
Ascomylactam C Induces an Immunogenic Cell Death Signature via Mitochondria-Associated ER Stress in Lung Cancer and Melanoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
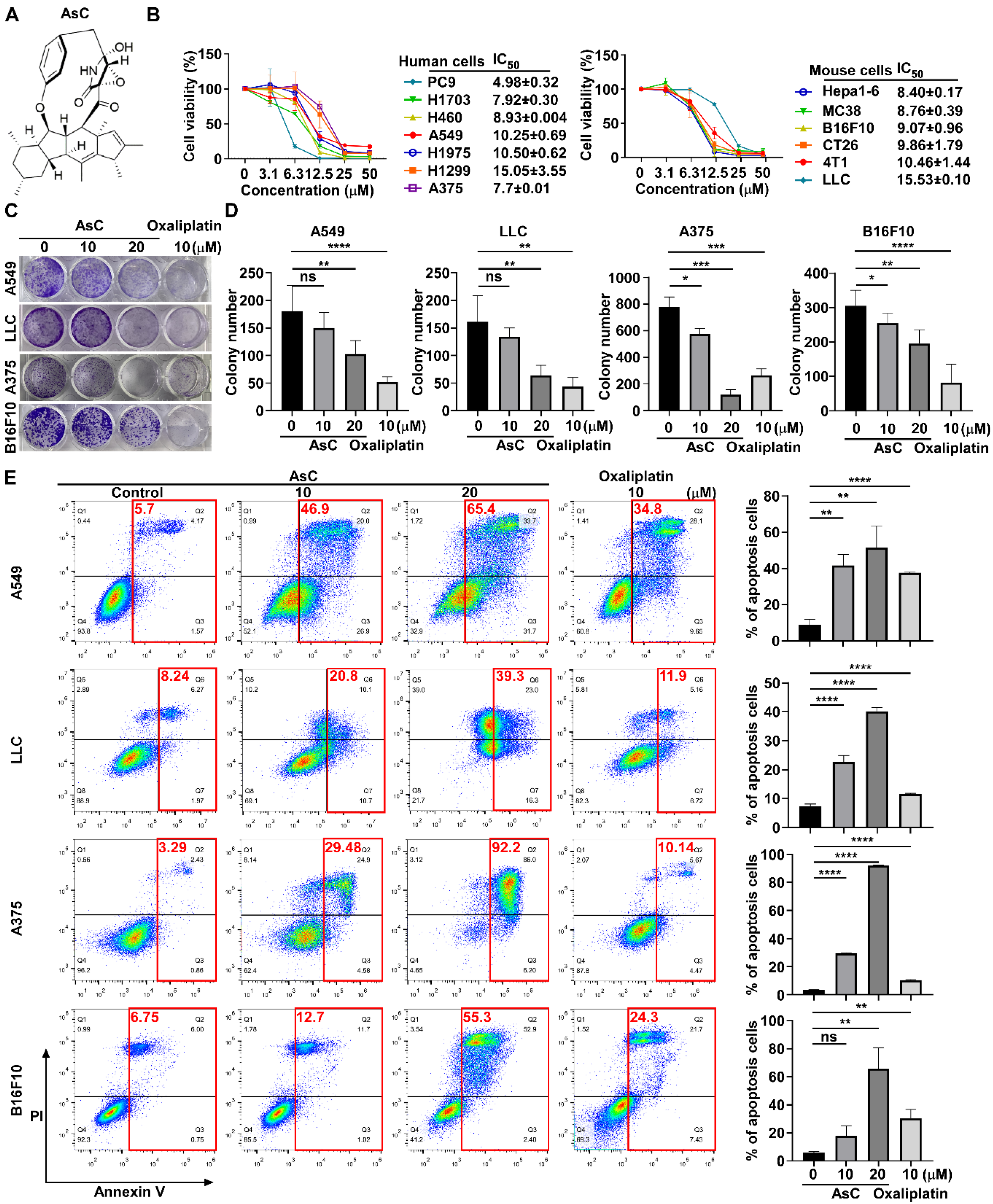
2.1. AsC Reduces Lung Cancer and Melanoma Cell Viability In Vitro
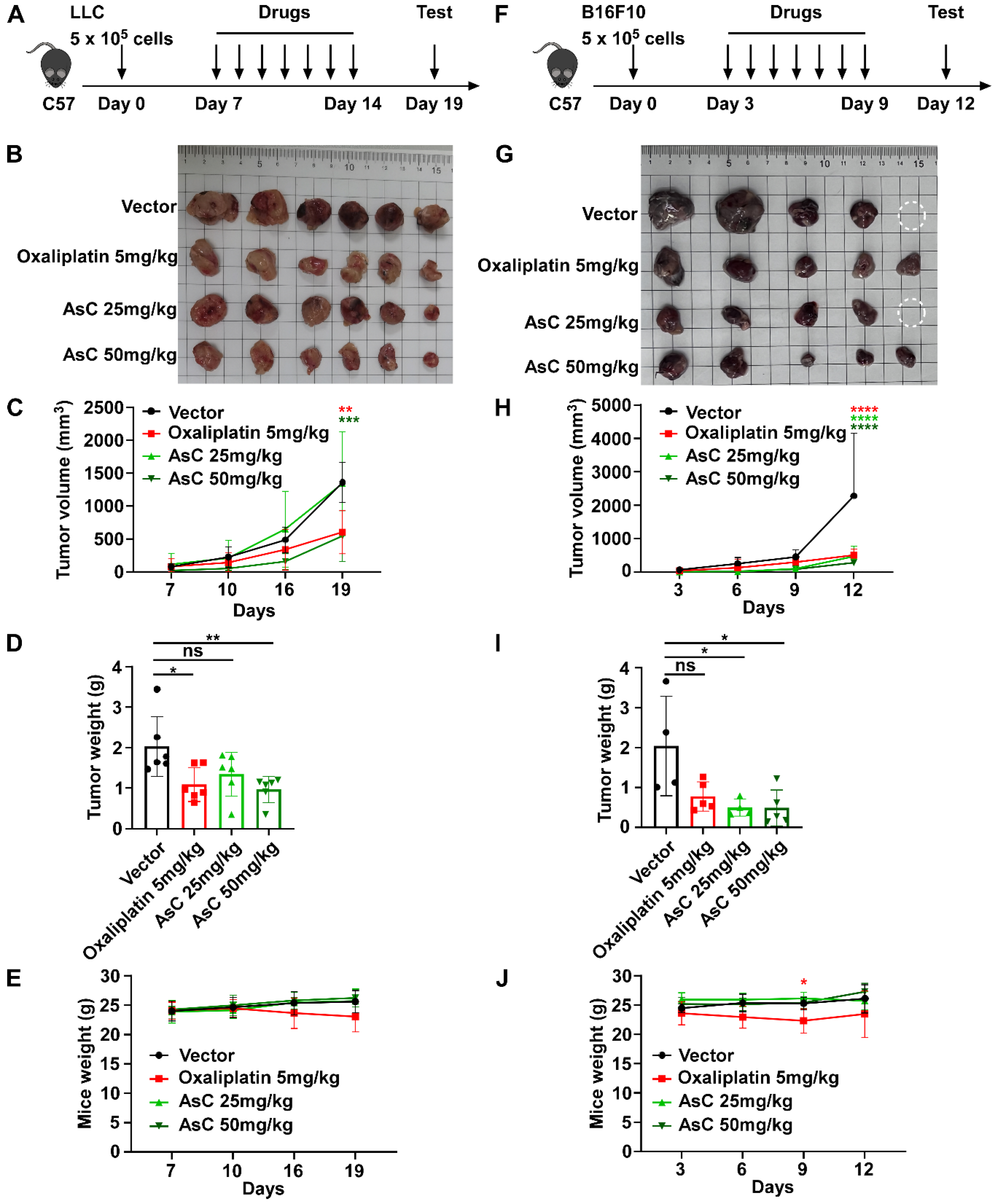
2.2. AsC Suppresses Lung Cancer and Melanoma Cell Viability In Vivo
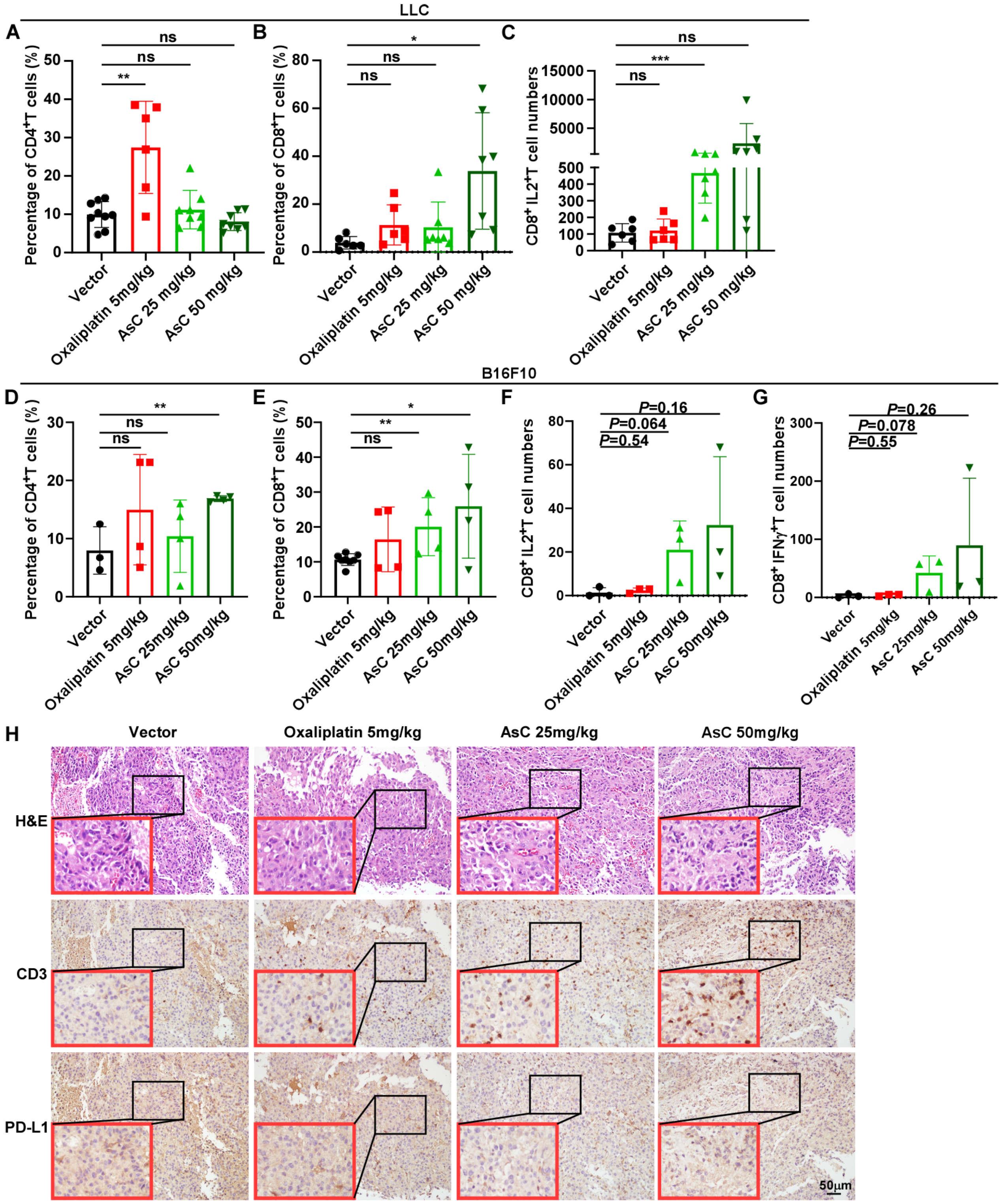
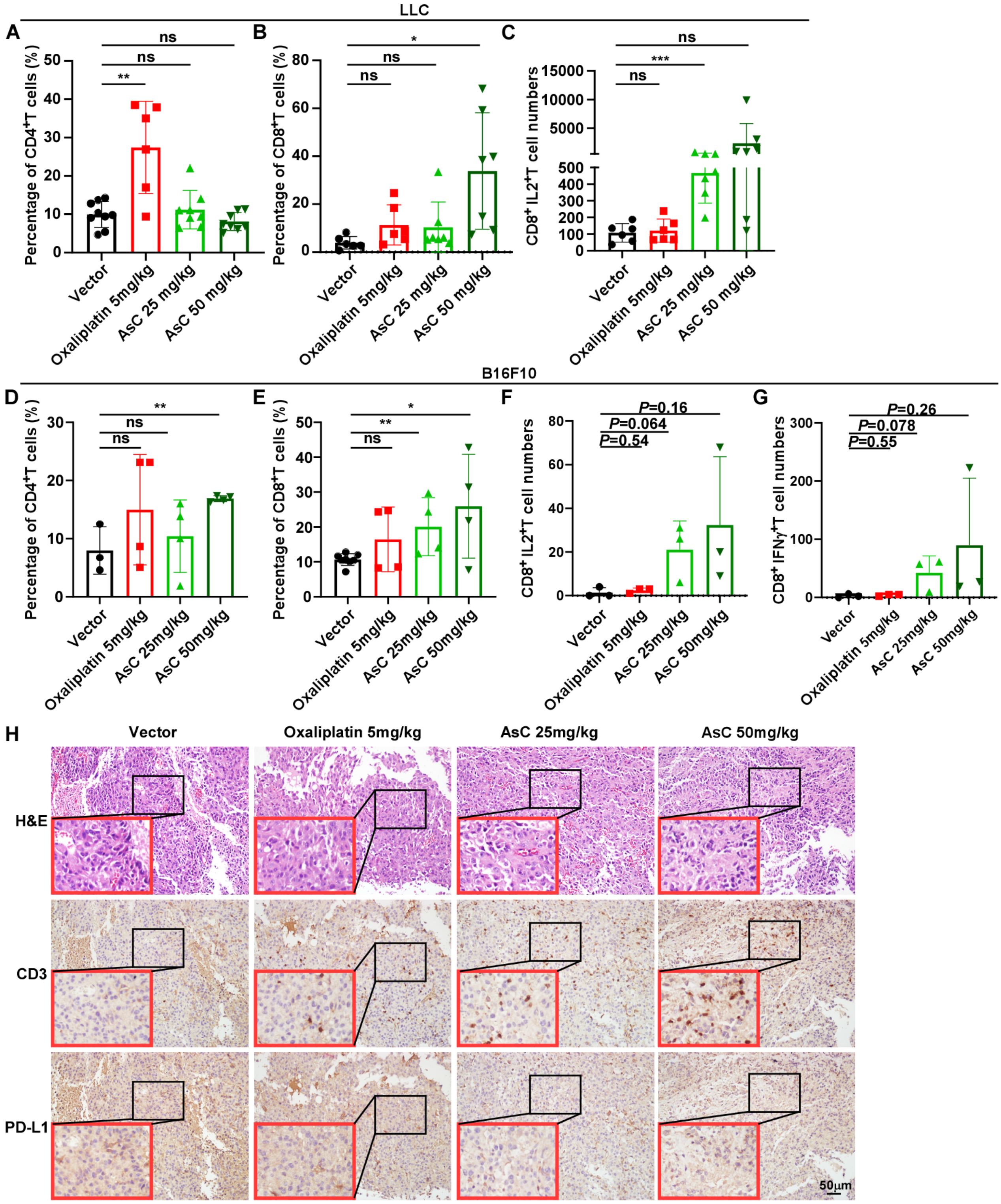
2.3. AsC Increases Infiltration of CD8+ T Cells in Lung Cancer and Melanoma Tissues
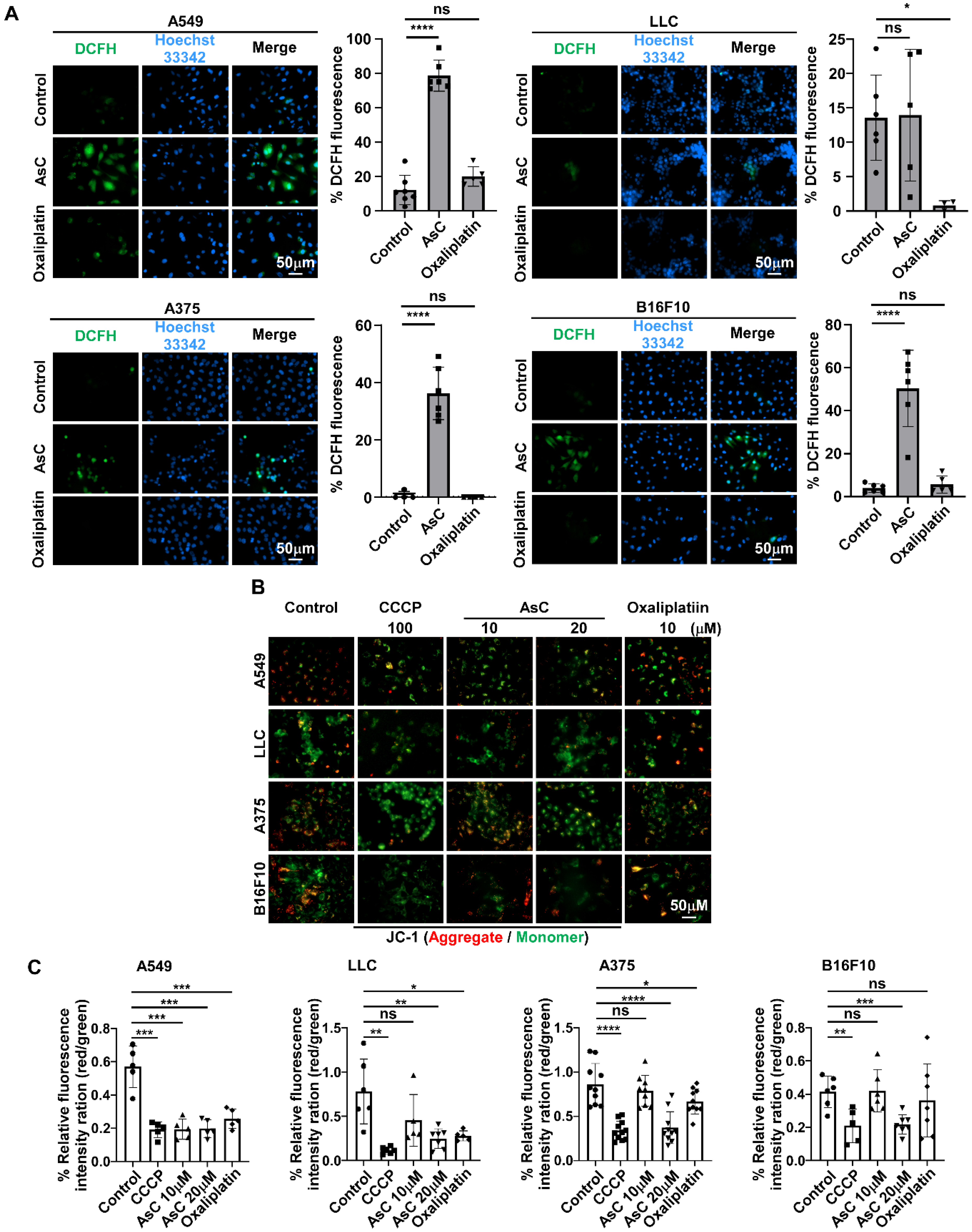
2.4. AsC Induces ROS Accumulation and Mitochondrial Membrane Depolarization in Lung Cancer and Melanoma Cells
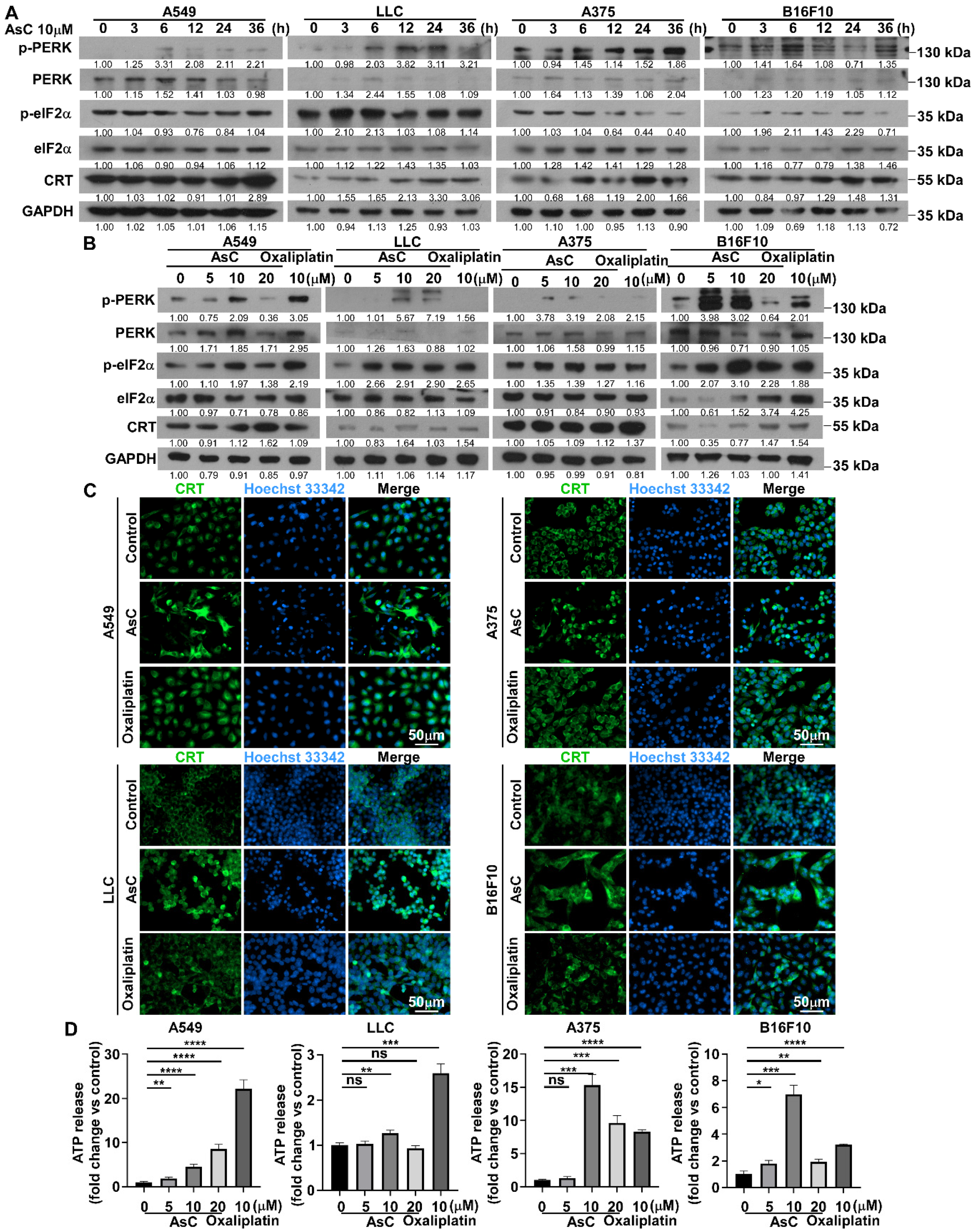
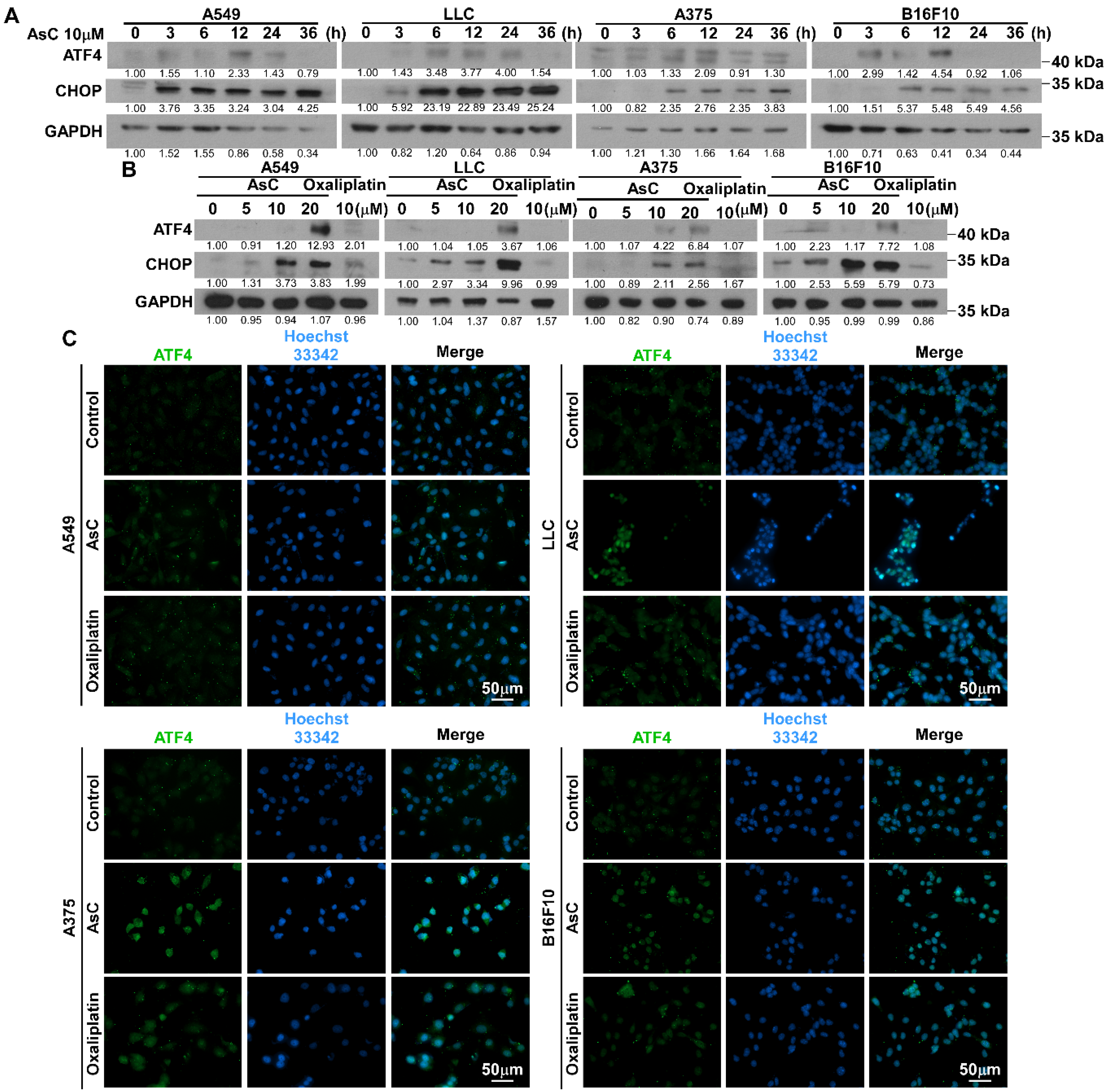
2.5. AsC Induces DAMPs and ICD in Lung Cancer and Melanoma Cells
2.6. AsC Promotes DC Maturation by Upregulating ROS Production
3. Discussion
4. Materials and Methods
4.1. Preparation of AsC
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Clonogenic Assay
4.5. Cell Apoptosis Analysis by Flow Cytometry
4.6. Western Blotting Analysis
4.7. Chemicals and Fluorescent Probes for Studying ROS Generation
4.8. In Vivo Assay
4.9. H&E Staining
4.10. Immunohistochemistry (IHC) Staining
4.11. Tumor-Infiltrating Lymphocytes (TIL) Analysis by Flow Cytometric
4.12. Immunofluorescence Staining
4.13. JC-1 Mitochondrial Membrane Potential Assay
4.14. ATP Release Assay
4.15. In Vitro DC Maturation Assays
4.16. Detection of Intracellular ROS
4.17. Statistical Analysis
4.18. Materials
