


Light-Mediated Transformation of Renieramycins and Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells

 ,
,  ,
,  , , , ,
, , , ,  and
and 
Abstract
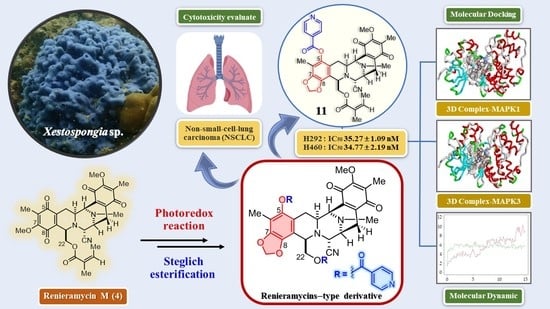
1. Introduction
2. Results and Discussion
2.1. Light-Induced Intramolecular Photoredox Reaction of Renieramycins
2.2. Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives
2.3. Cytotoxicity of Compounds 3–12 against NSCLC Cell Lines
2.4. In Silico Prediction of the Target Genes and Molecular Pathways
2.4.1. Investigation of the Potential Targets against NSCLC
2.4.2. Protein–Protein Interaction Network Construction and Core Target Identification
2.4.3. Molecular Docking of MAPK1 and MAPK3
2.4.4. Molecular Dynamics Analysis of MAPK1 and MAPK3 with 11
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Extraction and Isolation
3.3. Screening of the Photoredox Reaction Conditions with the Renieramycins
3.4. Procedure for the Semisynthesis of Compound 11
3.5. Procedure for the Semisynthesis of Compound 12
3.6. Cytotoxic Evaluations against NSCLC Cell Lines
3.7. Network Pharmacology Study
3.7.1. Database Mining and Identification of 10, 11, and NSCLC-Related Targets
3.7.2. Construction of Protein–Protein Interaction Network and Identification of Potential Core Targets
3.8. Molecular Docking Study
3.9. Molecular Dynamics Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fang, Y.; Li, H.; Ji, B.; Cheng, K.; Wu, B.; Li, Z.; Zheng, C.; Hua, H.; Li, D. Renieramycin-type alkaloids from marine-derived organisms: Synthetic chemistry, biological activity and structural modification. Eur. J. Med. Chem. 2021, 210, 113092. [Google Scholar] [CrossRef] [PubMed]
- Saito, N. Chemical Research on Antitumor Isoquinoline Marine Natural Products and Related Compounds. Chem. Pharm. Bull. 2021, 69, 155–177. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Galmarini, C.M. A Review of Trabectedin (ET-743): A Unique Mechanism of Action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef]
- Cesne, A.L.; Martín-Broto, J.; Grignani, G. A review of the efficacy of trabectedin as second-line treatment of advanced soft tissue sarcoma. Future Oncol. 2022, 18, 5–11. [Google Scholar] [CrossRef]
- Patel, S.; Petty, W.J.; Sands, J.M. An overview of lurbinectedin as a new second-line treatment option for small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211020529. [Google Scholar] [CrossRef]
- Manzo, A.; Sforza, V.; Carillio, G.; Palumbo, G.; Montanino, A.; Sandomenico, C.; Costanzo, R.; Esposito, G.; Laudato, F.; Mercadante, E.; et al. Lurbinectedin in small cell lung cancer. Front. Oncol. 2022, 12, 932105. [Google Scholar] [CrossRef]
- Frincke, J.M.; Faulkner, D.J. Antimicrobial metabolites of the sponge Reniera sp. J. Am. Chem. Soc. 1982, 104, 265–269. [Google Scholar] [CrossRef]
- He, H.Y.; Faulkner, D.J. Renieramycins E and F from the sponge Reniera sp.: Reassignment of the stereochemistry of the renieramycins. J. Org. Chem. 1989, 54, 5822–5824. [Google Scholar] [CrossRef]
- Pettit, G.R.; Knight, J.C.; Collins, J.C.; Herald, D.L.; Pettit, R.K.; Boyd, M.R.; Young, V.G. Antineoplastic Agents 430. Isolation and Structure of Cribrostatins 3, 4, and 5 from the Republic of Maldives Cribrochalina Species. J. Nat. Prod. 2000, 63, 793–798. [Google Scholar] [CrossRef]
- Suwanborirux, K.; Amnuoypol, S.; Plubrukarn, A.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of Renieramycins. Part 3. Isolation and Structure of Stabilized Renieramycin Type Derivatives Possessing Antitumor Activity from Thai Sponge Xestospongia Species, Pretreated with Potassium Cyanide. J. Nat. Prod. 2003, 66, 1441–1446. [Google Scholar] [CrossRef]
- Amnuoypol, S.; Suwanborirux, K.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of Renieramycins. Part 5. Structure Elucidation of Renieramycin-Type Derivatives O, Q, R, and S from Thai Marine Sponge Xestospongia Species Pretreated with Potassium Cyanide. J. Nat. Prod. 2004, 67, 1023–1028. [Google Scholar] [CrossRef]
