
Simplified Synthesis of Renieramycin T Derivatives to Target Cancer Stem Cells via β-Catenin Proteasomal Degradation in Human Lung Cancer

 , ,
, , 
Abstract
:1. Introduction
2. Results
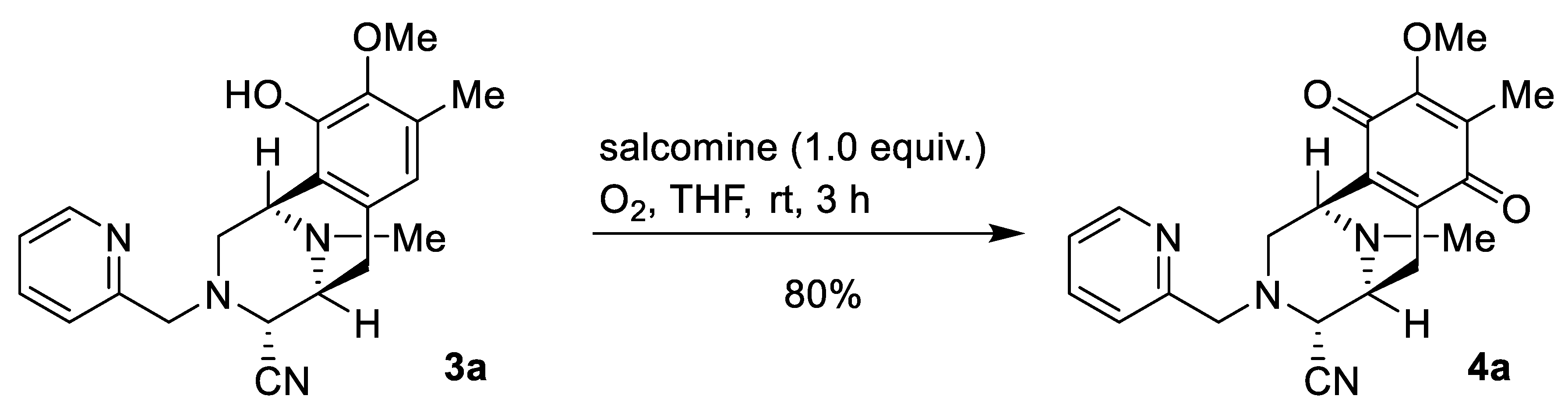
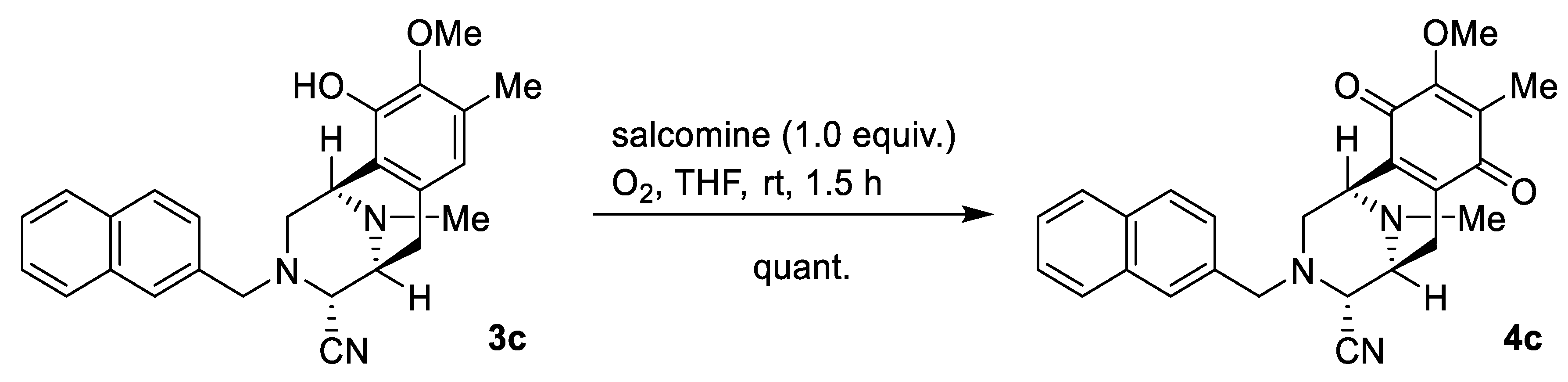
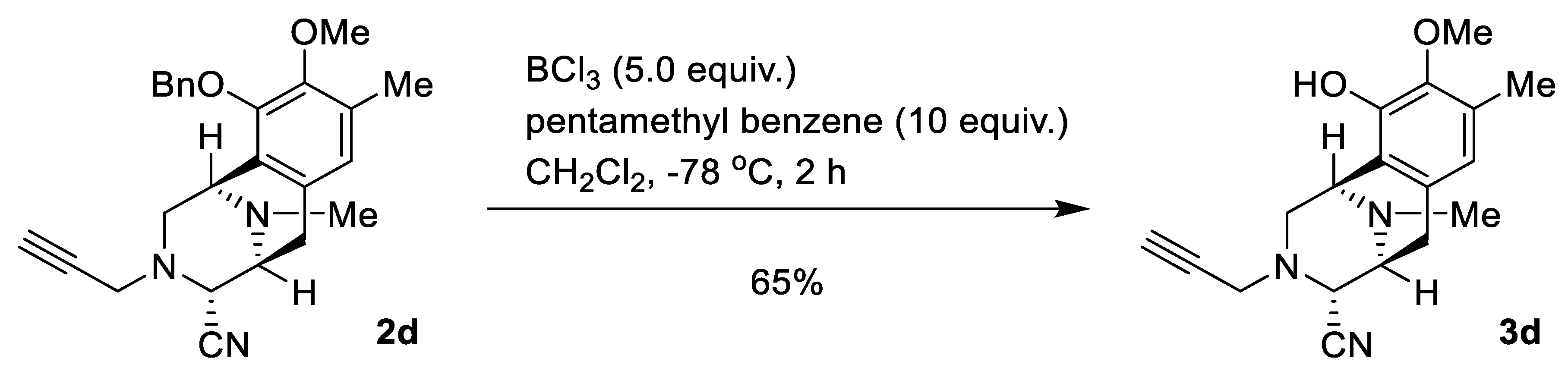
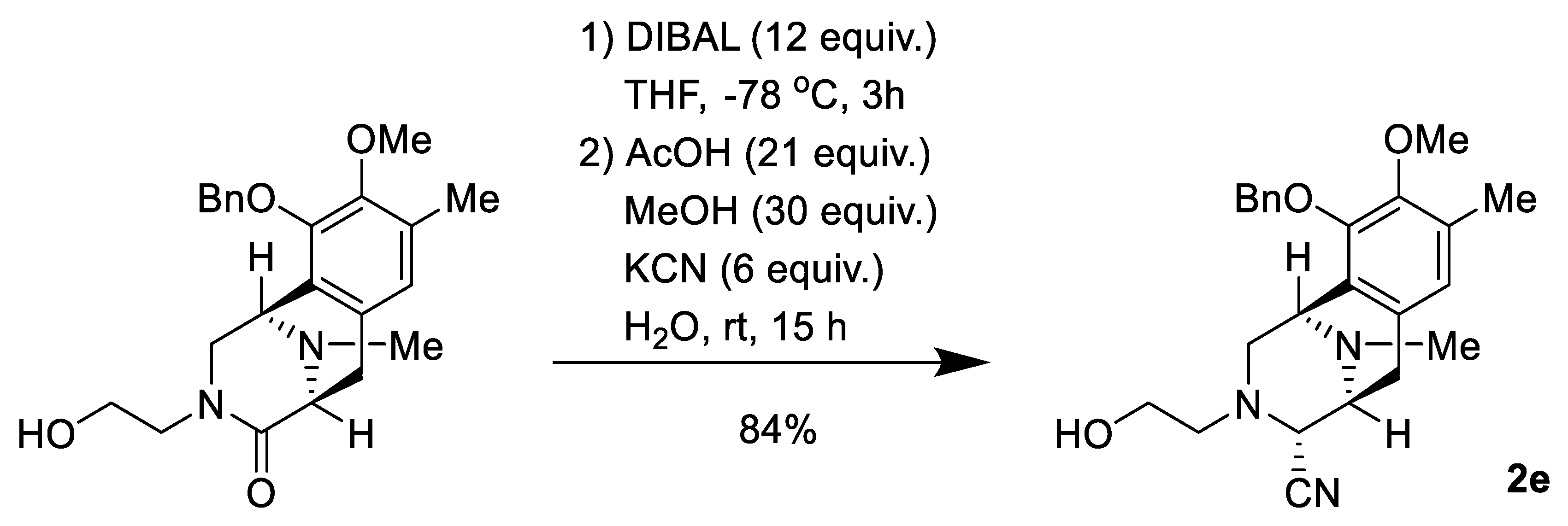
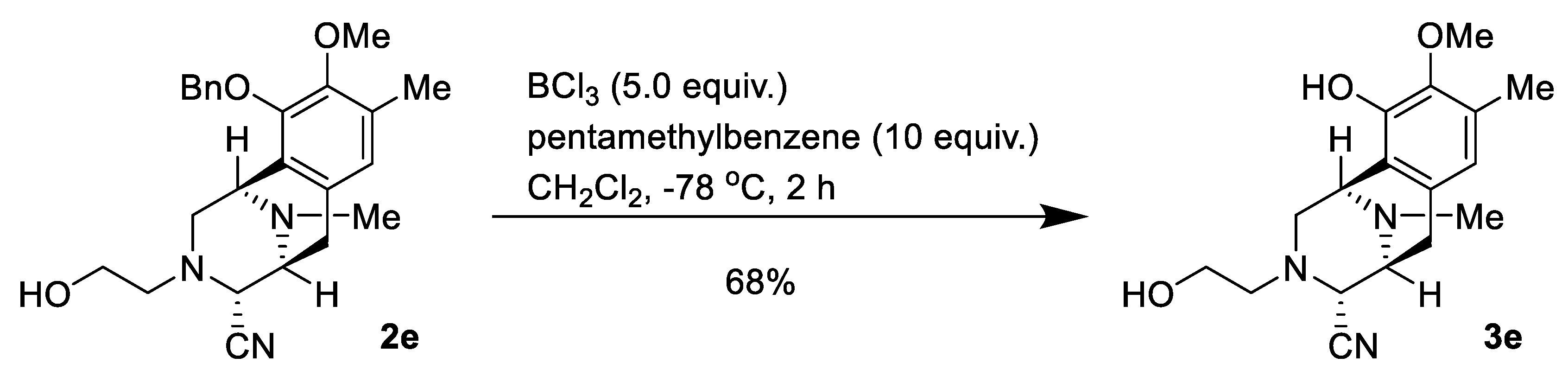
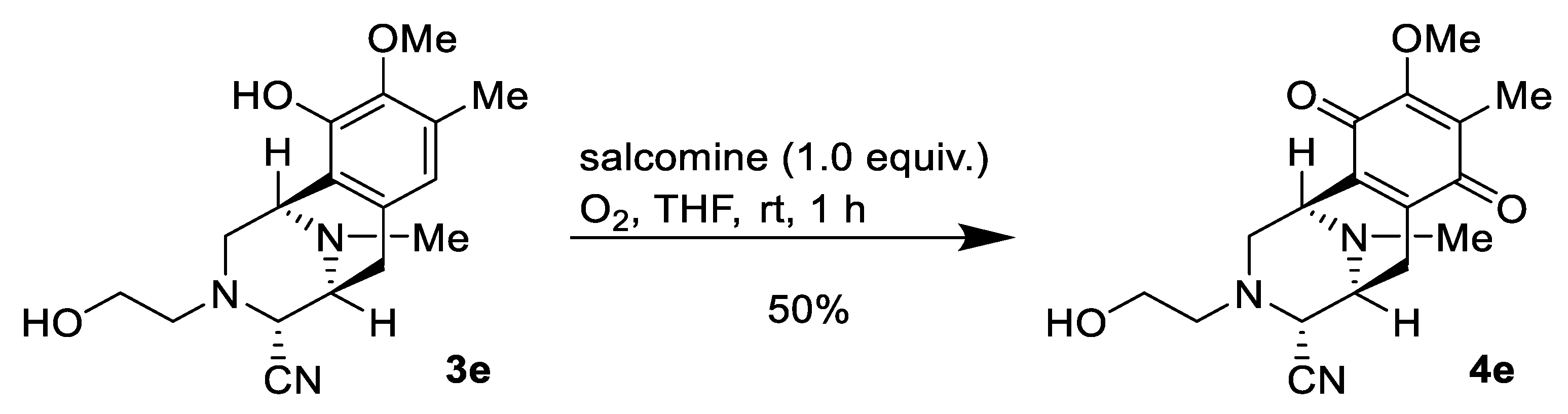
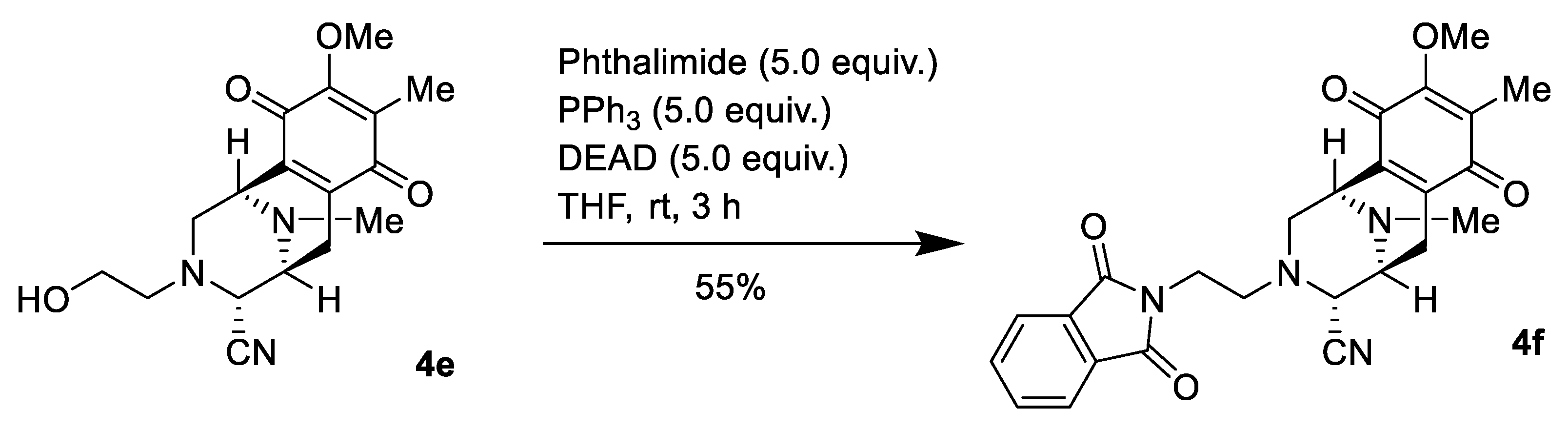
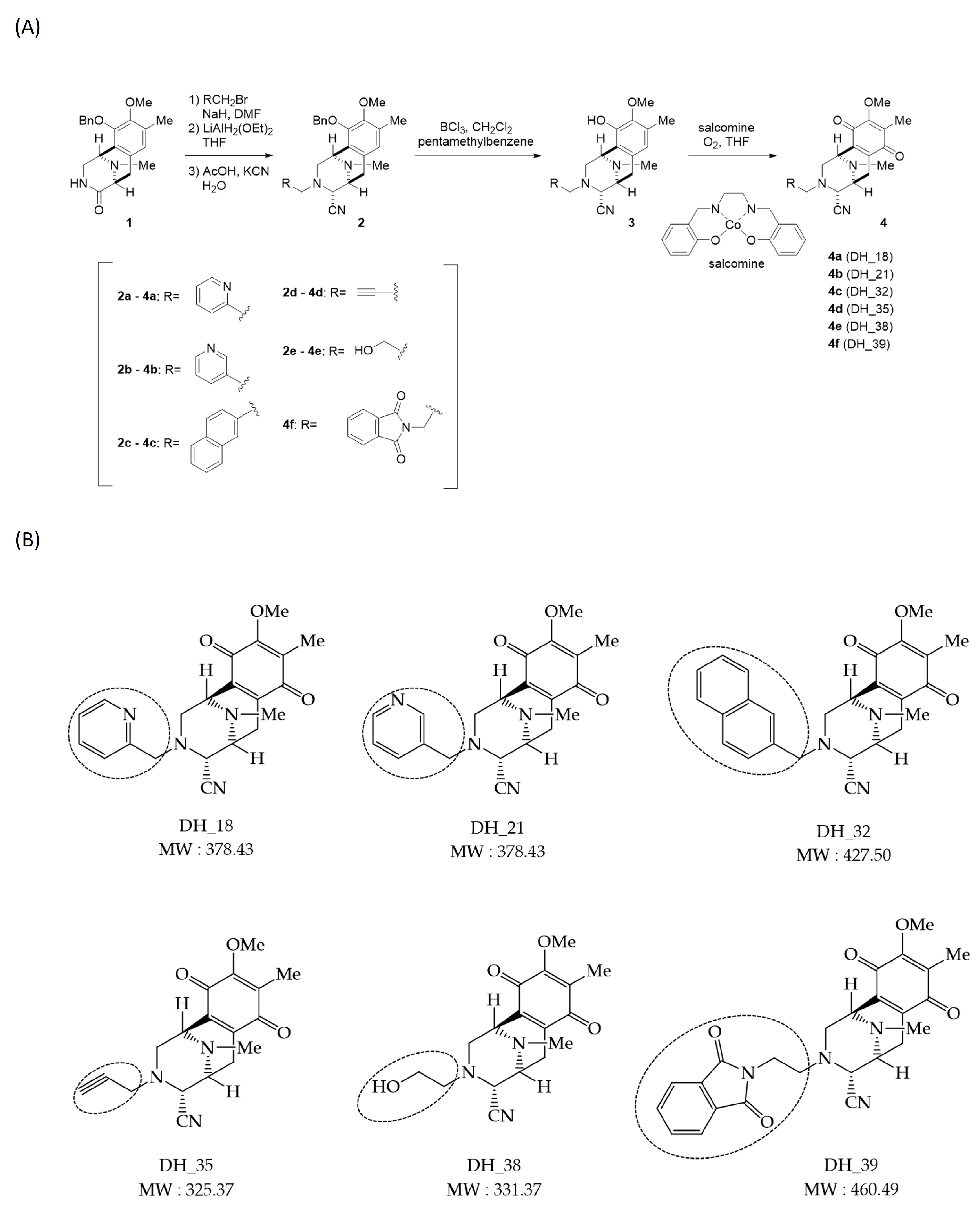
2.1. Synthesis of Right-Half RT Derivative DH_32
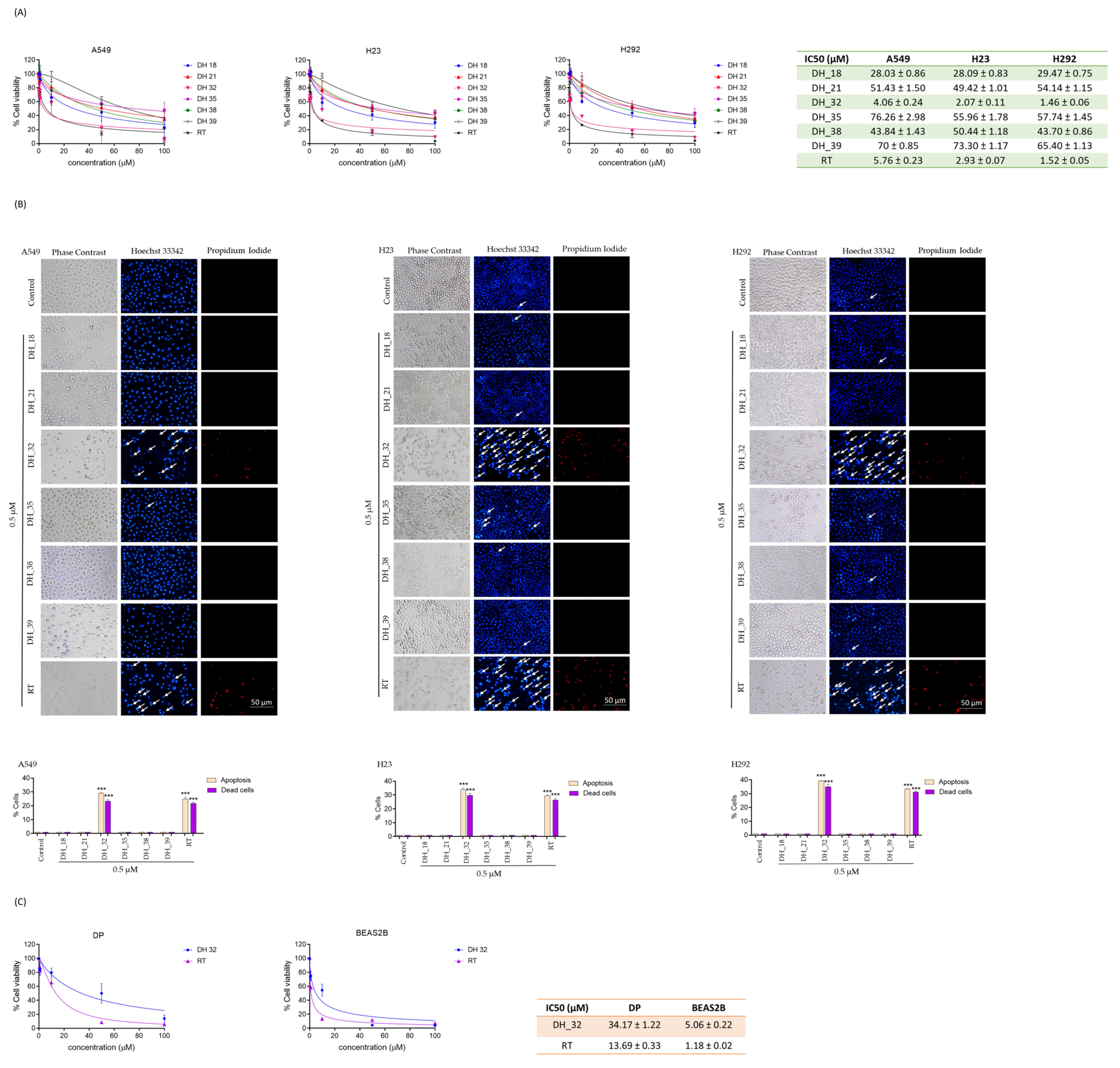
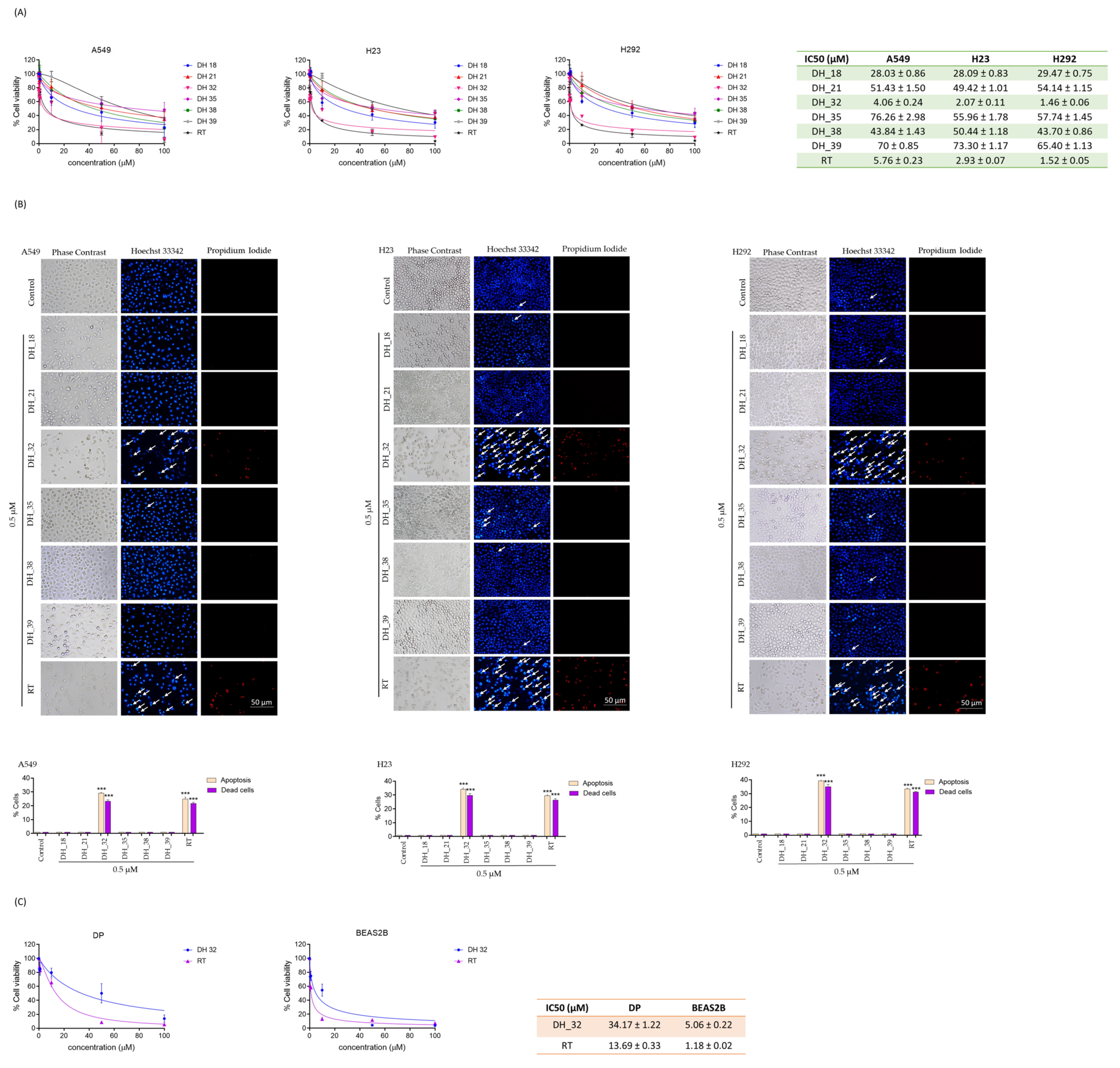
2.2. Assessing Cytotoxicity through an MTT Assay When Screening for Right-Half RT Analogs
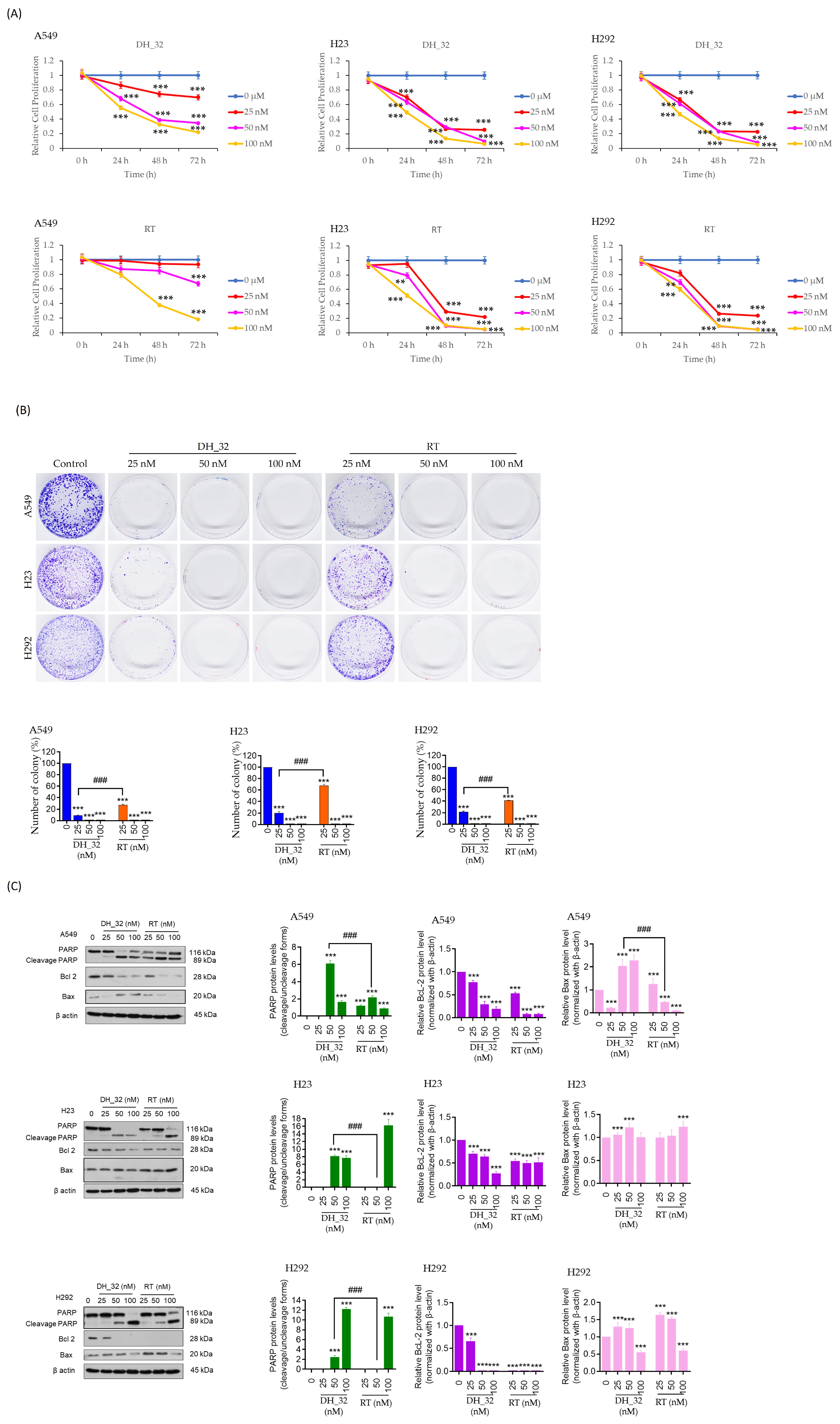
2.3. DH_32 Inhibits Proliferation, Decreases Colony Formation, and Affects Apoptosis-Related Proteins
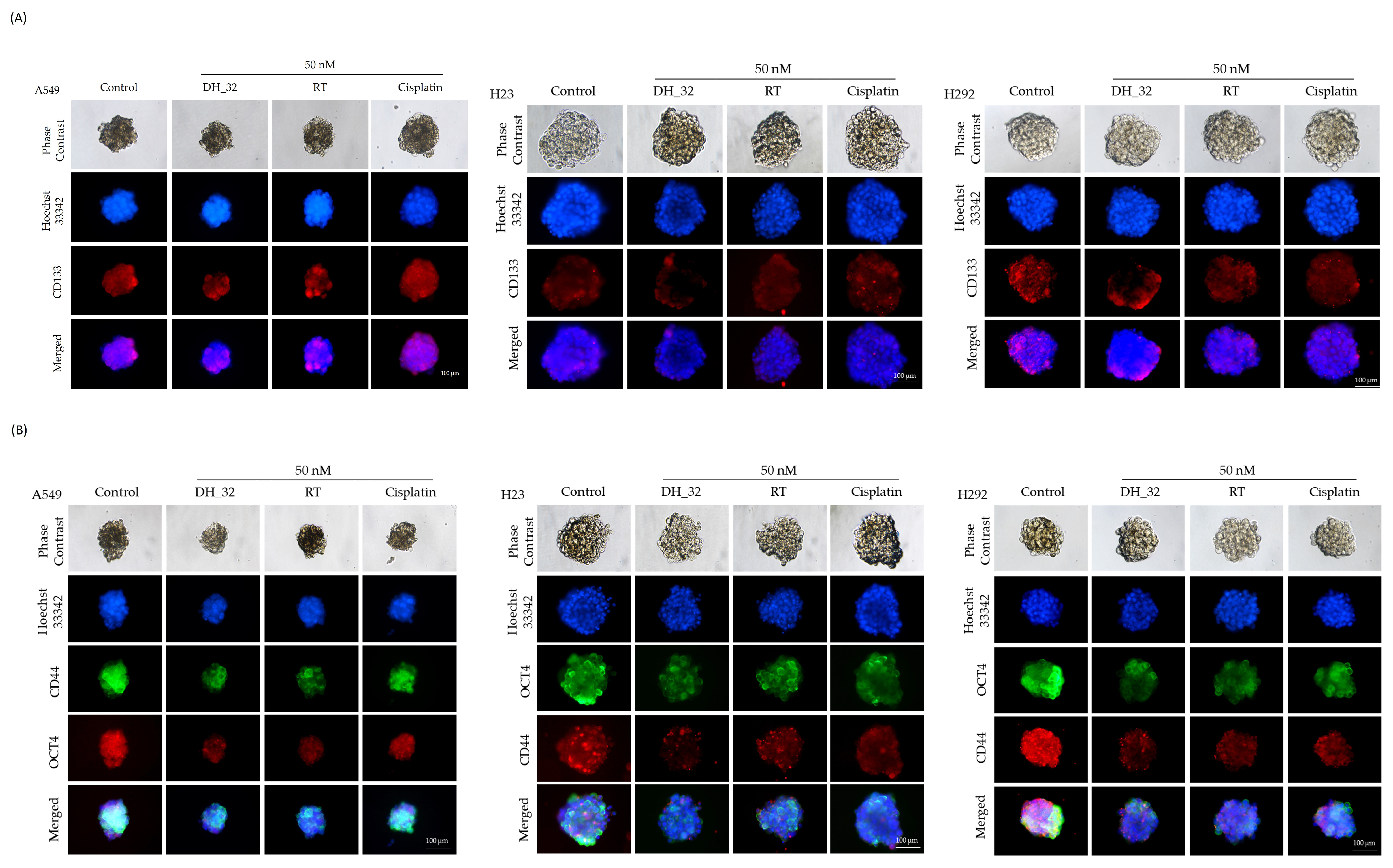
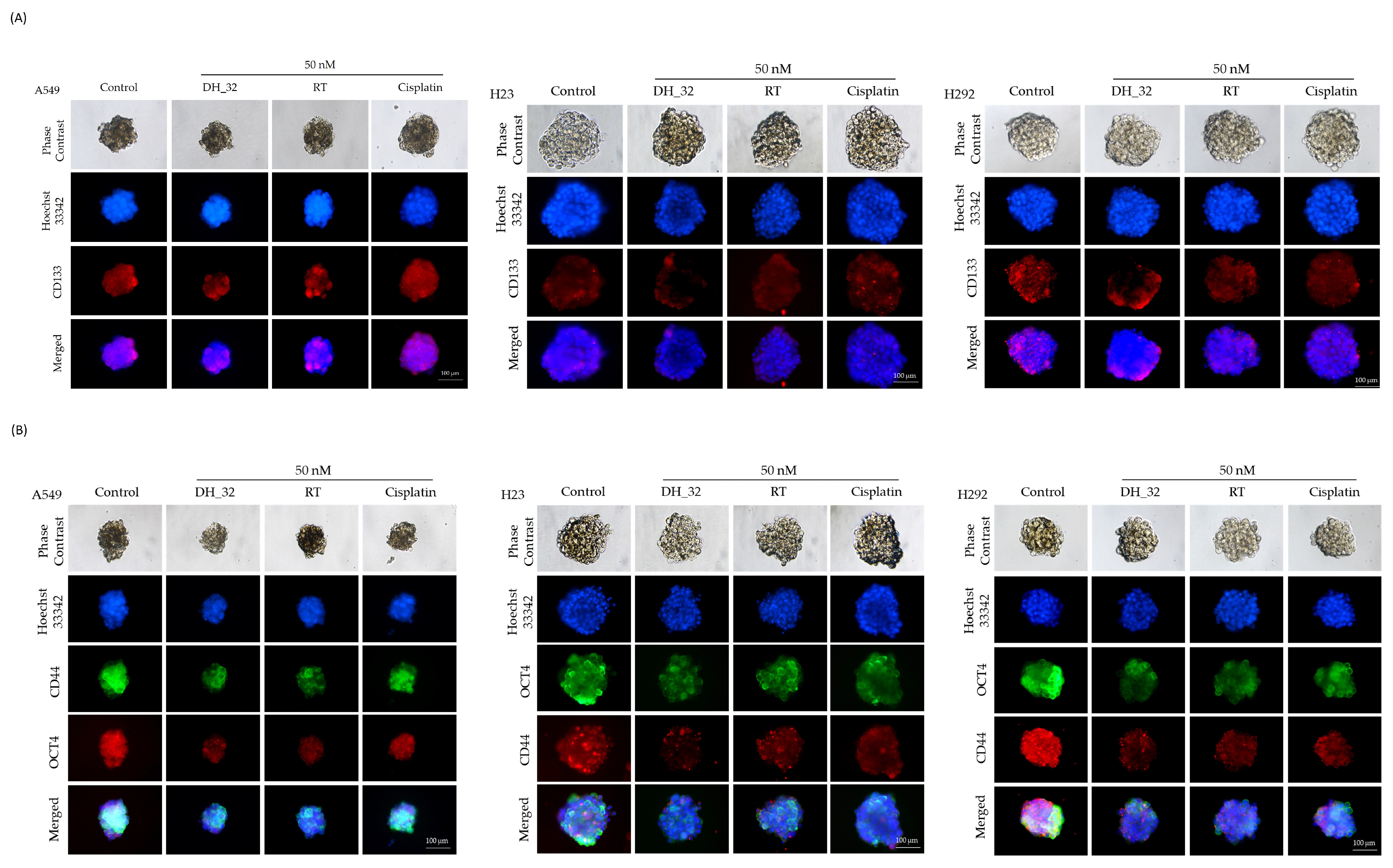
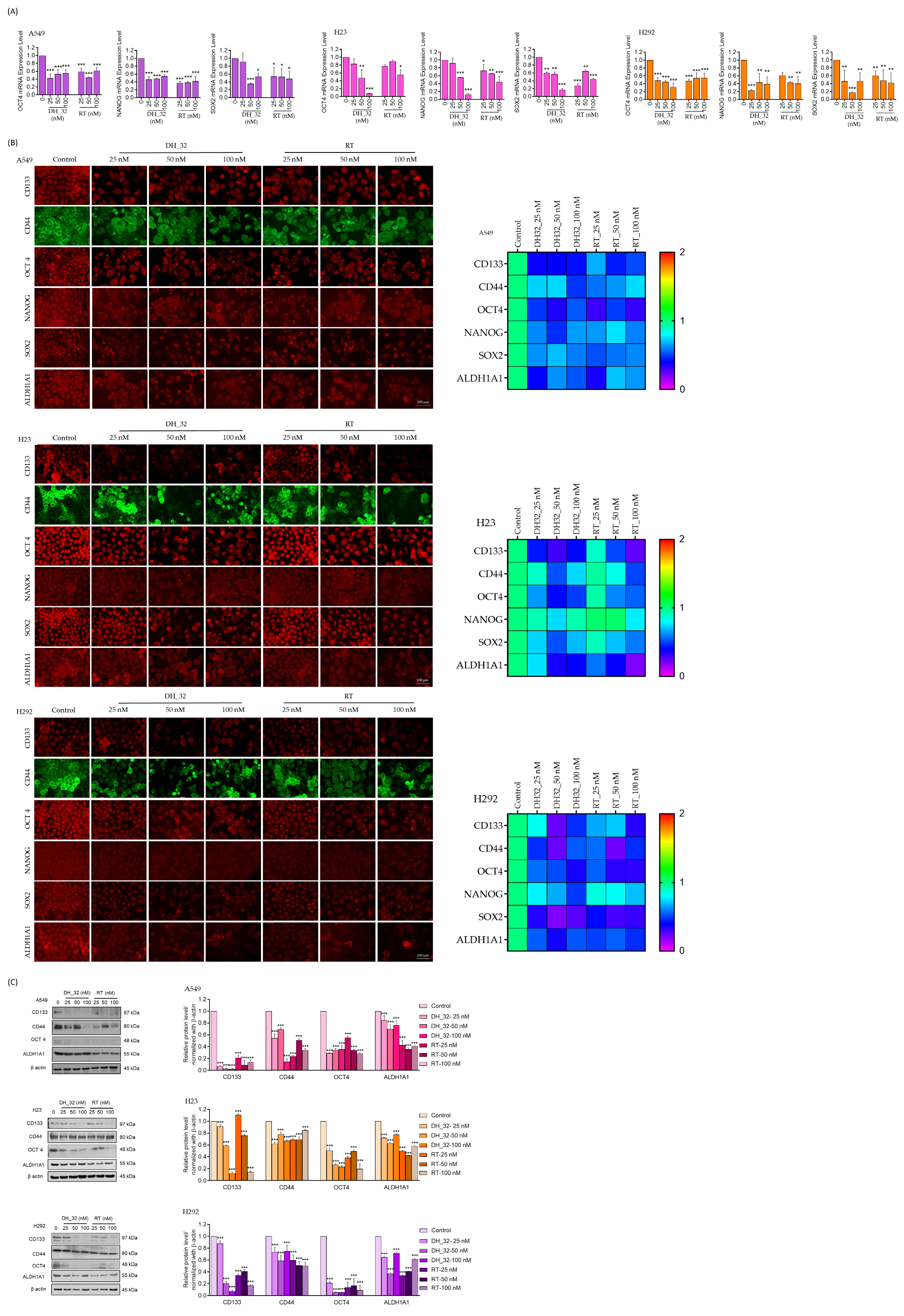
2.4. Inhibitory Effect of DH_32 on CSCs in Various Lung Cancer Cell Lines
2.5. DH_32 Suppression of CSC-like Phenotypes in Lung Cancer Cells
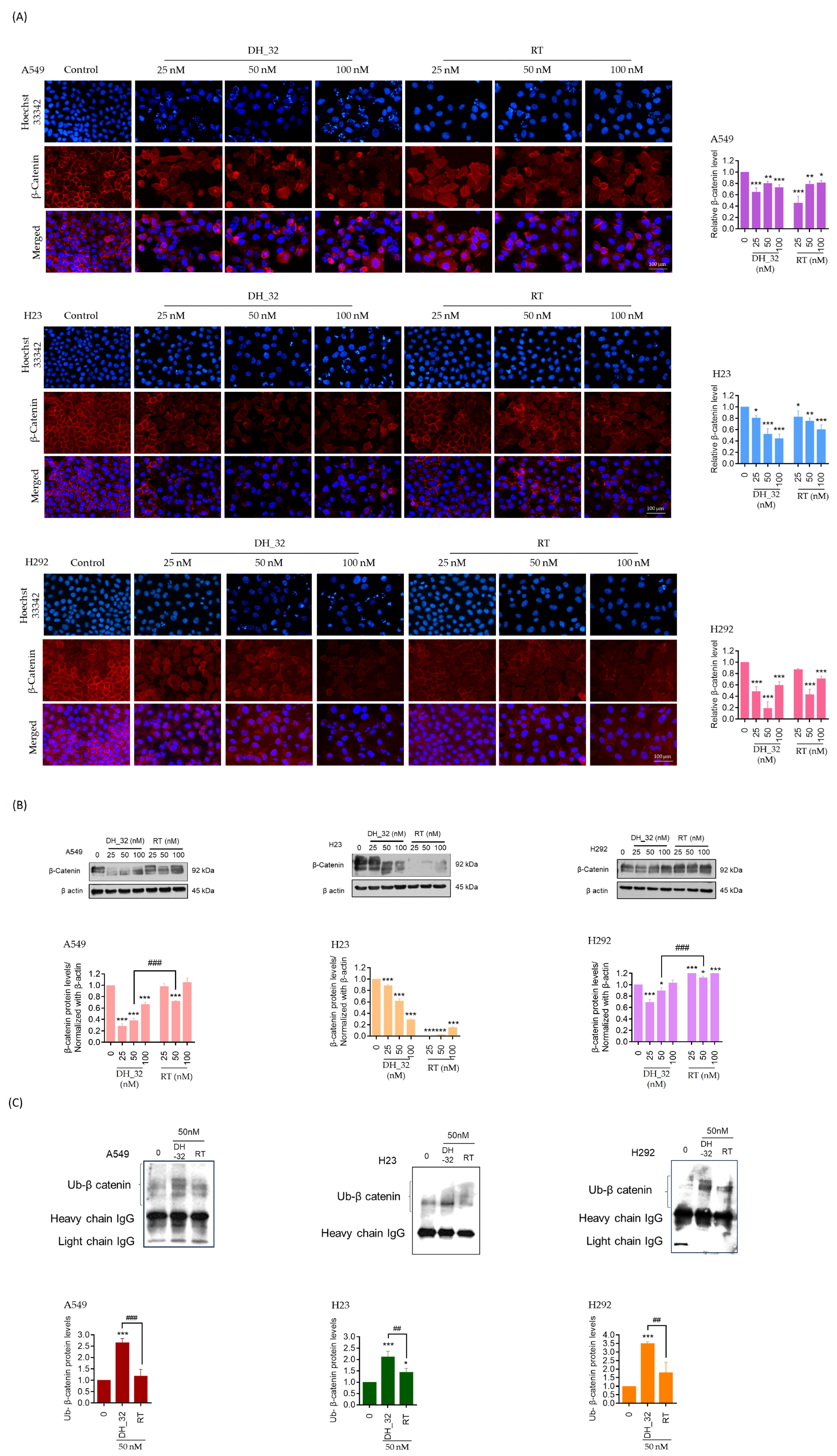
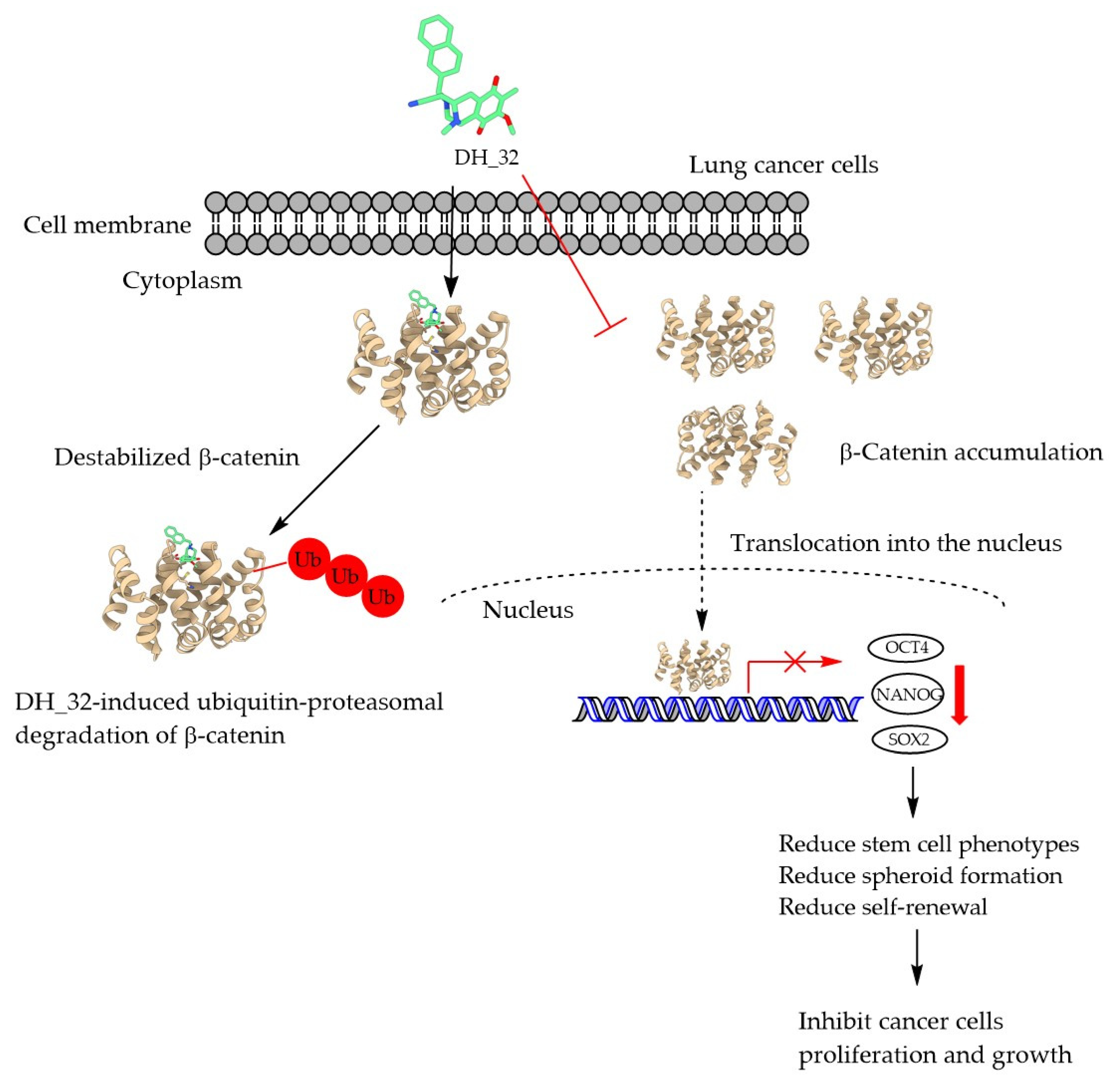
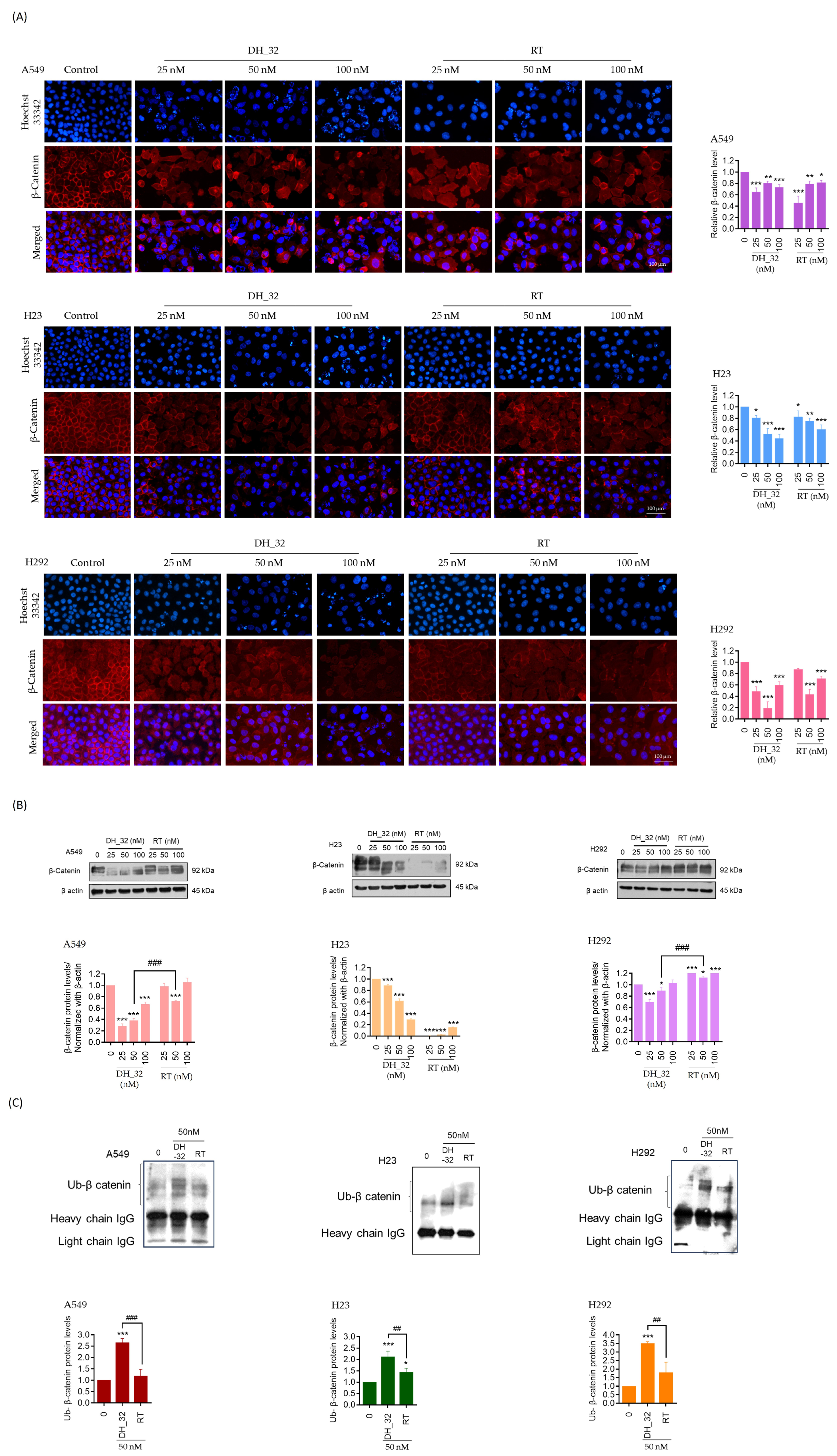
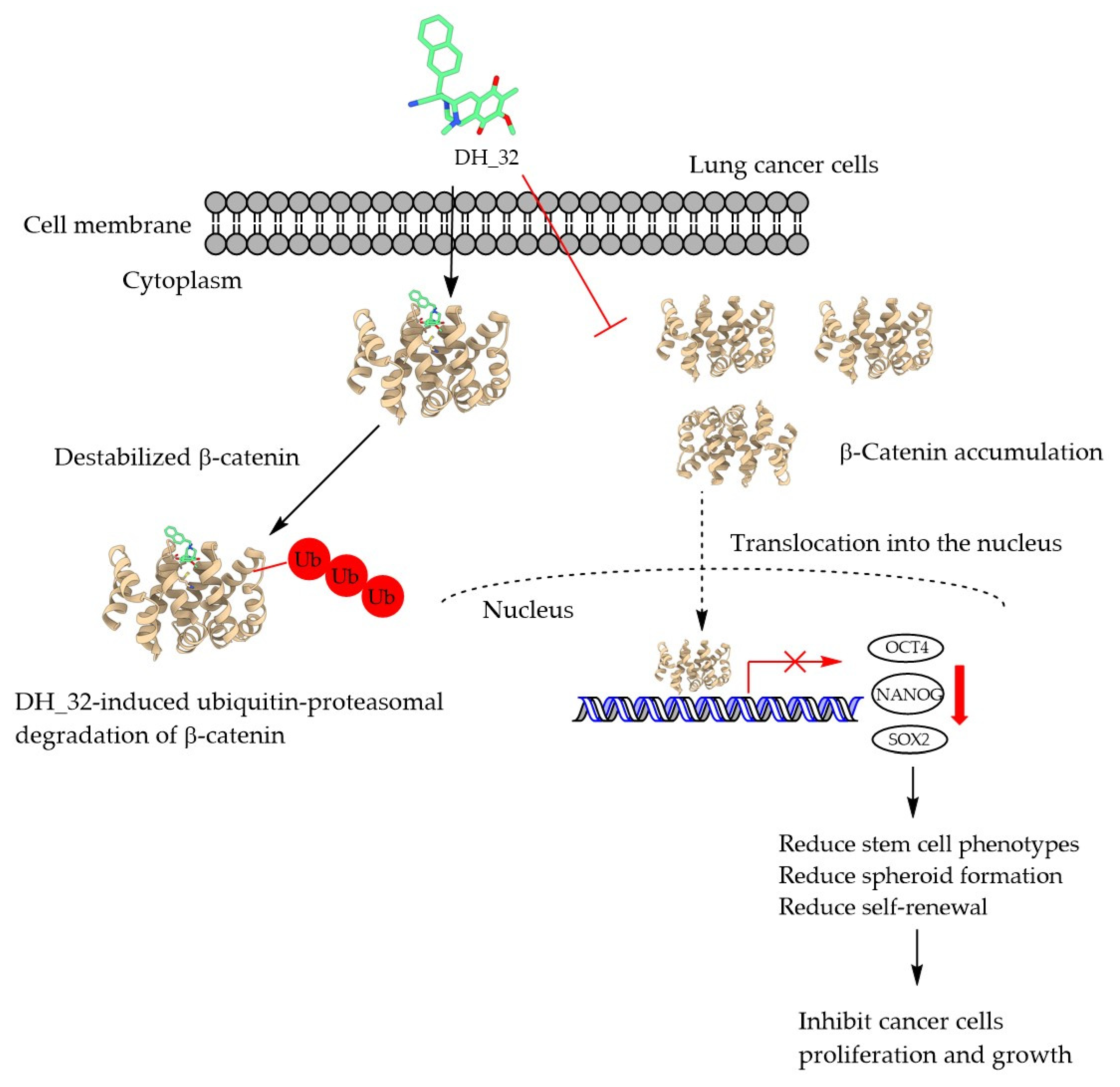
2.6. DH_32 Destabilizes β-Catenin and Facilitates Proteasomal Degradation
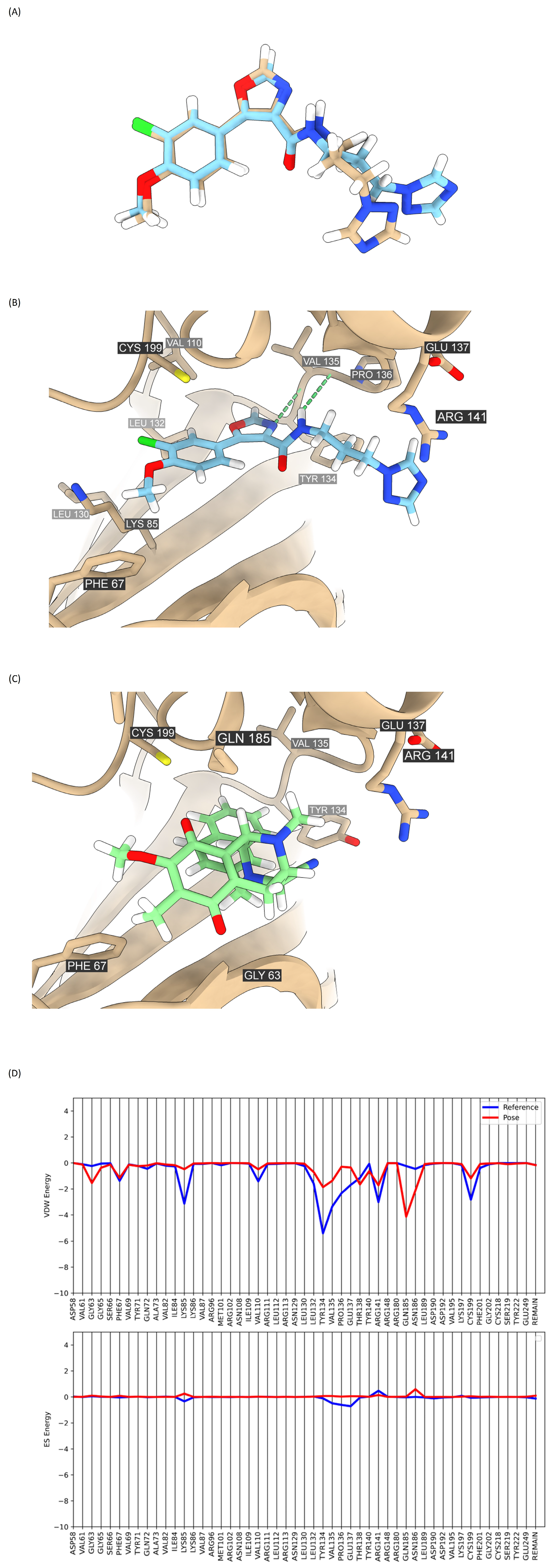
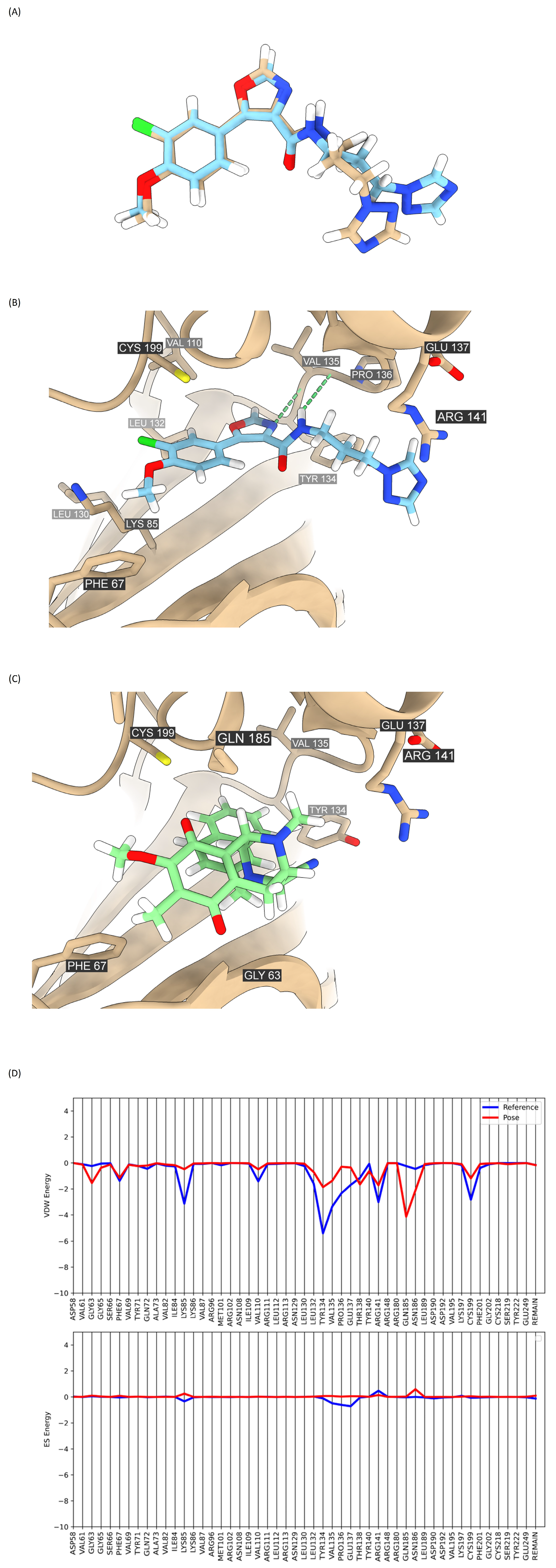
2.7. Analysis of Compound DH_32 Interactions with the GSK-3β Protein
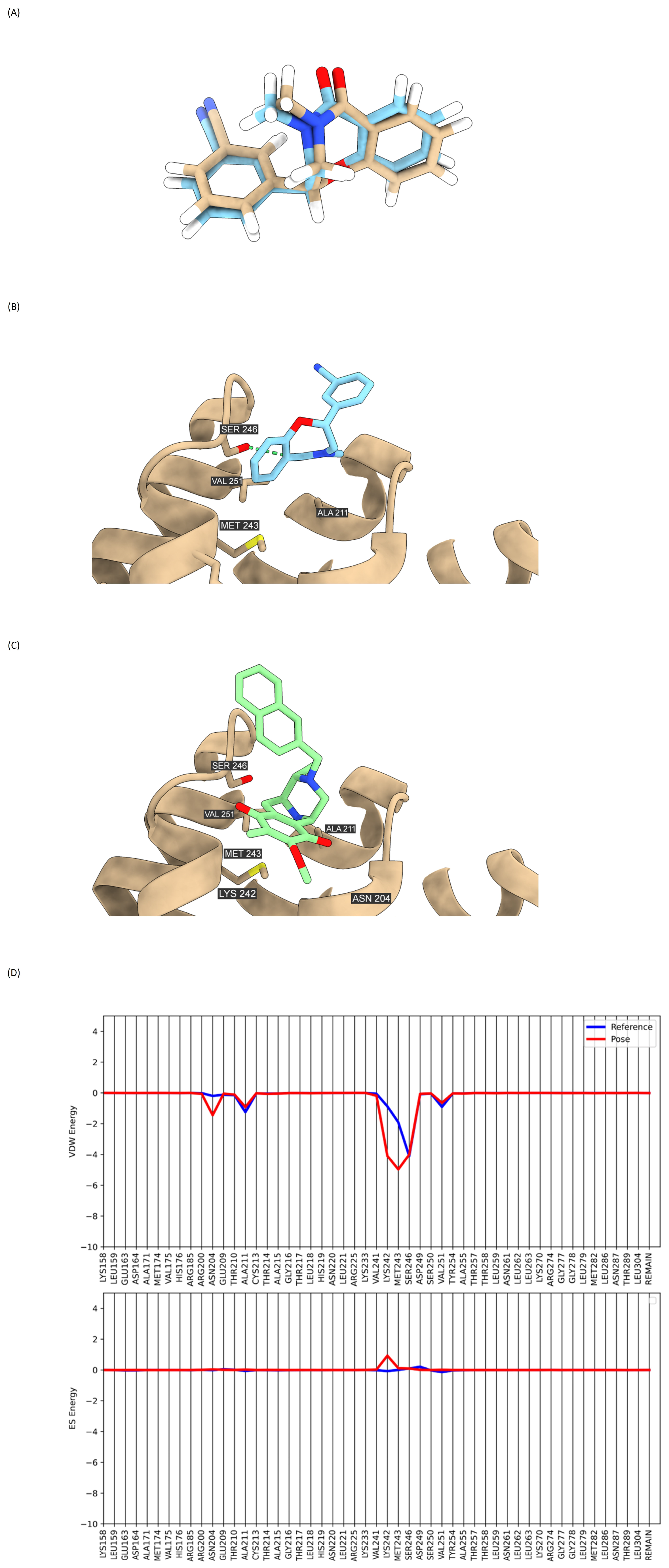
2.8. Binding Interaction of DH_32 with β-Catenin
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Antibodies
4.3. Synthesis of Right-Half RT Analogs
4.4. Preparation of Stock Solution for RT Derivatives
4.5. Cell Viability Assay
4.6. Nuclear Staining Assay
4.7. Proliferation Assay
4.8. Colony Formation Assay
4.9. Three-Dimensional (3D) CSC Spheroid Formation
4.10. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