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Won, M.; Levine, M.S.; Noh, Y.; Sedgwick, A.C.; Kim, J.S.; Sessler, J.L.; Arambula, J.F. Metal-based anticancer agents as immunogenic cell death inducers: The past, present, and future. Chem. Soc. Rev. 2022, 51, 1212–1233. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shan, Y.; Ge, K.; Lu, J.; Kong, W.; Jia, C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell. Oncol. 2020, 43, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S.; Yamazaki, T.; Iribarren, K.; et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer metal drugs and immunogenic cell death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef]
- Kepp, O.; Semeraro, M.; Bravo-San Pedro, J.M.; Bloy, N.; Buqué, A.; Huang, X.; Zhou, H.; Senovilla, L.; Kroemer, G.; Galluzzi, L. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin. Cancer Biol. 2015, 33, 86–92. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Martin, S.; Garg, A.D.; Agostinis, P. The PERKs of damage-associated molecular patterns mediating cancer immunogenicity: From sensor to the plasma membrane and beyond. Semin. Cancer Biol. 2015, 33, 74–85. [Google Scholar] [CrossRef]
- Li, X.; Zheng, J.; Chen, S.; Meng, F.D.; Ning, J.; Sun, S.L. Oleandrin, a cardiac glycoside, induces immunogenic cell death via the PERK/elF2α/ATF4/CHOP pathway in breast cancer. Cell Death Dis. 2021, 12, 314. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef] [PubMed]
