
New Rare Triterpene Glycosides from Pacific Sun Star, Solaster pacificus, and Their Anticancer Activity

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
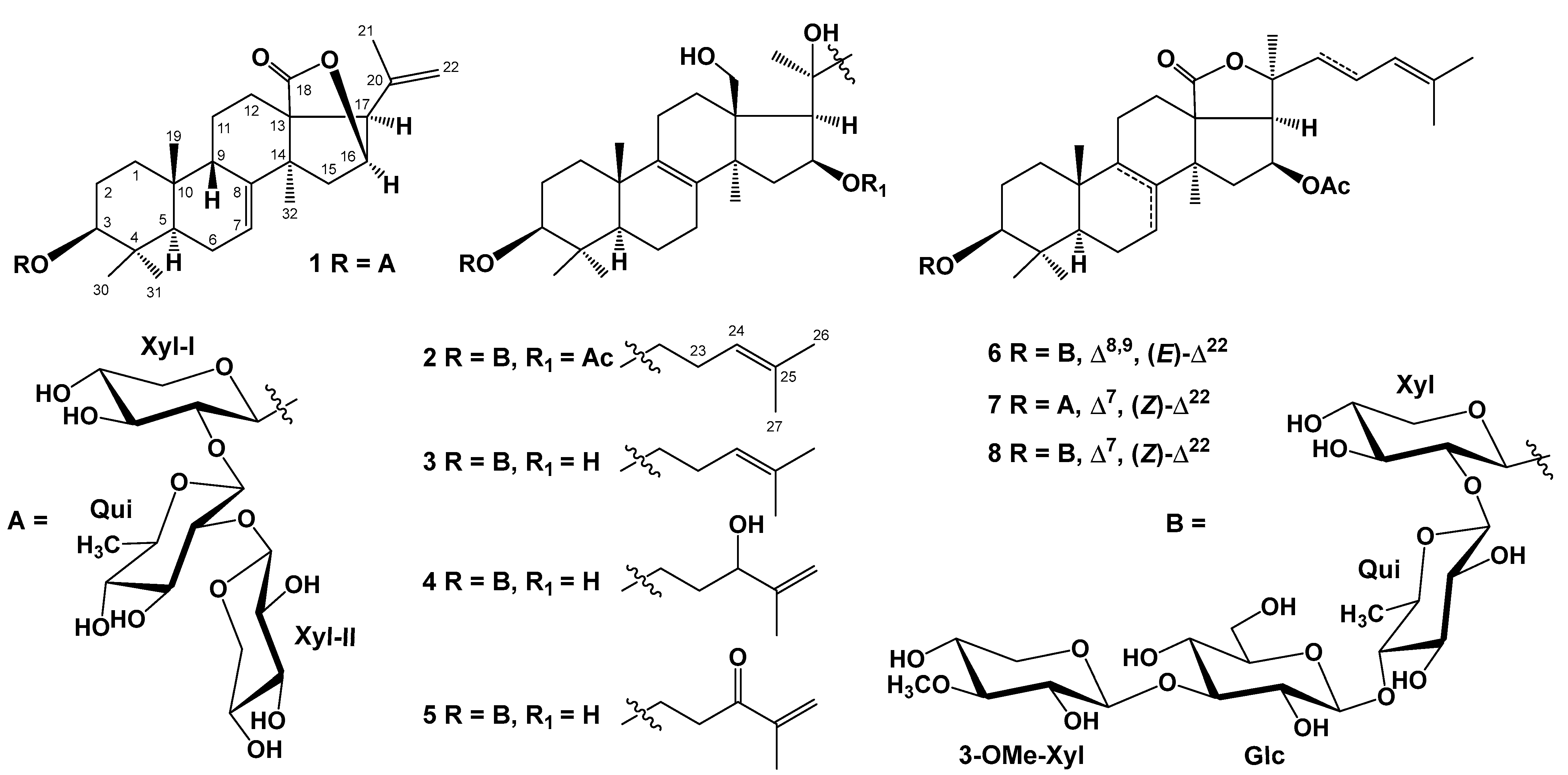
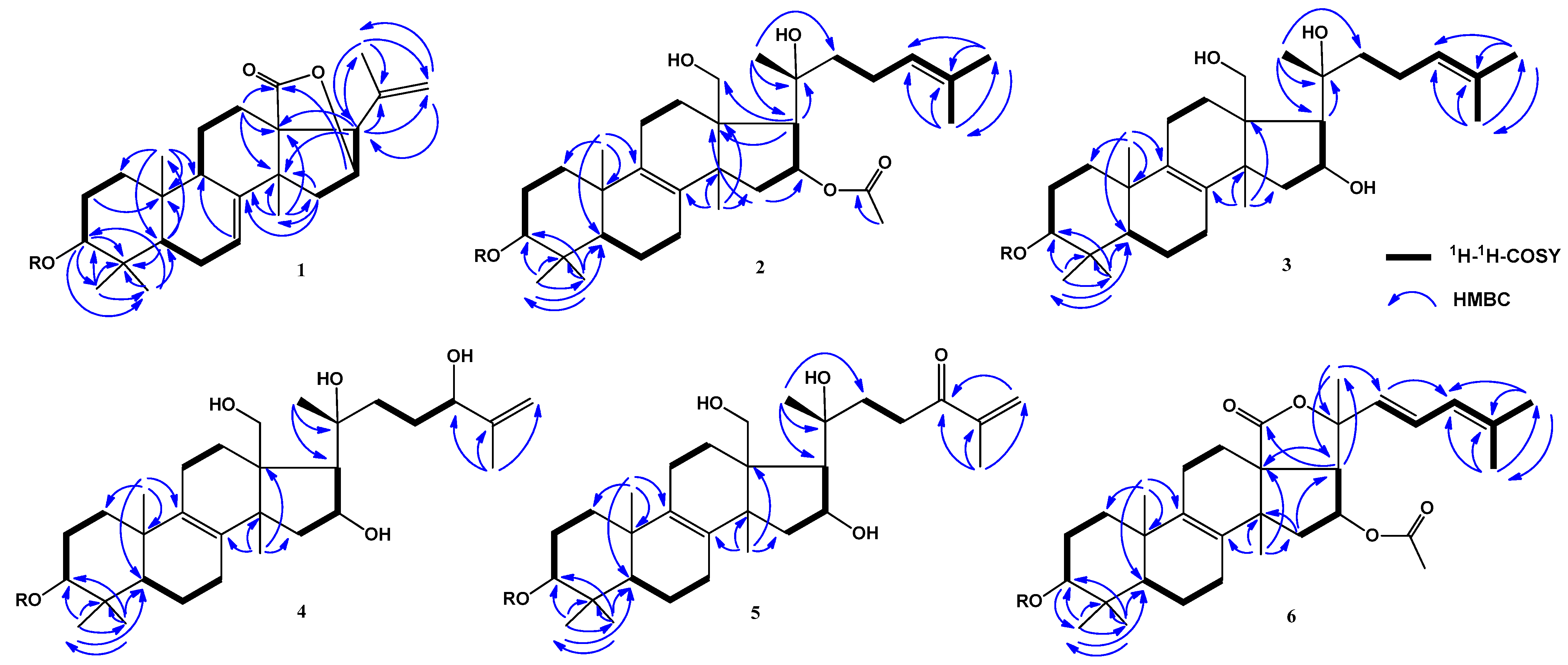
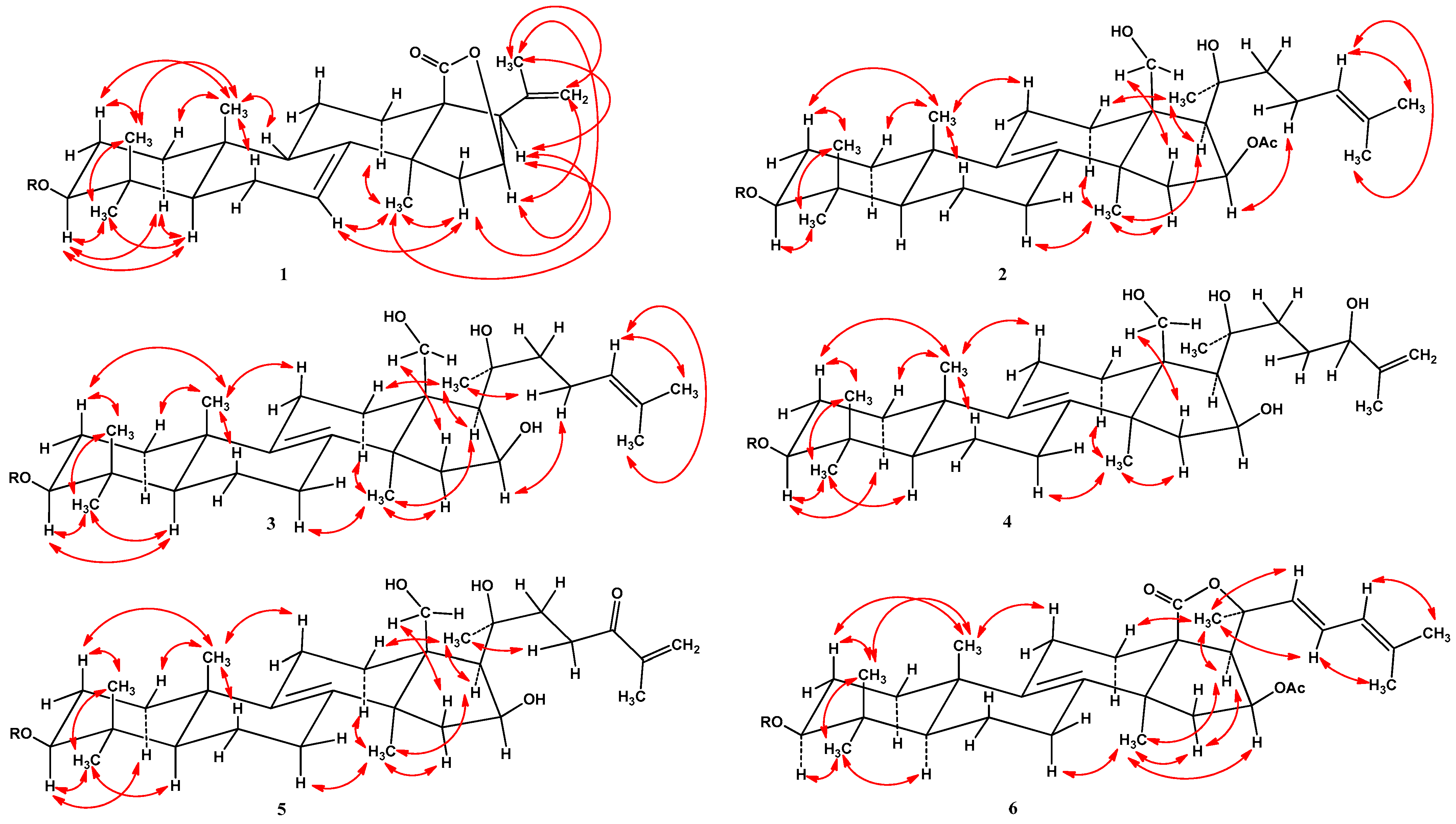
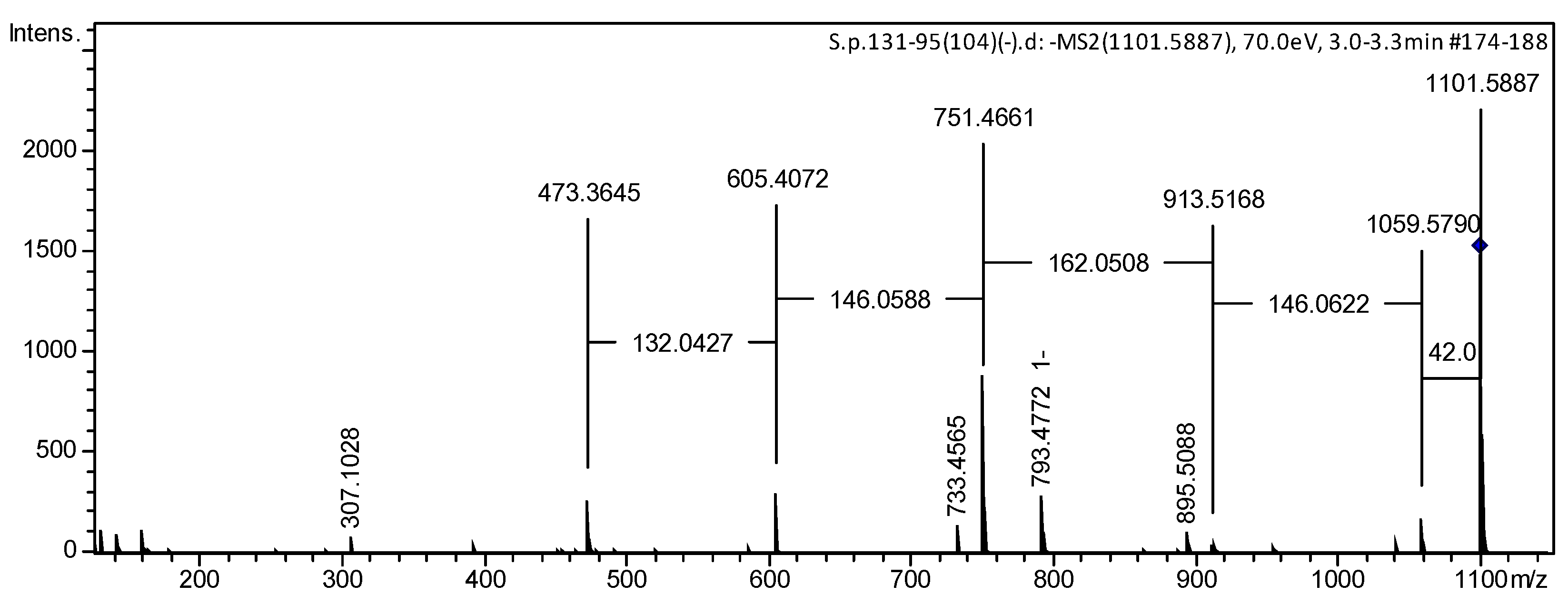
2.1. Isolation and Structure Elucidation of Compounds 1–8 from S. pacificus
2.2. Investigation of Biological Activities
2.2.1. Cytotoxicity of Compounds 1–3 and 6–8 against Normal and Cancer Cells
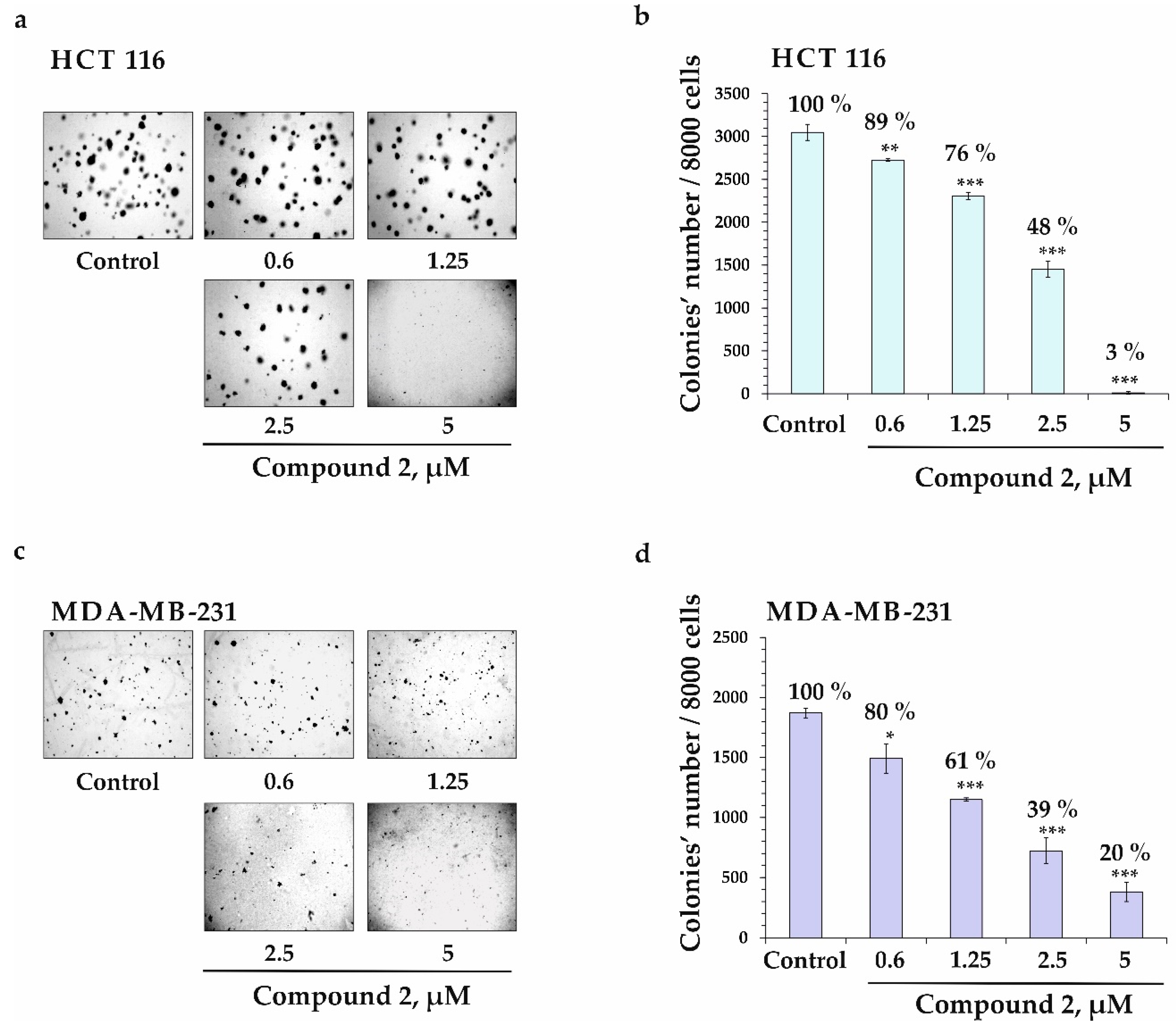
2.2.2. Colony-Inhibiting Activity of Compound 2 in Cancer Cells HCT 116 and MDA-MB-231
2.2.3. Anti-Invasive Activity of Compound 2 in Cancer Cells HCT 116 and MDA-MB-231
3. Materials and Methods
3.1. General Methods
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Spectral Data of New Compounds
3.5. Acid Hydrolysis and Determination of Absolute Configurations of Monosaccharides
3.6. Reagents
3.7. Cell Lines and Culture Conditions
3.8. Preparation of Compounds
3.9. MTS Assay
3.10. Soft Agar Assay
3.11. Scratch Assay