4.11. Immunofluorescence
4.12. Western Blot Analysis
4.13. Immunoprecipitation
4.14. Molecular Docking
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar]
- Yao, Y.; Fareed, R.; Zafar, A.; Saleem, K.; Huang, T.; Duan, Y.; Rehman, M.U. State-of-the-art combination treatment strategies for advanced stage non-small cell lung cancer. Front. Oncol. 2022, 12, 958505. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kuang, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; Yu, F.; et al. Mechanisms of drugs-resistance in small cell lung cancer: DNA-related, RNA-related, apoptosis-related, drug accumulation and metabolism procedure. Transl. Lung Cancer Res. 2020, 9, 768–786. [Google Scholar] [CrossRef]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Chacon Castro, M.D.C.; Deese, A.R.; Zhang, L. Current landscape of therapeutic resistance in lung cancer and promising strategies to overcome resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef] [PubMed]
- Shuel, S.L. Targeted cancer therapies: Clinical pearls for primary care. Can. Fam. Physician 2022, 68, 515–518. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [PubMed]
- Zulfiqar, B.; Farooq, A.; Kanwal, S.; Asghar, K. Immunotherapy and targeted therapy for lung cancer: Current status and future perspectives. Front. Pharmacol. 2022, 13, 1035171. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, L.; Yin, L.; Yao, Z.; Tong, R.; Xue, J.; Lu, Y. Lung cancer stem cell markers as therapeutic targets: An update on signaling pathways and therapies. Front. Oncol. 2022, 12, 873994. [Google Scholar] [CrossRef]
- Raniszewska, A.; Kwiecień, I.; Rutkowska, E.; Rzepecki, P.; Domagała-Kulawik, J. Lung cancer stem cells-origin, diagnostic techniques and perspective for therapies. Cancers 2021, 13, 2996. [Google Scholar] [CrossRef]