- Chuang, A.W.; Kepp, O.; Kroemer, G.; Bezu, L. Chapter One—Endoplasmic reticulum stress in the cellular release of damage-associated molecular patterns. In International Review of Cell and Molecular Biology; Kepp, O., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 350, pp. 1–28. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Huang, Y.; Liu, L.; He, J.; Wang, L.; Yuan, J.; She, Z. Ascomylactams A-C, Cytotoxic 12- or 13-Membered-Ring Macrocyclic Alkaloids Isolated from the Mangrove Endophytic Fungus Didymella sp. CYSK-4, and Structure Revisions of Phomapyrrolidones A and C. J. Nat. Prod. 2019, 82, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Bakshi, R.K.; Ingram, J.T.; Hendrickson, R.C.; Lefkowitz, E.J.; Crossman, D.K.; Harrington, L.E.; Weaver, C.T.; Zajac, A.J. Intrinsic IL-2 production by effector CD8 T cells affects IL-2 signaling and promotes fate decisions, stemness, and protection. Sci. Immunol. 2022, 7, eabl6322. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Yu, Z.; Li, P.; Oh, J.; Spolski, R.; Zhao, L.; Glassman, C.R.; Yamamoto, T.N.; Chen, Y.; Golebiowski, F.M.; et al. An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature 2021, 597, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.S.; Chen, Y.; Hou, C.; Liu, Y.H.; Su, G.F.; Liang, H.; Chen, Z.F. Potent Zinc(II)-Based Immunogenic Cell Death Inducer Triggered by ROS-Mediated ERS and Mitochondrial Ca2+ Overload. J. Med. Chem. 2023, 66, 10497–10509. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Ren, J.; He, X.; Shi, H.; Zhang, F.; Li, H.; Tong, R. ROS-triggered nanoinducer based on dermatan sulfate enhances immunogenic cell death in melanoma. J. Control. Release Off. J. Control. Release Soc. 2022, 348, 22–33. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, J.; Sun, S.; Lin, W.; Li, Y.; Lu, Q.; Li, F.; Yang, Z.; Lu, Y.; Liu, W. Mecheliolide elicits ROS-mediated ERS driven immunogenic cell death in hepatocellular carcinoma. Redox Biol. 2022, 54, 102351. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.Y.; Liu, X.S.; Chen, H.Z.; Ai, Y.L.; Cheng, K.; Sun, R.Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef]
- Guo, L.; Gong, H.; Tang, T.L.; Zhang, B.K.; Zhang, L.Y.; Yan, M. Crizotinib and Sunitinib Induce Hepatotoxicity and Mitochondrial Apoptosis in L02 Cells via ROS and Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 620934. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 2019, 10, 3349. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Kim, D.Y.; Pyo, A.; Yun, M.; Thangam, R.; You, S.H.; Zhang, Y.; Jung, Y.R.; Nguyen, D.H.; Venu, A.; Kim, H.S.; et al. Imaging Calreticulin for Early Detection of Immunogenic Cell Death During Anticancer Treatment. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 956–960. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Sáez, P.J.; Vargas, P.; Shoji, K.F.; Harcha, P.A.; Lennon-Duménil, A.M.; Sáez, J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci. Signal. 2017, 10, eaah7107. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Ganesan, J.; Müller, T.; Dürr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Jüttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, F.; Hasegawa, K.; Ando, K.; Ogawa, T.; Hara, A. Antibiotic Antitumor GKK1032 Manufacture with Penicillium. Kokat Tokkyo Koho Japanese Patent 2001247574 A2, 11 September 2001. [Google Scholar]
