
Synaptamide Modulates Astroglial Activity in Mild Traumatic Brain Injury

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
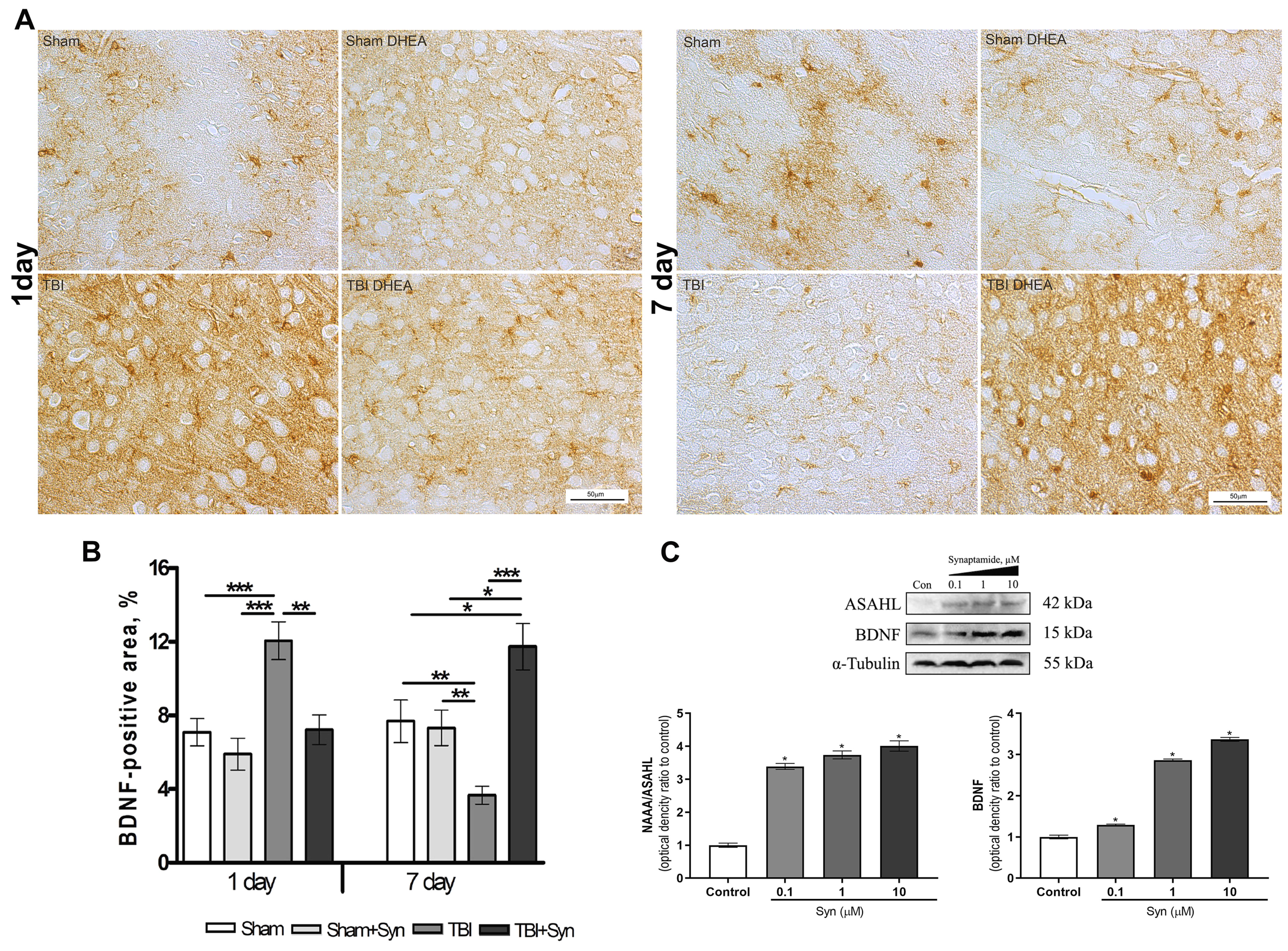{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
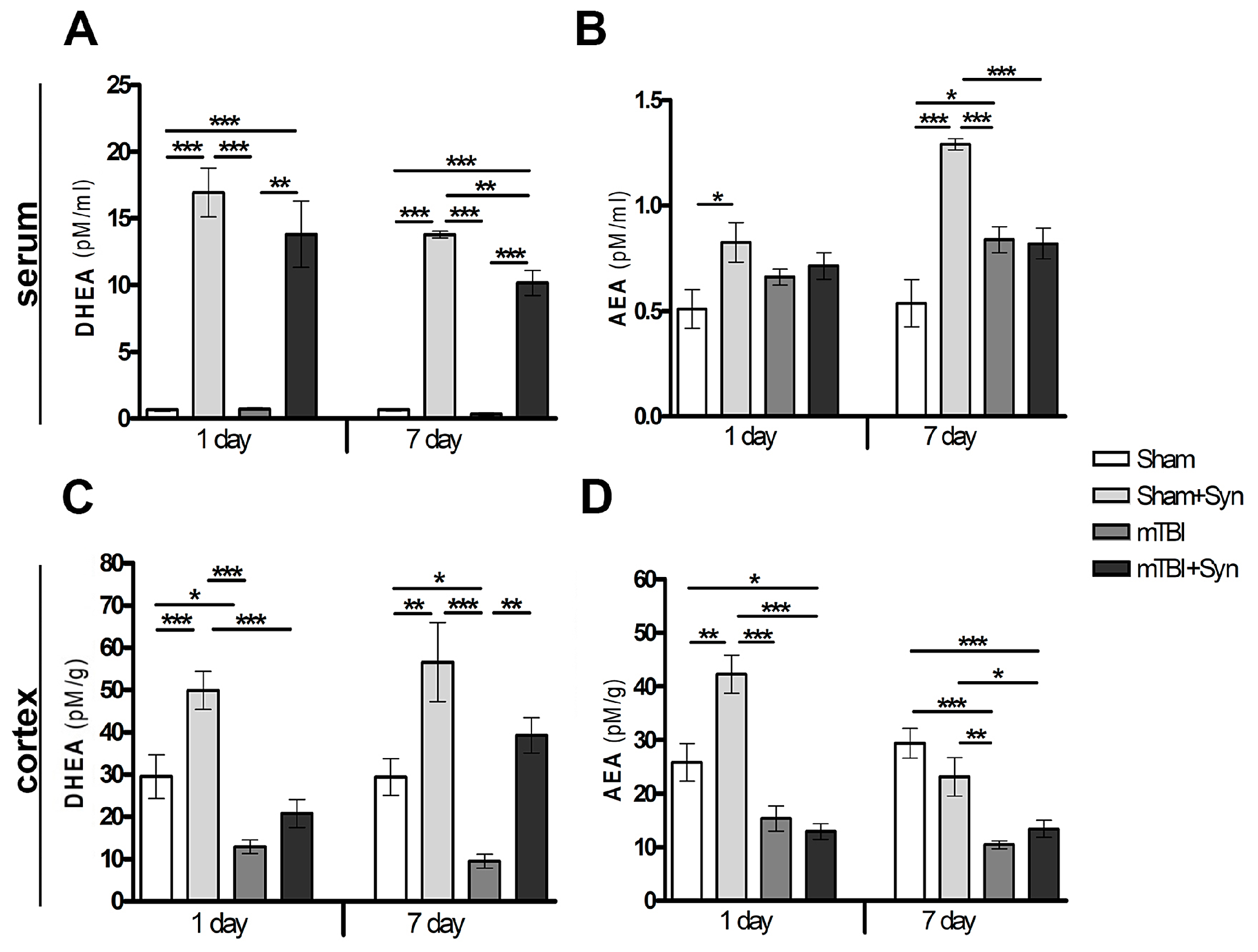
2.1. Synaptamide Increases Serum and Brain Levels of DHEA and AEA
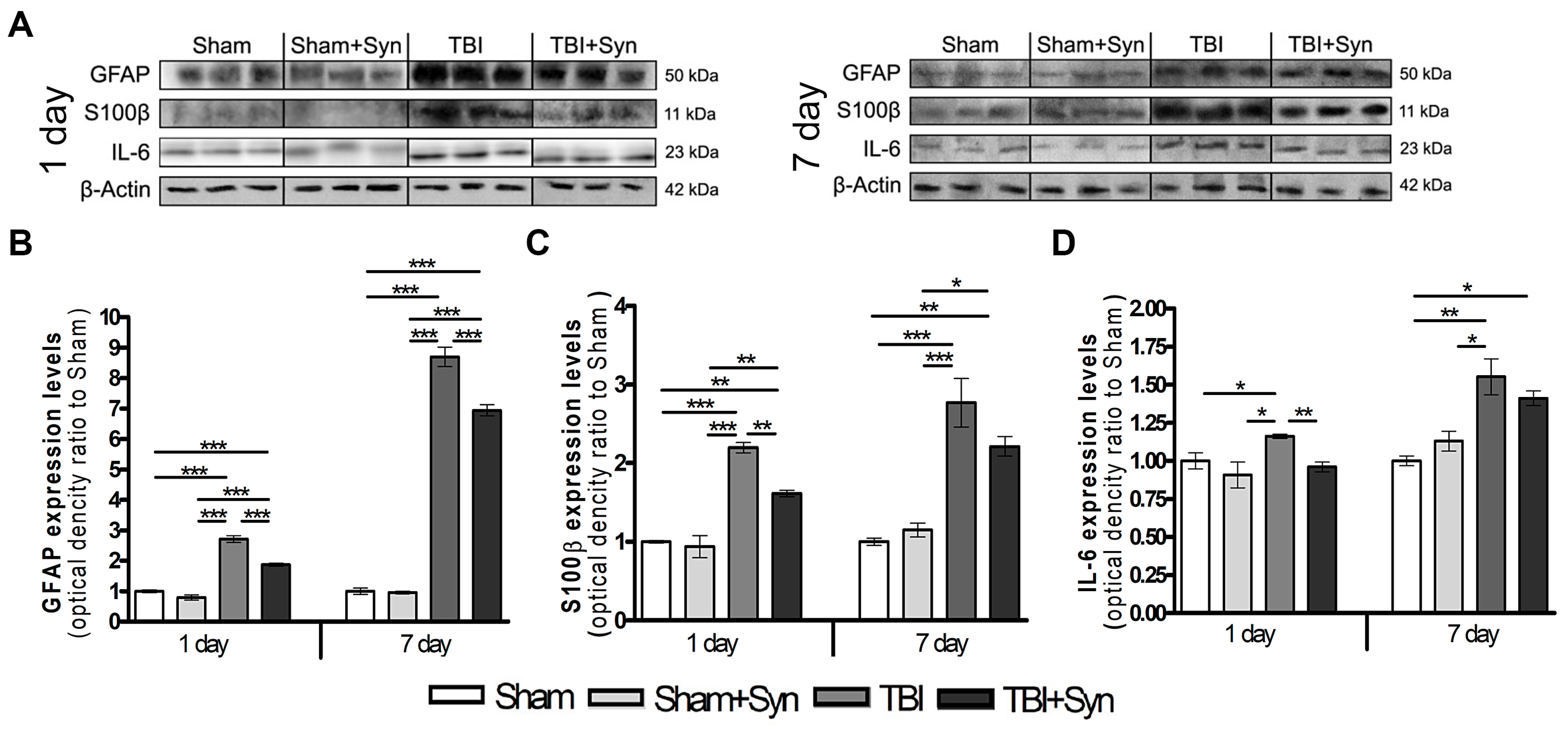
2.2. Synaptamide Reduces Serum Biomarkers after mTBI
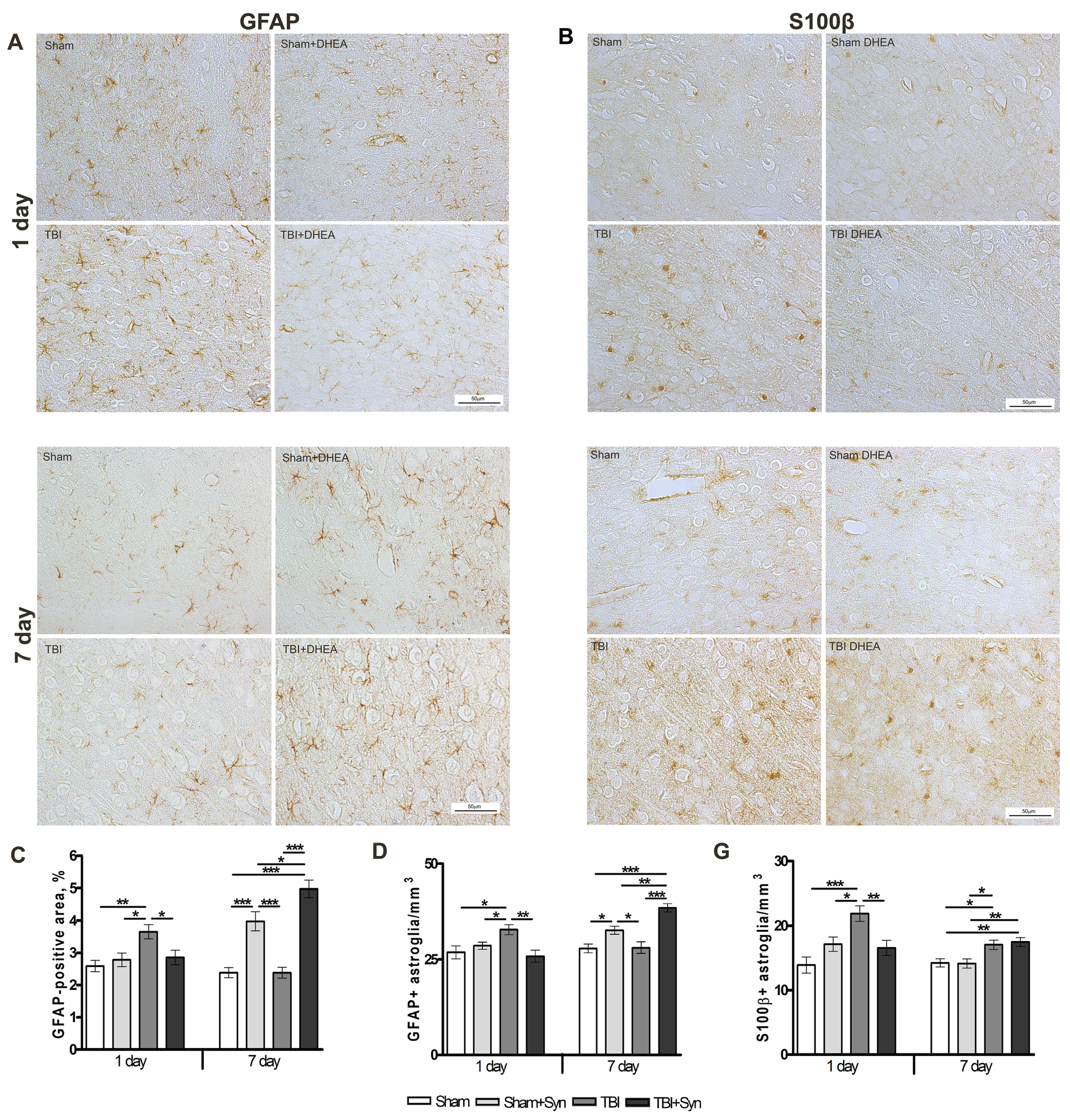
2.3. Synaptamide Regulates Reactive Astrogliosis in mTBI Conditions
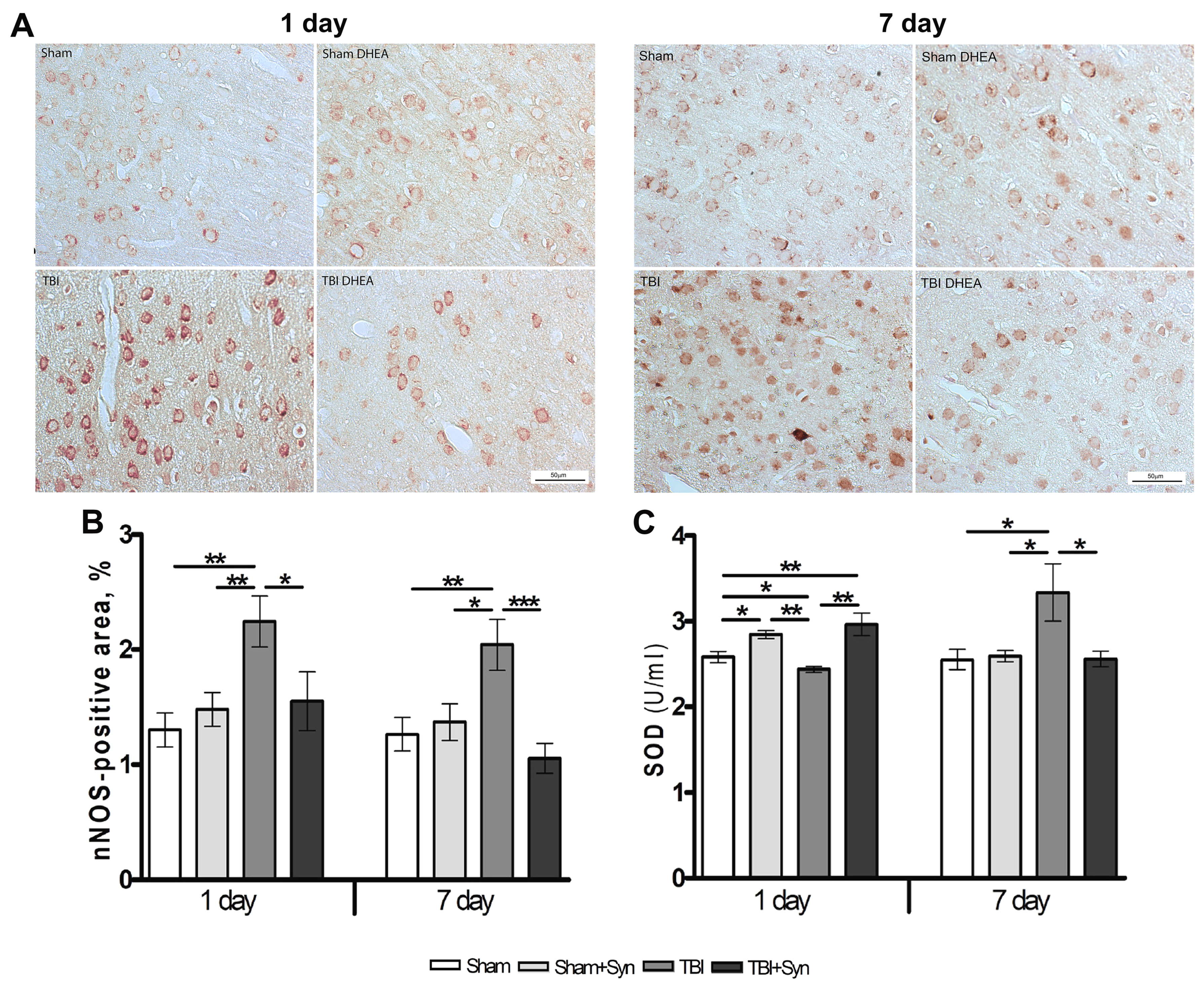