- Romeo, H.E.; Barreiro Arcos, M.L. Clinical relevance of stem cells in lung cancer. World J. Stem Cells 2023, 15, 576–588. [Google Scholar] [CrossRef]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95 (Suppl. S1), S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Hoque, M.O. Targeting cancer stem cells: A strategy for effective eradication of cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Zeng, F.; Xu, L.; Liu, X.; Huang, Y. The role of CD133 expression in the carcinogenesis and prognosis of patients with lung cancer. Mol. Med. Rep. 2013, 8, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Sodja, E.; Rijavec, M.; Koren, A.; Sadikov, A.; Korošec, P.; Cufer, T. The prognostic value of whole blood SOX2, NANOG and OCT4 mRNA expression in advanced small-cell lung cancer. Radiol. Oncol. 2016, 50, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ma, Y.; Yang, Y.; Zhang, L.; Han, H.; Chen, J. CD44 promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2018, 15, 5627–5633. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, H.; Zhu, D. Wnt/β-catenin signaling pathway in lung cancer. Med. Drug Discov. 2022, 13, 100113. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; Graveel, C.R.; Zylstra-Diegel, C.R.; Zhong, Z.; Williams, B.O. Wnt/β-catenin signaling in normal and cancer stem cells. Cancers 2011, 3, 2050–2079. [Google Scholar] [CrossRef]
- Sousa, E.M.F.; Vermeulen, L. Wnt signaling in cancer stem cell biology. Cancers 2016, 8, 60. [Google Scholar]
- Teng, Y.; Wang, X.; Wang, Y.; Ma, D. Wnt/β-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem. Biophys. Res. Commun. 2010, 392, 373–379. [Google Scholar] [CrossRef]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef]
- Daikuhara, N.; Tada, Y.; Yamaki, S.; Charupant, K.; Amnuoypol, S.; Suwanborirux, K.; Saito, N. Chemistry of renieramycins. Part 7: Renieramycins T and U, novel renieramycin–ecteinascidin hybrid marine natural products from Thai sponge Xestospongia sp. Tetrahedron Lett. 2009, 50, 4276–4278. [Google Scholar] [CrossRef]