- Uchiro, H.; Kato, R.; Sakuma, Y.; Takagi, Y.; Arai, Y.; Hasegawa, D. Synthetic studies of GKK1032s: The asymmetric synthesis of the decahydrofluorene skeleton via a novel cyclization of silyl enol ether and sequential retro Diels–Alder and intramolecular Diels–Alder reactions. Tetrahedron Lett. 2011, 52, 6242–6245. [Google Scholar] [CrossRef]
- Shiono, Y.; Shimanuki, K.; Hiramatsu, F.; Koseki, T.; Tetsuya, M.; Fujisawa, N.; Kimura, K. Pyrrospirones A and B, apoptosis inducers in HL-60 cells, from an endophytic fungus, Neonectria ramulariae Wollenw KS-246. Bioorg. Med. Chem. Lett. 2008, 18, 6050–6053. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, S.; Fujisawa, N.; Yoshida, J.; Watanabe, M.; Dan, S.; Yamori, T.; Shiono, Y.; Kimura, K. Pyrrocidine A, a metabolite of endophytic fungi, has a potent apoptosis-inducing activity against HL60 cells through caspase activation via the Michael addition. J. Antibiot. 2016, 69, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, W.; Aly, A.H.; Wray, V.; Mándi, A.; Teiten, M.H.; Gaascht, F.; Orlikova, B.; Kassack, M.U.; Lin, W.; Diederich, M.; et al. Embellicines A and B: Absolute configuration and NF-κB transcriptional inhibitory activity. J. Med. Chem. 2013, 56, 2991–2999. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Huang, C.H.; Yu, J.C.; Zheng, Y.C.; Chen, Y.; She, Z.G.; Yuan, J. A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway. Mar. Drugs 2020, 18, 494. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, S.; Huang, L.; Li, Y.; Lu, Y.; Li, H.; Chen, G.; Meng, F.; Liu, G.L.; Yang, X.; et al. Reactive oxygen species-responsive and Raman-traceable hydrogel combining photodynamic and immune therapy for postsurgical cancer treatment. Nat. Commun. 