3.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A.; Islam, M.T. Sea cucumber glycosides: Chemical structures, producing species and important biological properties. Mar. Drugs 2017, 15, 317. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Progress in the studies of triterpene glycosides from sea cucumbers (Holothuroidea, Echinodermata) between 2017 and 2021. Nat. Prod. Commun. 2021, 16, 1–24. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Non-holostane aglycones of sea cucumber triterpene glycosides. Structure, biosynthesis, evolution. Steroids 2019, 147, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Careaga, V.P.; Maier, M.S. Cytotoxic triterpene glycosides from sea cucumbers. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.-K., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 515–528. [Google Scholar]
- Malyarenko, T.V.; Kicha, A.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Malyarenko, O.S.; Kuzmich, A.S.; Stonik, V.A.; Ivanchina, N.V. New triterpene glycosides from the Far Eastern starfish Solaster pacificus and their biological activity. Biomolecules 2021, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Malyarenko, T.V.; Malyarenko, O.S.; Kicha, A.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Ivanchina, N.V. In vitro anticancer and cancer-preventive activity of new triterpene glycosides from the Far Eastern starfish Solaster pacificus. Mar. Drugs 2022, 20, 216. [Google Scholar] [CrossRef]
- Chludil, H.D.; Murray, A.P.; Seldes, A.M.; Maier, M.S. Biologically active triterpene glycosides from sea cucumbers (Holothuroidea, Echinodermata). In Studies in Natural Products Chemistry; Rahman, A.-u., Ed.; Elsevier Science Publisher: Amsterdam, The Netherlands, 2003; Volume 28, pp. 587–615. [Google Scholar] [CrossRef]
- Kim, S.K.; Himaya, S.W.A. Chapter 20-Triterpene glycosides from sea cucumbers and their biological activities. Adv. Food Nutr. Res. 2012, 63, 297–319. [Google Scholar] [CrossRef]
- Maier, M.S.; Roccatagliata, A.J.; Kuriss, A.; Chludil, H.; Seldes, A.M.; Pujol, C.A.; Damonte, E.B. Two new cytotoxic and virucidal trisulfated triterpene glycosides from the Antarctic sea cucumber Staurocucumis liouvillei. J. Nat. Prod. 2001, 64, 732–736. [Google Scholar] [CrossRef]
- Aminin, D.L.; Pislyagin, E.A.; Menchinskaya, E.S.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Immunomodulatory and anticancer activity of sea cucumber triterpene glycosides. In Studies in Natural Products Chemistry (Bioactive Natural Products); Rahman, A.-u., Ed.; Elsevier Science Publisher: Amsterdam, The Netherlands, 2014; Volume 41, pp. 75–94. [Google Scholar]