- Yokoya, M.; Toyoshima, R.; Suzuki, T.; Le, V.H.; Williams, R.M.; Saito, N. Stereoselective total synthesis of (-)-renieramycin T. J. Org. Chem. 2016, 81, 4039–4047. [Google Scholar] [CrossRef]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Saito, N.; Suwanborirux, K. Chemistry of Renieramycins. 17. A new generation of Renieramycins: Hydroquinone 5-O-monoester analogues of Renieramycin M as potential cytotoxic agents against non-small-cell lung cancer cells. J. Nat. Prod. 2017, 80, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Kubo, K.; Yokoya, M.; Saito, N. Preparation of renieramycin left-half model compounds. Tetrahedron 2014, 70, 6529–6545. [Google Scholar] [CrossRef]
- Matsubara, T.; Yokoya, M.; Sirimangkalakitti, N.; Saito, N. Asymmetric synthesis and cytotoxicity evaluation of right-half models of antitumor renieramycin marine natural products. Mar. Drugs 2018, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Tokuyama, H.; Fukuyama, T. Total synthesis of (+)-yatakemycin. Chem. Asian J. 2008, 3, 296–309. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate-triphenylphosphine system in the synthesis and transformation of natural products. In Phosphorus Chemistry Directed towards Biology; Pergamon: Oxford, UK, 1980; pp. 213–218. [Google Scholar]
- Lopez-Ayllon, B.D.; Moncho-Amor, V.; Abarrategi, A.; Ibañez de Cáceres, I.; Castro-Carpeño, J.; Belda-Iniesta, C.; Perona, R.; Sastre, L. Cancer stem cells and cisplatin-resistant cells isolated from non-small-lung cancer cell lines constitute related cell populations. Cancer Med. 2014, 3, 1099–1111. [Google Scholar] [CrossRef]
- Barr, M.P.; Gray, S.G.; Hoffmann, A.C.; Hilger, R.A.; Thomale, J.; O’Flaherty, J.D.; Fennell, D.A.; Richard, D.; O’Leary, J.J.; O’Byrne, K.J. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef]
- Hartz, R.A.; Ahuja, V.T.; Sivaprakasam, P.; Xiao, H.; Krause, C.M.; Clarke, W.J.; Kish, K.; Lewis, H.; Szapiel, N.; Ravirala, R.; et al. Design, structure–activity relationships, and in vivo evaluation of potent and brain-penetrant imidazo[1,2-b]pyridazines as glycogen synthase kinase-3β (GSK-3β) inhibitors. J. Med. Chem. 2023, 66, 4231–4252. [Google Scholar] [CrossRef]