- Nakao, Y.; Shiroiwa, T.; Murayama, S.; Matsunaga, S.; Goto, Y.; Matsumoto, Y.; Fusetani, N. Identification of Renieramycin A as an Antileishmanial Substance in a Marine Sponge Neopetrosia sp. Mar. Drugs 2004, 2, 55–62. [Google Scholar] [CrossRef]
- Daikuhara, N.; Tada, Y.; Yamaki, S.; Charupant, K.; Amnuoypol, S.; Suwanborirux, K.; Saito, N. Chemistry of renieramycins. Part 7: Renieramycins T and U, novel renieramycin–ecteinascidin hybrid marine natural products from Thai sponge Xestospongia sp. Tetrahedron Lett. 2009, 50, 4276–4278. [Google Scholar] [CrossRef]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Saito, N.; Suwanborirux, K. Chemistry of Renieramycins. 17. A New Generation of Renieramycins: Hydroquinone 5-O-Monoester Analogues of Renieramycin M as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells. J. Nat. Prod. 2017, 80, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Petsri, K.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. Renieramycin T Induces Lung Cancer Cell Apoptosis by Targeting Mcl-1 Degradation: A New Insight in the Mechanism of Action. Mar. Drugs 2019, 17, 301. [Google Scholar] [CrossRef] [PubMed]
- Petsri, K.; Yokoya, M.; Tungsukruthai, S.; Rungrotmongkol, T.; Nutho, B.; Vinayanuwattikun, C.; Saito, N.; Matsubara, T.; Sato, R.; Chanvorachote, P. Structure–Activity Relationships and Molecular Docking Analysis of Mcl-1 Targeting Renieramycin T Analogues in Patient-derived Lung Cancer Cells. Cancers 2020, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liang, J.; Li, X.; Liu, L.; Yao, J.; Chen, X.; Chen, R. Renieramycin T Inhibits Melanoma B16F10 Cell Metastasis and Invasion via Regulating Nrf2 and STAT3 Signaling Pathways. Molecules 2022, 27, 5337. [Google Scholar] [CrossRef]
- Chantarawong, W.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. 5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis. Mar. Drugs 2019, 17, 109. [Google Scholar] [CrossRef]
- Suksamai, D.; Racha, S.; Sriratanasak, N.; Chaotham, C.; Aphicho, K.; Lin, A.C.K.; Chansriniyom, C.; Suwanborirux, K.; Chamni, S.; Chanvorachote, P. 5-O-(N-Boc-l-Alanine)-Renieramycin T Induces Cancer Stem Cell Apoptosis via Targeting Akt Signaling. Mar. Drugs 2022, 20, 235. [Google Scholar] [CrossRef]
- Charupant, K.; Daikuhara, N.; Saito, E.; Amnuoypol, S.; Suwanborirux, K.; Owa, T.; Saito, N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin M–jorunnamycin A analogues. Bioorg. Med. Chem. 2009, 17, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Charupant, K.; Chanvorachote, P.; Mori, N.; Saito, N.; Suwanborirux, K. Chemistry of Renieramycins. 15. Synthesis of 22-O-Ester Derivatives of Jorunnamycin A and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cells. J. Nat. Prod. 2016, 79, 2089–2093. [Google Scholar] [CrossRef]
- Yokoya, M.; Yamazaki-Nakai, M.; Nakai, K.; Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Saito, N. Transformation of Renieramycin M into Renieramycins T and S by Intramolecular Photoredox Reaction of 7-Methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-5,8-dione Derivatives. J. Nat. Prod. 2023, 86, 222–231. [Google Scholar] [CrossRef]
- Saito, N.; Tanaka, C.; Koizumi, Y.-I.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of renieramycin M into jorumycin and renieramycin J including oxidative degradation products, mimosamycin, renierone, and renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Suwanborirux, K.; Saito, N. Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines. Mar. Drugs 2020, 18, 418. [Google Scholar] [CrossRef] [PubMed]
- Xi, P.; Niu, Y.; Zhang, Y.; Li, W.; Gao, F.; Gu, W.; Kui, F.; Liu, Z.; Lu, L.; Du, G. The mechanism of dioscin preventing lung cancer based on network pharmacology and experimental validation. J. Ethnopharmacol. 2022, 292, 115138. [Google Scholar] [CrossRef]
