C to eliminate any cellular debris. The mixture of supernatants and cell lysates was promptly subjected to analysis for TNF levels using the Mouse TNF-α ELISA Kit SEA133Mu TNF-α (Cloud-Clone Corp., Katy, TX, USA).
4.7. COX-2 Activity Assay Kit
RAW 264.7 cells were seeded in 96-well plates at a density of 2 × 104 cells in a volume of 200 µL per well. The plates were incubated for 2 h at 37 °C in a 5% CO2 incubator to allow the cells to adhere. After the adhesion period, the cells were treated with compounds U-443 or U-573 at concentrations ranging from 0.1 to 1.0 µM for 1 h. Subsequently, LPS was added to each well at a concentration of 1.0 μg/mL, and the cells were incubated for 24 h. Positive and negative controls were included where cells were incubated without LPS and compounds and with LPS alone, respectively.
To prepare the cell lysate, the cells were washed once with 0.2 mL of PBS and resuspended in the same volume of PBS. Cells were then centrifuged at 500× g for 3 min. The cell pellet was resuspended in 0.1 mL of lysis buffer containing a protease inhibitor cocktail. After vortexing and incubating on ice for 5 min, the lysate was again centrifuged at 12,000× g, 4 °C for 3 min. The supernatant was collected and kept on ice for further use. The activity of cyclooxygenase-2 (COX-2) was determined using the Cyclooxygenase Activity Assay kit (BN00779, Assay Genie, Ireland) according to the manufacturer’s protocol.
4.8. Western Blotting
Western blotting was used to determine the expression of IL-1β and pro-IL-1β. RAW 264.7 cells were incubated with the test compounds at concentrations of 0.1 and 1.0 μM for 24 h. Afterward, LPS was added and the cells were further incubated for another 24 h. The cells were then washed with cold PBS (BioloT, Russia) and lysed using the RIPA buffer (Sigma-Aldrich, St Louis, MO, USA). The protein concentration was measured using the Bradford method. Electrophoresis with SDS on a 12% polyacrylamide gel was performed to separate the proteins. The separated proteins were transferred onto Immobilon®-P polyvinylidene difluoride (PVDF) membrane using a semi-dry transfer cell (Bio-Rad, Hercules, CA, USA). Specific primary polyclonal antibodies against IL-1β (Invitrogen, Waltham, MA, USA) (1:1000) were used to detect the IL-1β protein zones, while specific monoclonal antibodies against β-actin were used to detect the β-actin protein zones as a loading control. Horseradish peroxidase-conjugated (Sigma-Aldrich, St. Louis, MO, USA) secondary antibodies were used. Protein zones on the membrane were visualized using the PierceTM ECL kit (Thermo Fisher Scientific, Waltham, MA, USA) and a Versa Doc imaging system (Bio-Rad, Hercules, CA, USA). Densitometry analysis of the protein zones was carried out using Image Lab 6.0.1 software (Bio-Rad, Hercules, CA, USA).
4.9. Determination of Antioxidant Activity in Brain Homogenate
The brains of male CD1 mice were rinsed with a cold buffer solution containing 140 mM KCl and 10 mM K2HPO4 at pH 7.4. The brains were then homogenized using a Potter-Elvehjem tissue homogenizer, with the buffer added to the brain tissue at a weight ratio of 1:4. To the resulting homogenate, 10 mL of buffer was added, and the mixture was centrifuged for 10 min at 900 rpm using a Labofuge 44R centrifuge (Thermo Scientific, Germany). The supernatant was transferred to a volumetric flask and adjusted to a final volume of 25 mL with the buffer. The Lowry protein concentration in the homogenate was found to be within the range of 0.27–0.3 mg/mL.
To evaluate the antioxidant activity of the compounds in the absence of enzymatic reactions, 0.5 mL of distilled water or a solution of the tested compound in water was combined with 0.5 mL of brain homogenate. The mixture was then incubated for 10 min, followed by the addition of 20 μL of a 2 mM FeSO4 solution. The reaction was terminated by introducing 1 mL of 20% trichloroacetic acid (TCA, Reachim, Russia) into the system. Subsequently, the homogenate was subjected to centrifugation for 10 min at 4000 rpm using a Labofuge 44R centrifuge. To the 1 mL of supernatant, 1 mL of a freshly made solution of 0.7% thiobarbituric acid (TBA, Sigma, USA) in 50% glacial acetic acid was added. This mixture was then incubated for 30 min in a water bath at 100 °C to allow the reaction with TBA to occur. The resulting products that react with TBA were assessed by determining the content of TBA-reacting substances (TBARS) using a spectrophotometric method. A PHERAstar FS plate reader (MG Labtech, Germany) was utilized to measure the fluorescence intensity at wavelengths of 480/520 nm (excitation/emission). TBA-reacting products were determined in the initial homogenate of the mouse brain (spontaneous level, Fsp), in the homogenate after incubation with an oxidizing agent (induced level, Fin), and in the homogenate after incubation with an oxidizing agent and a natural compound in the experiment (experimental level, Fexp). The content of TBARS after iron-induced oxidation was Fin − Fsp and was taken as 100%. The content of TBARS (%) after oxidation in the presence of compounds was (Fexp − Fsp )/(Fin − Fsp) × 100%. Ionol (2,6-di-tert-butyl-4-methylphenol, Sigma-Aldrich, St Louis, MO, USA) was used as a reference antioxidant compound.