- Amnuoypol, S.; Suwanborirux, K.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 5. Structure elucidation of renieramycin-type derivatives O, Q, R, and S from Thai marine sponge Xestospongia species pretreated with potassium cyanide. J. Nat. Prod. 2004, 67, 1023–1028. [Google Scholar] [CrossRef]
- Saito, N.; Hiramatsu, A.; Hirade, H.; Kubota, M.; Toyoshima, R.; Fujino, A.; Sirimangkalakitti, N.; Concepcion, G.P. Chemistry of Renieramycins. 16. Structure of 7-Desmethylrenieramycin O (14α-Hydroxyrenieramycin S) from blue sponge, Xestospongia sp. Heterocycles 2017, 95, 748–752. [Google Scholar] [CrossRef]
- Frincke, J.M.; Faulkner, D.J. Antimicrobial metabolites of the sponge Reniera sp. J. Am. Chem. Soc. 1982, 104, 265–269. [Google Scholar] [CrossRef]
- Parameswaran, P.S.; Naik, C.G.; Kamat, S.Y.; Pramanik, B.N. Renieramycins H and I, Two Novel Alkaloids from the Sponge Haliclona cribricutis Dendy. Cheminform 1998, 37, 1258–1263. [Google Scholar]
- Oku, N.; Matsunaga, S.; van Soest, R.; Fusetani, N. Renieramycin J, a highly cytotoxic tetrahydroisoquinoline alkaloid, from a marine sponge Neopetrosia sp. J. Nat. Prod. 2003, 66, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Petsri, K.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. Renieramycin T induces lung cancer cell apoptosis by targeting Mcl-1 degradation: A new insight in the mechanism of action. Mar. Drugs 2019, 17, 301. [Google Scholar] [CrossRef] [PubMed]
- Chantarawong, W.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. 5-O-Acetyl-Renieramycin T from blue sponge Xestospongia sp. induces lung cancer stem cell apoptosis. Mar. Drugs 2019, 17, 109. [Google Scholar] [CrossRef] [PubMed]