2022, 13, 4553. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Wu, Y.; Xu, X.; Qian, X.; Lei, Y.; Liu, H.; Zhang, Z.; Li, Y. ROS-Responsive Nanocomplex of aPD-L1 and Cabazitaxel Improves Intratumor Delivery and Potentiates Radiation-Mediated Antitumor Immunity. Nano Lett. 2022, 22, 8312–8320. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Yamashita, N.; Ogawa, T.; Koseki, K.; Warabi, E.; Ohue, T.; Komatsu, M.; Matsushita, H.; Kakimi, K.; Kawakami, E.; et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J. Immunother. Cancer 2021, 9, e002954. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.; Jitkova, Y.; et al. Venetoclax enhances T cell-mediated antileukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Gandhi, S.; Yan, L.; Tokumaru, Y.; Wu, R.; Yamada, A.; Matsuyama, R.; Endo, I.; Takabe, K. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res. Treat. 2022, 194, 231–241. [Google Scholar] [CrossRef]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Torrente, L.; Prieto-Farigua, N.; Falzone, A.; Elkins, C.M.; Boothman, D.A.; Haura, E.B.; DeNicola, G.M. Inhibition of TXNRD or SOD1 overcomes NRF2-mediated resistance to β-lapachone. Redox Biol. 2020, 30, 101440. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Gsottberger, F.; Meier, C.; Ammon, A.; Parker, S.; Wendland, K.; George, R.; Petkovic, S.; Mellenthin, L.; Emmerich, C.; Lutzny-Geier, G.; et al. Targeted inhibition of protein synthesis renders cancer cells vulnerable to apoptosis by unfolded protein response. Cell Death Dis. 2023, 14, 561. [Google Scholar] [CrossRef] [PubMed]