4.10. Molecular Modeling
The spatial structures of 1,4-NQs used in the work were obtained and optimized using the potential of the forces MFF94 using the MOE program [
56]. The crystal structure of mouse cyclooxygenase-2 (mCOX-2) PDB code 1CX2 [
44] was used for 1,4-NQ molecular docking. Molecular docking was performed with the docking module of the MOE program using a selective inhibitor SC-558 in a complex with mCOX-2 (PDB code 1CX2) as a template for the binding site. For each compound, 30 complexes were calculated using the London dG parameters, for 5 of which the optimization was performed with the GBVI/WSA dG parameters. The analysis of contacts in the complexes was carried out using the ligand interaction module. The calculations were performed using the Center for Collective Use of the IAPU FEB RAS "Far Eastern Computational Resource".
4.11. Animals
Experiments involving female CD1 mice aged two months (weighing 20–22 g) were carried out using mice obtained from the Russian National Center for Genetic Resources of Laboratory Animals. The mice were housed in a specific pathogen-free (SPF) vivarium at the ICiG SB RAS in Novosibirsk, Russia. They were provided with food and water in an environment with a 12 h light/dark cycle at a temperature of 22 ± 1 °C. All experimental procedures adhered to the International Recommendations for Biomedical Research Using Animals established by the International Council of Medical Scientific Societies (CIOMS), as well as the Laboratory Practice Rules approved by the Ministry of Health and Social Development of Russia on 23.08.2010 (Order No. 708n) and approved by the local ethics committee of the G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences (protocol No 01/20 of September 6, 2020).
4.12. Parkinson Disease Induction
The early stage of Parkinson’s disease was induced in mice according to [
10,
57] with modifications. Rotenone (Sigma-Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulfoxide and olive oil (1:9
v/
v). Dissolved rotenone was injected subcutaneously during 8 consecutive days with a dose of 6 mg/kg to construct the model.
L-DOPA (3,4-dihydroxy-L-phenylalanine, Sigma-Aldrich, St Louis, MO, USA) was used as a positive control in an experiment at a dose of 5 mg/kg per oral, once, one day after the last dose of rotenone. After 40 min, the effect of L-DOPA was assessed.
Studied compounds U-443 and U-573 were dissolved in dimethyl sulfoxide and olive oil (1:9 v/v). Compounds were administrated intraperitoneally at doses 0.1, 1.0, or 10.0 mg/kg, three times every second day. The solvent dimethyl sulfoxide and olive oil (1:9 v/v) were used instead of naphthoquinone in the control group of mice treated with rotenone only.
4.13. Experimental Procedure
The experimental procedure proceeded as outlined below. Once the mice had acclimated, they were randomly divided into six groups: the first group consisted of control mice in their normal state; the second group served as the control for the model (receiving a solvent); the third group received L-DOPA at a dosage of 5 mg/kg; and the fourth to sixth groups constituted the model groups exposed to 1,4-naphthoquinones at dosages of 0.1, 1.0, or 10.0 mg/kg. Behavioral assessments were conducted one day after the final injection using the “Cylinder” and “Open field” tests.
4.14. “Cylinder” Test
The transparent glass cylinder, measuring 19.5 cm in height and 15.0 cm in diameter, was partially covered with black cardboard on three sides to minimize the impact of ambient lighting. The side of the cylinder facing the camera was left uncovered for video recording purposes. The camera SONY (Carl Zeiss, VirioTessar, 70×) was used during the experiment. Mouse behavior was recorded for 3 min. To prevent any disturbances in the behavior of the mice, measures were taken to eliminate noise and minimize changes in light during the process. Afterward, the cylinder was thoroughly cleaned using water, followed by sanitization of the inner wall using 70% ethanol to eliminate mouse scents. It was then dried before introducing another mouse. The changes in the behavioral reactions of mice were assessed against the background of the development of the rotenone model. The number of mouse climbs (rearings) was calculated. A rearing behavior in mice is characterized by the act of the mouse standing on its hind limbs, lifting both forelimbs above shoulder level, and subsequently descending. The elevation of a single front limb is not considered in this test.
4.15. “Open field” Test
For the open field test, a green opaque plastic box measuring 100 × 100 × 70 cm was employed. Prior to the assessment, a preadaptation period of 5 min was allotted for all mice within the box. Subsequently, a pair of mice was gently placed in the center of the open field, and their movements were recorded for a duration of 3 min using a digital camera (SONY, Carl Zeiss, VirioTessar, 70×). To assess changes in animal behavior, the “ToxTrac” v.2.84 software (UMEA University, LinneaausVag, SE-901 87, Sweden) was used for data collection, plotting, and data analysis. The average speed (sm/min), total distance (mm), total time frozen (min), and frozen events (number of times) were determined. Between each trial, the box was thoroughly cleaned using a solution of 10% ethanol and water.
4.16. Acute Toxicity
To determine acute toxicity of 1,4-NQs CD1, female mice, weighing 20–22 g, 6 animals per group, were used. The mice were kept on a standard vivarium diet. The substances were diluted with olive oil and injected intraperitoneally into the mice in a volume of 0.1 mL. The control and experimental animals were observed for two weeks.
The acute toxicity was assessed using the Kerber formula [
58]:
LD50 = LD100 – Σ (z × d)/m;
where LD50 is the death rate of 50% of mice; LD100 is the death rate of all animals; z is a half of the sum of the animal number that died from the last two doses; d is the interval between every two last doses; m is the number of animals for each dose.
4.17. Data Analysis and Statistics
The data were collected in three separate replicates, and the calculated values were presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using SigmaPlot 14.0 (Systat Software Inc., San Jose, CA, USA), utilizing one-way analysis of variance (ANOVA) followed by the post hoc Student–Newman–Keuls test.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}