- Suksamai, D.; Racha, S.; Sriratanasak, N.; Chaotham, C.; Aphicho, K.; Lin, A.C.K.; Chansriniyom, C.; Suwanborirux, K.; Chamni, S.; Chanvorachote, P. 5-O-(N-Boc-l-Alanine)-Renieramycin T induces cancer stem cell apoptosis via targeting Akt signaling. Mar. Drugs 2022, 20, 235. [Google Scholar] [CrossRef]
- Liu, M.; Wu, H.; Xu, C. Targeting cancer stem cell pathways for lung cancer therapy. Curr. Opin. Oncol. 2023, 35, 78–85. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc. Pharmacol. 2013, 61, 14–25. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Entezari, M.; Reza Mirmazloomi, S.; Hassanpoor, A.; Aboutalebi, M.; Rezaei, S.; Hejazi, E.S.; Kakavand, A.; Heidari, H.; et al. SOX2 function in cancers: Association with growth, invasion, stemness and therapy response. Biomed. Pharmacother. 2022, 156, 113860. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.; Ahmed, M.; Lorenzi, F.; Nateri, A.S. Spheroid-formation (colonosphere) assay for in vitro assessment and expansion of stem cells in colon cancer. Stem Cell Rev. Rep. 2016, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-formation assay: Three-dimensional in vitro culturing of prostate cancer stem/progenitor sphere-forming cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288. [Google Scholar] [CrossRef]
- Santarpia, M.; Ciappina, G.; Spagnolo, C.C.; Squeri, A.; Passalacqua, M.I.; Aguilar, A.; Gonzalez-Cao, M.; Giovannetti, E.; Silvestris, N.; Rosell, R. Targeted therapies for KRAS-mutant non-small cell lung cancer: From preclinical studies to clinical development-a narrative review. Transl. Lung Cancer Res. 2023, 12, 346–368. [Google Scholar] [CrossRef]
- Karimi, N.; Moghaddam, S.J. KRAS-mutant lung cancer: Targeting molecular and immunologic pathways, therapeutic advantages and restrictions. Cells 2023, 12, 749. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Chan, S.H.; Chiang, J.; Ngeow, J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hered. Cancer Clin. Pract. 2021, 19, 21. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Yoon, J.; Cho, Y.H.; Cha, P.H.; Park, J.C.; Choi, K.Y. A mutant KRAS-induced factor REG4 promotes cancer stem cell properties via Wnt/β-catenin signaling. Int. J. Cancer 2020, 146, 2877–2890. [Google Scholar] [CrossRef]