- Sansone, C.; Bruno, A.; Piscitelli, C.; Baci, D.; Fontana, A.; Brunet, C.; Noonan, D.M.; Albini, A. Natural Compounds of Marine Origin as Inducers of Immunogenic Cell Death (ICD): Potential Role for Cancer Interception and Therapy. Cells 2021, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvær, E.; Gjertsen, B.T.; Bruserud, O. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556. [Google Scholar] [CrossRef]
- Bley, K.; Boorman, G.; Mohammad, B.; McKenzie, D.; Babbar, S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol. Pathol. 2012, 40, 847–873. [Google Scholar] [CrossRef]
- D’Eliseo, D.; Manzi, L.; Velotti, F. Capsaicin as an inducer of damage-associated molecular patterns (DAMPs) of immunogenic cell death (ICD) in human bladder cancer cells. Cell Stress Chaperones 2013, 18, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Humphreys, I.; Sahu, R.P.; Shi, Y.; Srivastava, S.K. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis Int. J. Program. Cell Death 2008, 13, 1465–1478. [Google Scholar] [CrossRef]
- Upadhaya, S.; Neftelinov, S.T.; Hodge, J.; Campbell, J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 2022, 21, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Saez-Ibanez, A.R.; Upadhaya, S.; Campbell, J. Immuno-oncology clinical trials take a turn beyond PD1/PDL1 inhibitors. Nat. Rev. Drug Discov. 2023, 22, 442–443. [Google Scholar] [CrossRef] [PubMed]