- Aminin, D.L.; Menchinskaya, E.S.; Pisliagin, E.A.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Anticancer activity of sea cucumber triterpene glycosides. Mar. Drugs 2015, 13, 1202–1223. [Google Scholar] [CrossRef]
- Li, X.; Roginsky, A.B.; Ding, X.; Woodward, C.; Collin, P.; Newman, R.A.; Bell, R.H., Jr.; Adrian, T.E. Review of the apoptosis pathways in pancreatic cancer and the anti-apoptotic effects of the novel sea cucumber compound, frondoside A. Ann. N.Y. Acad. Sci. 2008, 1138, 181–198. [Google Scholar] [CrossRef]
- Al Marzouqi, N.; Iratni, R.; Nemmar, A.; Arafat, K.; Ahmed Al Sultan, M.; Yasin, J.; Collin, P. Frondoside A inhibits human breast cancer cell survival, migration, invasion and the growth of breast tumor xenografts. Eur. J. Pharmacol. 2011, 668, 25–34. [Google Scholar] [CrossRef]
- Ma, X.; Kundu, N.; Collin, P.D.; Goloubeva, O.; Fulton, A.M. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res. Treat. 2012, 132, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Andrijaschenko, P.V.; Dmitrenok, P.S.; Kalinin, V.I.; Stonik, V.A. 3β-O-Glycosylated 16β-acetoxy-9β-H-lanosta-7,24-diene-3β,18,20β-triol, an intermediate metabolite from the sea cucumber Eupentacta fraudatrix and its biosynthetic significance. Biochem. Syst. Ecol. 2012, 44, 53–60. [Google Scholar] [CrossRef]
- Wang, X.-H.; Li, L.; Yi, Y.-H.; Sun, P.; Yan, B.; Pan, M.-X.; Han, H.; Wang, X.-D. Two new triterpene glycosides from sea cucumber Stichopus variegatus. Chin. J. Nat. Med. 2006, 4, 177–180. [Google Scholar]
- Wang, X.-H.; Zou, Z.-R.; Yi, Y.-H.; Han, H.; Li, L.; Pan, M.-X. Variegatusides: New non-sulphated triterpene glycosides from the sea cucumber Stichopus variegates Semper. Mar. Drugs 2014, 12, 2004–2018. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ravi, S.; Alencar, A.M., Jr.; Arakelyan, J.; Xu, W.; Stauber, R.; Wang, C.I.; Papyan, R.; Ghazaryan, N.; Pereira, R.M. An update to hallmarks of cancer. Cureus 2022, 14, e24803. [Google Scholar] [CrossRef] [PubMed]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer—The new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef] [PubMed]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- AAT Bioquest. Available online: https://www.aatbio.com/tools/ic50-calculator (accessed on 10 November 2020).