- Iksen, I.; Witayateeraporn, W.; Wirojwongchai, T.; Suraphan, C.; Pornputtapong, N.; Singharajkomron, N.; Nguyen, H.M.; Pongrakhananon, V. Identifying molecular targets of Aspiletrein-derived steroidal saponins in lung cancer using network pharmacology and molecular docking-based assessments. Sci. Rep. 2023, 13, 1545. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Iksen, I.; Sinsook, S.; Wattanathamsan, O.; Buaban, K.; Chamni, S.; Pongrakhananon, V. Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis. Molecules 2022, 27, 8948. [Google Scholar] [CrossRef]
- Petsri, K.; Yokoya, M.; Racha, S.; Thongsom, S.; Thepthanee, C.; Innets, B.; Ei, Z.Z.; Hotta, D.; Zou, H.; Chanvorachote, P. Novel Synthetic Derivative of Renieramycin T Right-Half Analog Induces Apoptosis and Inhibits Cancer Stem Cells via Targeting the Akt Signal in Lung Cancer Cells. Int. J. Mol. Sci. 2023, 24, 5345. [Google Scholar] [CrossRef]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Pithi, C.; Supakarn, C.; Chuanpit, N.; Preeyaporn Plaimee, P. Potential Anti-metastasis Natural Compounds for Lung Cancer. Anticancer Res. 2016, 36, 5707. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Light Source a | Solvent | Renieramycin Product | Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 | 4 | 18 W | CH2Cl2 | 7 | 64 |
| 2 | 4 | 18 W | CHCl3 | 7 | 46 |
| 3 | 4 | 18 W | THF | 7 | 47 |
| 4 | 4 | 4 W | CH2Cl2 | 7 | 81 |
| 5 | 4 | 4 W | CHCl3 | 7 | 51 |
| 6 | 4 | 4 W | THF | 7 | 54 |
| 7 | 5 | 4 W | CH2Cl2 | 6:8 = 1:2 b | 18:37 c |
| 8 | 6 | 4 W | CH2Cl2 | 8 | 48 |
| Entry | Compound | 5-O-Substituents | 22-O-Substituents | IC50 ± S.D. (nM) | |
|---|---|---|---|---|---|
| H292 | H460 | ||||
| 1 | 3 | carbonyl | OH | 97.85 ± 6.75 | 157.53 ± 6.65 |
| 2 | 4 | carbonyl | angeloyl | 35.36 ± 4.51 | 33.86 ± 2.16 |
| 3 | 5 | carbonyl | angeloyl | 170.03 ± 10.07 | 104.36 ± 22.02 |
| 4 | 6 | carbonyl | angeloyl | 111.74 ± 12.94 | 99.74 ± 0.13 |
| 5 | 7 | OH | angeloyl | 72.64 ± 2.55 | 83.32 ± 4.72 |
| 6 | 8 | OH | angeloyl | 183.37 ± 37.04 | 167.97 ± 3.53 |
| 7 | 9 | carbonyl | 4′-pyridinecarbonyl | 3.52 ± 0.62 | 3.98 ± 0.38 |
| 8 | 10 | 4′-pyridinecarbonyl | angeloyl | 33.79 ± 0.40 | 35.77 ± 2.11 |
| 9 | 11 | 4′-pyridinecarbonyl | angeloyl | 35.27 ± 1.09 | 34.77 ± 2.19 |
| 10 | 12 | OH | 4′-pyridinecarbonyl | 1.27 ± 0.20 µM | 1.83 ± 0.83 µM |
| 11 | Cisplatin | 4.23 ± 0.40 µM | 3.86 ± 0.46 µM | ||
| 12 | Doxorubicin | 37.54 ± 4.41 | 43.37 ± 5.60 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinsook, S.; Buaban, K.; Iksen, I.; Petsri, K.; Innets, B.; Chansriniyom, C.; Suwanborirux, K.; Yokoya, M.; Saito, N.; Pongrakhananon, V.; et al. Light-Mediated Transformation of Renieramycins and Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells. Mar. Drugs 2023, 21, 400. https://doi.org/10.3390/md21070400
Sinsook S, Buaban K, Iksen I, Petsri K, Innets B, Chansriniyom C, Suwanborirux K, Yokoya M, Saito N, Pongrakhananon V, et al. Light-Mediated Transformation of Renieramycins and Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells. Marine Drugs. 2023; 21(7):400. https://doi.org/10.3390/md21070400
Chicago/Turabian StyleSinsook, Suwimon, Koonchira Buaban, Iksen Iksen, Korrakod Petsri, Bhurichaya Innets, Chaisak Chansriniyom, Khanit Suwanborirux, Masashi Yokoya, Naoki Saito, Varisa Pongrakhananon, and et al. 2023. "Light-Mediated Transformation of Renieramycins and Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells" Marine Drugs 21, no. 7: 400. https://doi.org/10.3390/md21070400
APA StyleSinsook, S., Buaban, K., Iksen, I., Petsri, K., Innets, B., Chansriniyom, C., Suwanborirux, K., Yokoya, M., Saito, N., Pongrakhananon, V., Chanvorachote, P., & Chamni, S. (2023). Light-Mediated Transformation of Renieramycins and Semisynthesis of 4′-Pyridinecarbonyl-Substituted Renieramycin-Type Derivatives as Potential Cytotoxic Agents against Non-Small-Cell Lung Cancer Cells. Marine Drugs, 21(7), 400. https://doi.org/10.3390/md21070400