- Offringa, R.; Kötzner, L.; Huck, B.; Urbahns, K. The expanding role for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2022, 21, 821–840. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yu, J.; Chen, T.; Liu, W.; Huang, Y.; Li, J.; Zhang, B.; Zhu, G.; He, Z.; Long, Y.; et al. Design, synthesis, and biological evaluation of a series of new anthraquinone derivatives as anti-ZIKV agents. Eur. J. Med. Chem. 2023, 258, 115620. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Yan, H.; Zhang, B.; Zhu, G.; Yu, J.; Xiao, X.; He, W.; Chen, Y.; Gao, X.; She, Z.; et al. Ascomylactam C Induces an Immunogenic Cell Death Signature via Mitochondria-Associated ER Stress in Lung Cancer and Melanoma. Mar. Drugs 2023, 21, 600. https://doi.org/10.3390/md21120600
Huang Y, Yan H, Zhang B, Zhu G, Yu J, Xiao X, He W, Chen Y, Gao X, She Z, et al. Ascomylactam C Induces an Immunogenic Cell Death Signature via Mitochondria-Associated ER Stress in Lung Cancer and Melanoma. Marine Drugs. 2023; 21(12):600. https://doi.org/10.3390/md21120600
Chicago/Turabian StyleHuang, Yun, Hongmei Yan, Bingzhi Zhang, Ge Zhu, Jianchen Yu, Xuhan Xiao, Wenxuan He, Yan Chen, Xiaoxia Gao, Zhigang She, and et al. 2023. "Ascomylactam C Induces an Immunogenic Cell Death Signature via Mitochondria-Associated ER Stress in Lung Cancer and Melanoma" Marine Drugs 21, no. 12: 600. https://doi.org/10.3390/md21120600
APA StyleHuang, Y., Yan, H., Zhang, B., Zhu, G., Yu, J., Xiao, X., He, W., Chen, Y., Gao, X., She, Z., Li, M., & Yuan, J. (2023). Ascomylactam C Induces an Immunogenic Cell Death Signature via Mitochondria-Associated ER Stress in Lung Cancer and Melanoma. Marine Drugs, 21(12), 600. https://doi.org/10.3390/md21120600