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of in-vitro bioassay methods: Application in herbal drug research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 1.47 m | 1.68 m 1.18 m | 1.68 m 1.18 m | 1.69 m 1.18 m | 1.69 m 1.18 m | 1.72 m 1.22 m |
| 2 | 2.18 m 1.91 m | 2.16 m 1.89 m | 2.17 m 1.89 m | 2.17 m 1.89 m | 2.17 m 1.89 m | 2.23 m 1.97 m |
| 3 | 3.32 dd (12.0; 3.3) | 3.27 dd (12.0, 4.3) | 3.27 dd (12.0, 3.7) | 3.27 m | 3.27 m | 3.32 dd (11.5, 3.9) |
| 4 | ||||||
| 5 | 1.00 dd (12.0; 3.3) | 1.12 m | 1.13 brd (12.6) | 1.13 m | 1.13 m | 1.81 m |
| 6 | 2.04 m 1.97 m | 1.73 m 1.53 m | 1.72 m 1.51 m | 1.72 m 1.51 m | 1.72 m 1.51 m | 1.72 m 1.68 m |
| 7 | 5.62 brd (7.0) | 1.96 m 0.96 m | 2.07 m 1.03 m | 2.03 m 1.04 m | 2.03 m 1.04 m | 2.15 m 1.18 m |
| 8 | ||||||
| 9 | 3.00 brd (14.0) | |||||
| 10 | ||||||
| 11 | 1.99 m 1.46 m | 2.28 m 2.13 m | 2.28 m 2.13 m | 2.28 m 2.15 m | 2.28 m 2.15 m | 2.30 m 2.08 m |
| 12 | 2.37 m 1.88 m | 2.77 m 1.70 m | 2.77 m 1.70 m | 2.77 m 1.70 m | 2.77 m 1.70 m | 2.23 m 1.75 m |
| 13 | ||||||
| 14 | ||||||
| 15 | 2.15 dd (13.5, 2.3) 1.97 m | 2.26 m 1.76 m | 2.07 m | 2.10 m 2.04 m | 2.10 m 2.04 m | 2.03 dd (11.8, 6.9) 1.82 m |
| 16 | 4.74 s | 5.85 m | 5.02 m | 5.03 m | 5.03 m | 5.81 m |
| 17 | 2.93 s | 2.32 brd (7.0) | 2.15 m | 2.17 m | 2.10 m | 2.75 d (9.3) |
| 18 | 4.17 m 3.93 m | 4.41 brd (11.2) 3.97 brd (11.2) | 4.42 brd (11.2) 3.97 brd (11.2) | 4.42 brd (11.2) 3.97 brd (11.2) | ||
| 19 | 1.03 s | 1.05 s | 1.04 s | 1.04 s | 1.04 s | 1.31 s |
| 20 | ||||||
| 21 | 1.74 s | 1.62 s | 1.63 s | 1.67 s | 1.60 s | 1.56 s |
| 22 | 5.06 s 4.99 s | 1.92 m 1.84 m | 2.15 m | 2.42 m 2.28 m | 2.72 m 2.28 m | 5.93 d (15.8) |
| 23 | 2.32 m 2.22 m | 2.35 m 2.25 m | 2.18 m 2.03 m | 3.17 m 2.97 m | 6.58 dd (15.8, 11.3) | |
| 24 | 5.30 m | 5.28 m | 4.47 brd (9.2) | 5.90 d (11.3) | ||
| 25 | ||||||
| 26 | 1.67 s | 1.64 s | 5.26 brs 4.94 brs | 6.08 brs 5.67 brs | 1.64 s | |
| 27 | 1.67 s | 1.69 s | 1.93 s | 1.94 s | 1.72 s | |
| 30 | 1.18 s | 1.11 s | 1.10 s | 1.12 s | 1.12 s | 1.15 s |
| 31 | 1.32 s | 1.32 s | 1.32 s | 1.34 s | 1.34 s | 1.34 s |
| 32 | 1.33 s | 1.02 s | 1.02 s | 1.00 s | 1.00 s | 1.07 s |
| CH3-CO | 2.17 s | 2.06 s |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 35.9 | 35.6 | 35.8 | 35.6 | 35.6 | 35.8 |
| 2 | 27.0 | 27.1 | 27.3 | 27.1 | 27.1 | 27.0 |
| 3 | 89.0 | 88.7 | 88.9 | 88.7 | 88.7 | 88.8 |
| 4 | 39.4 | 39.6 | 39.7 | 39.6 | 39.6 | 39.6 |
| 5 | 47.4 | 50.7 | 51.1 | 50.9 | 50.9 | 50.9 |
| 6 | 23.3 | 18.1 | 18.1 | 18.1 | 18.1 | 17.9 |
| 7 | 122.7 | 26.3 | 26.3 | 26.3 | 26.3 | 27.7 |
| 8 | 147.3 | 132.7 | 133.5 | 133.5 | 133.5 | 134.2 |