- Merkle, J.; Breunig, M.; Schmid, M.; Allgöwer, C.; Krüger, J.; Melzer, M.K.; Bens, S.; Siebert, R.; Perkhofer, L.; Azoitei, N.; et al. CDKN2A-mutated pancreatic ductal organoids from induced pluripotent stem cells to model a cancer predisposition syndrome. Cancers 2021, 13, 5139. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M.; Chan, C.S.; Hainaut, P. The role of the p53 protein in stem-cell biology and epigenetic regulation. Cold Spring Harb. Perspect. Med. 2016, 6, a026153. [Google Scholar] [CrossRef]
- Chiou, S.H.; Wang, M.L.; Chou, Y.T.; Chen, C.J.; Hong, C.F.; Hsieh, W.J.; Chang, H.T.; Chen, Y.S.; Lin, T.W.; Hsu, H.S.; et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010, 70, 10433–10444. [Google Scholar] [CrossRef]
- Karachaliou, N.; Rosell, R.; Viteri, S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl. Lung Cancer Res. 2013, 2, 172–179. [Google Scholar] [PubMed]
- Li, Y.J.; Wei, Z.M.; Meng, Y.X.; Ji, X.R. Beta-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: Relationships with carcinogenesis and metastasis. World J. Gastroenterol. 2005, 11, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Chen, J.M.; Normandin, M.D.; Chang, J.S.; Chang, G.C.; Taylor, C.K.; Trapa, P.; Plummer, M.S.; Para, K.S.; Conn, E.L.; et al. Discovery of a highly selective glycogen synthase kinase-3 inhibitor (PF-04802367) that modulates tau phosphorylation in the brain: Translation for pet neuroimaging. Angew. Chem. Int. Ed. Engl. 2016, 55, 9601–9605. [Google Scholar] [CrossRef]
- Kessler, D.; Mayer, M.; Zahn, S.K.; Zeeb, M.; Wöhrle, S.; Bergner, A.; Bruchhaus, J.; Ciftci, T.; Dahmann, G.; Dettling, M.; et al. Getting a grip on the undrugged: Targeting β-catenin with fragment-based methods. Chem. Med. Chem. 2021, 16, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB protein data bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIRE Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]