| 9 | 46.4 | 136.5 | 136.4 | 136.4 | 136.4 | 136.0 |
| 10 | 35.5 | 36.9 | 37.0 | 36.9 | 36.9 | 37.0 |
| 11 | 21.7 | 20.9 | 21.0 | 20.8 | 20.8 | 21.2 |
| 12 | 20.0 | 25.4 | 25.5 | 25.3 | 25.3 | 28.3 |
| 13 | 56.7 | 51.2 | 51.3 | 51.1 | 51.1 | 45.0 |
| 14 | 46.0 | 46.5 | 47.0 | 46.9 | 46.9 | 59.3 |
| 15 | 43.8 | 41.5 | 44.1 | 44.0 | 44.0 | 40.8 |
| 16 | 80.4 | 76.6 | 73.5 | 73.2 | 73.2 | 73.6 |
| 17 | 59.0 | 54.0 | 54.1 | 54.0 | 55.2 | 53.4 |
| 18 | 180.7 | 62.2 | 62.8 | 62.6 | 62.6 | 176.5 |
| 19 | 23.9 | 19.2 | 19.2 | 19.1 | 19.1 | 19.0 |
| 20 | 139.9 | 74.6 | 76.6 | 76.6 | 76.1 | 82.4 |
| 21 | 23.0 | 27.6 | 27.9 | 26.2 | 25.5 | 30.5 |
| 22 | 113.9 | 42.9 | 43.2 | 35.9 | 36.8 | 134.2 |
| 23 | 23.7 | 23.8 | 31.1 | 32.8 | 122.2 | |
| 24 | 125.0 | 125.5 | 75.8 | 201.9 | 125.3 | |
| 25 | 131.1 | 131.0 | 148.7 | 144.4 | 134.5 | |
| 26 | 17.4 | 17.5 | 110.0 | 124.2 | 18.2 | |
| 27 | 25.6 | 25.7 | 17.5 | 17.7 | 25.6 | |
| 30 | 17.3 | 16.3 | 16.3 | 16.3 | 16.3 | 16.3 |
| 31 | 28.6 | 27.7 | 27.7 | 27.7 | 27.7 | 27.1 |
| 32 | 33.9 | 26.4 | 26.2 | 26.2 | 26.2 | 26.5 |
| CO | 170.1 | 170.5 | ||||
| CH3-CO | 21.4 | 21.0 |
| Position | 1 | 2–6 | ||||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | HMBC | ROESY | δH | δC | HMBC | ROESY | |
| Xyl-I | Xyl | |||||||
| 1 | 4.88 d (7.1) | 105.1 | C3-agl; C5-Xyl-I | H3, H31-agl, H3, H5-Xyl-I | 4.79 d (7.5) | 105.5 | C3-agl | H3, H31-agl, H3, H5-Xyl |
| 2 | 4.01 dd (8.7, 7.4) | 83.3 | C1, C3-Xyl-I, C1-Qui | H1-Qui | 4.05 dd (8.2, 7.2) | 84.0 | C1-Xyl, C1-Qui | H1-Qui |
| 3 | 4.26 t (9.1) | 77.9 | C2, C4-Xyl-I | H1-Xyl-I | 4.19 t (9.0) | 78.0 | C1, C2-Xyl | H1-Xyl |
| 4 | 4.18 m | 70.2 | 4.16 m | 70.7 | ||||
| 5 | 4.35 dd (11.2, 5.1) 3.74 m | 66.5 | C1, C3-Xyl-I C1, C3-Xyl-I | H1-Xyl-I | 4.30 dd (11.6, 5.1) 3.69 t (10.0) | 66.6 | C3, C4-Xyl | H1-Xyl |
| Qui | Qui | |||||||
| 1 | 5.31 d (7.5) | 103.4 | C2-Xyl-I | H3, H5-Qui, H2-Xyl-I | 5.16 d (7.5) | 105.5 | C2-Xyl | H3, H5-Qui, H2-Xyl |
| 2 | 4.14 m | 84.8 | C1, C3-Qui, C1-Xyl-II | H4-Qui, H1-Xyl-II | 4.06 m | 76.3 | C1, C3-Qui | |
| 3 | 4.19 m | 77.5 | C2,C4-Qui | H1, H5-Qui | 4.11 t (9.1) | 75.8 | C2, C4-Qui | H1-Qui |
| 4 | 3.70 m | 76.3 | C2, C3, C5-Qui | H2, H6-Qui | 3.67 t (9.1) | 87.3 | C3-Qui, C1-Glc | H1-Glc, H6-Qui |
| 5 | 3.70 m | 72.7 | H1, H3-Qui | 3.80 m | 71.6 | H1-Qui | ||
| 6 | 1.64 d (5.0) | 18.3 | C4, C5-Qui | H4-Qui | 1.76 d (6.0) | 18.1 | C4, C5-Qui | H4-Qui |
| Xyl-II | Glc | |||||||
| 1 | 5.35 d (7.2) | 106.4 | C2-Qui | H3, H5-Xyl-II; H2-Qui | 4.97 d (8.0) | 104.9 | C4-Qui | H4-Qui, H3, H5-Glc |
| 2 | 4.08 m | 75.8 | C1, C3-Xyl-II | 4.02 m | 73.9 | C1, C3-Glc | ||
| 3 | 4.10 m | 77.6 | C4-Xyl-II | H1-Xyl-II | 4.20 m | 87.2 | C4-Glc | H1-3-OMe-Xyl, H1, H5-Glc |
| 4 | 4.11 m | 70.5 | 4.02 m | 69.5 | C3, C5-Glc | |||
| 5 | 4.31 dd (11.6, 5.0) 3.67 m | 67.3 | C1, C3, C4-Xyl-II C1, C3, C4-Xyl-II | H1-Xyl-II | 4.01 m | 77.9 | H1, H3-Glc | |
| 6 | 4.50 brd (12.2) 4.20 m | 62.1 | ||||||
| 3-OMe-Xyl | ||||||||
| 1 | 5.20 d (7.7) | 106.0 | C3-Glc | H3, H5-3-OMe-Xyl; H3-Glc | ||||
| 2 | 3.93 t (8.1) | 74.6 | C1, C3-3-OMe-Xyl | H4-3-OMe-Xyl | ||||
| 3 | 3.59 t (8.9) | 87.7 | C2, C4-3-OMe-Xyl, OMe | H1-3-OMe-Xyl; OMe | ||||
| 4 | 4.06 m | 70.0 | H2-3-OMe-Xyl | |||||
| 5 | 4.20 dd (10.6, 5.0) 3.63 m | 66.9 | C3, C4-3-OMe-Xyl C1, C3-3-OMe-Xyl | H1-3-OMe-Xyl | ||||
| 3-OMe | 3.85 s | 60.5 | C3-3-OMe-Xyl | H3-3-OMe-Xyl | ||||
| Compound | HEK 293 | HCT 116 | MDA-MB-231 | ||
|---|---|---|---|---|---|
| IC50, µM | IC50, µM | SI | IC50, µM | SI | |
| Cisplatin | 64.6 ± 1.5 * | 11.4 ± 1.3 * | 1.6 | 34.5 ± 0.5 * | 1.9 |
| 1 | 27 ± 0.3 | 81 ± 3.2 | ns | 51.2 ± 0.8 | ns |
| 2 | 18.5 ± 2.5 | 18.6 ± 1.4 | 1 | 15.5 ± 1.6 | 1.2 |
| 3 | 52.8 ± 0.8 | 42.2 ± 0.4 | 1.25 | 36.7 ± 3.3 | 1.4 |
| 6 | 15.3 ± 1.2 | 25.5 ± 1.4 | ns | 29.8 ± 2.1 | ns |
| 7 | 28.5 ± 1.5 | 35.6 ± 0.6 | ns | 34 ± 2.9 | ns |
| 8 | 6.2 ± 0.7 | 11.4 ± 2.7 | ns | 13.1 ± 4.2 | ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyarenko, T.V.; Kicha, A.A.; Kuzmich, A.S.; Malyarenko, O.S.; Kalinovsky, A.I.; Popov, R.S.; Dmitrenok, P.S.; Ivanchina, N.V.; Stonik, V.A. New Rare Triterpene Glycosides from Pacific Sun Star, Solaster pacificus, and Their Anticancer Activity. Mar. Drugs 2024, 22, 19. https://doi.org/10.3390/md22010019
Malyarenko TV, Kicha AA, Kuzmich AS, Malyarenko OS, Kalinovsky AI, Popov RS, Dmitrenok PS, Ivanchina NV, Stonik VA. New Rare Triterpene Glycosides from Pacific Sun Star, Solaster pacificus, and Their Anticancer Activity. Marine Drugs. 2024; 22(1):19. https://doi.org/10.3390/md22010019
Chicago/Turabian StyleMalyarenko, Timofey V., Alla A. Kicha, Alexandra S. Kuzmich, Olesya S. Malyarenko, Anatoly I. Kalinovsky, Roman S. Popov, Pavel S. Dmitrenok, Natalia V. Ivanchina, and Valentin A. Stonik. 2024. "New Rare Triterpene Glycosides from Pacific Sun Star, Solaster pacificus, and Their Anticancer Activity" Marine Drugs 22, no. 1: 19. https://doi.org/10.3390/md22010019
APA StyleMalyarenko, T. V., Kicha, A. A., Kuzmich, A. S., Malyarenko, O. S., Kalinovsky, A. I., Popov, R. S., Dmitrenok, P. S., Ivanchina, N. V., & Stonik, V. A. (2024). New Rare Triterpene Glycosides from Pacific Sun Star, Solaster pacificus, and Their Anticancer Activity. Marine Drugs, 22(1), 19. https://doi.org/10.3390/md22010019