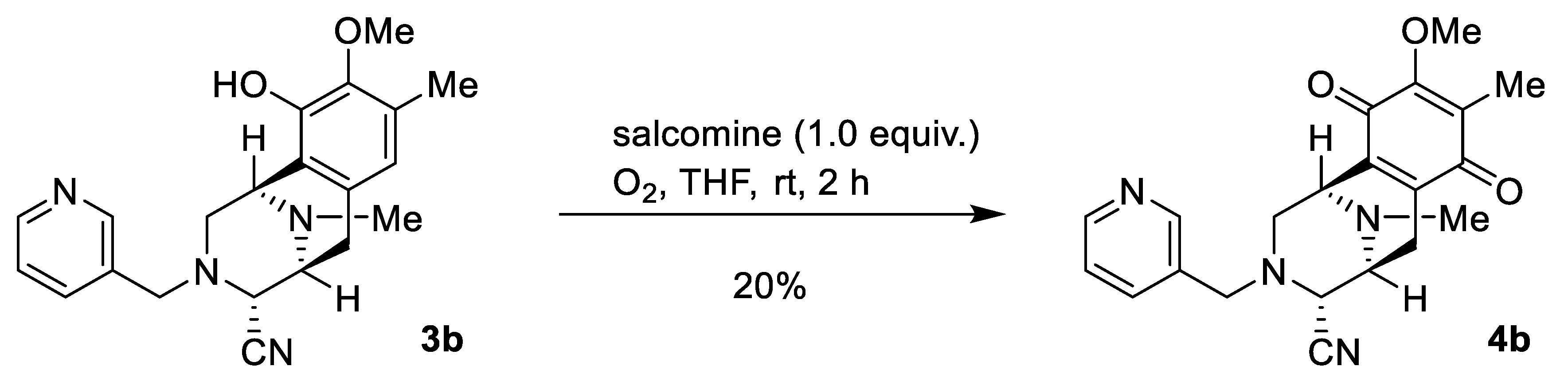
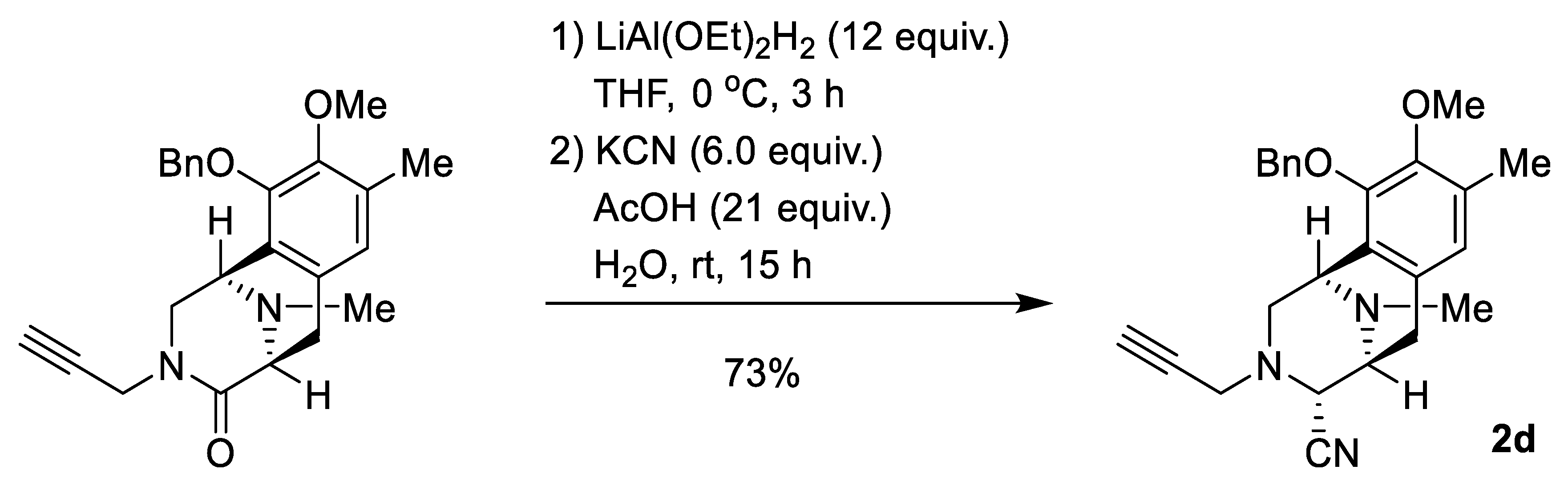


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
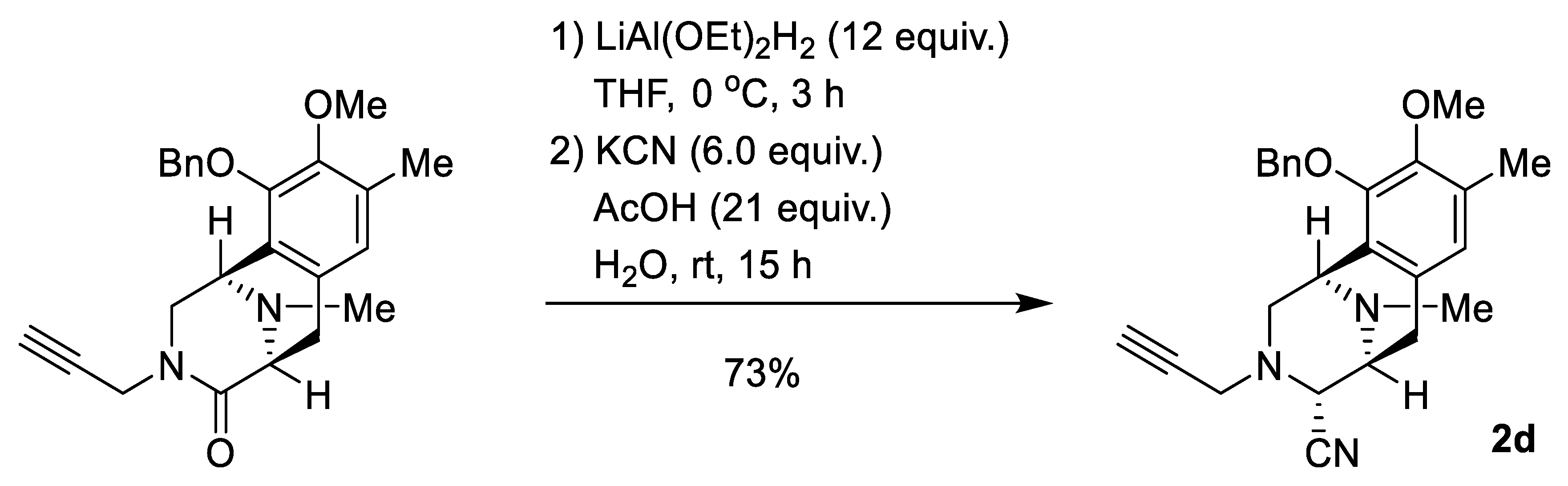{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Grid Score (kcal/mol) | VDW Energy (kcal/mol) | ES Energy (kcal/mol) |
|---|---|---|---|
| DH_32 | −38.421 | −37.933 | −0.489 |
| PF-04802367 (reference compound) | −49.199 | −46.755 | −2.444 |
| Compounds | Grid Score (kcal/mol) | VDW Energy (kcal/mol) | ES Energy (kcal/mol) |
|---|---|---|---|
| DH_32 | −35.559 | −35.623 | 0.064 |
| R9Q (reference compound) | −29.044 | −27.517 | −1.527 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ei, Z.Z.; Racha, S.; Yokoya, M.; Hotta, D.; Zou, H.; Chanvorachote, P. Simplified Synthesis of Renieramycin T Derivatives to Target Cancer Stem Cells via β-Catenin Proteasomal Degradation in Human Lung Cancer. Mar. Drugs 2023, 21, 627. https://doi.org/10.3390/md21120627
Ei ZZ, Racha S, Yokoya M, Hotta D, Zou H, Chanvorachote P. Simplified Synthesis of Renieramycin T Derivatives to Target Cancer Stem Cells via β-Catenin Proteasomal Degradation in Human Lung Cancer. Marine Drugs. 2023; 21(12):627. https://doi.org/10.3390/md21120627
Chicago/Turabian StyleEi, Zin Zin, Satapat Racha, Masashi Yokoya, Daiki Hotta, Hongbin Zou, and Pithi Chanvorachote. 2023. "Simplified Synthesis of Renieramycin T Derivatives to Target Cancer Stem Cells via β-Catenin Proteasomal Degradation in Human Lung Cancer" Marine Drugs 21, no. 12: 627. https://doi.org/10.3390/md21120627
APA StyleEi, Z. Z., Racha, S., Yokoya, M., Hotta, D., Zou, H., & Chanvorachote, P. (2023). Simplified Synthesis of Renieramycin T Derivatives to Target Cancer Stem Cells via β-Catenin Proteasomal Degradation in Human Lung Cancer. Marine Drugs, 21(12), 627. https://doi.org/10.3390/md21120627