2.4. Synaptamide Reduce nNOS Production and Contributes to the Antioxidant Protection of Neurons
2.5. Synaptamide Regulates Trophic Support of Neurons in mTBI through the Production of Brain-Derived Neurotrophic Factor (BDNF)
3. Discussion
4. Materials and Methods
4.1. N-Docosahexaenoylethanolamine (DHEA, Synaptamide)
4.2. Animals
4.3. mTBI Procedure
4.4. Immunohistochemistry and Microscopy
4.5. Assessment of SOD Enzyme Activity
4.6. Cells for Bioassay
4.7. Biomarker Measurement
4.8. Western Blotting
4.9. Quantitation of NAE in Brain Lipids
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.V.; Dustin, M.L. Innate response to focal necrotic injury inside the blood-brain barrier. J. Immunol. 2006, 177, 5269–5277. [Google Scholar] [CrossRef]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- TTyzack, G.; Sitnikov, S.; Barson, D.; Adams-Carr, K.; Lau, N.K.; Kwok, J.C.; Zhao, C.; Franklin, R.; Karadottir, R.T.; Fawcett, J.; et al. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 2014, 5, 4294. [Google Scholar] [CrossRef]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Senapati, S.; Malampati, S.; Zhu, Z.; Su, C.-F.; Liu, J.; Guan, X.-J.; Tong, B.C.-K.; et al. Protopine promotes the proteasomal degradation of pathological tau in Alzheimer’s disease models via HDAC6 inhibition. Phytomedicine 2022, 96, 153887. [Google Scholar] [CrossRef]
- Scheff, S.W.; Ansari, M.A. Natural Compounds as a Therapeutic Intervention following Traumatic Brain Injury: The Role of Phytochemicals. J. Neurotrauma 2017, 34, 1491–1510. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J. Neurotrauma 2011, 28, 2113–2122. [Google Scholar] [CrossRef]
- Joffre, C.; Rey, C.; Layé, S. N-3 polyunsaturated fatty acids and the resolution of neuroinflammation. Front. Pharmacol. 2019, 10, 1022. [Google Scholar] [CrossRef]
- Horrocks, L.A.; Farooqui, A.A. Docosahexaenoic acid in the diet: Its importance in maintenance and restoration of neural membrane function. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Axelrod, J. Enzymatic synthesis of anandamide, an endogenous ligand for the cannabinoid receptor, by brain membranes. Proc. Natl. Acad. Sci. USA 1994, 91, 6698–6701. [Google Scholar] [CrossRef] [PubMed]
- De Bus, I.; Witkamp, R.; Zuilhof, H.; Albada, B.; Balvers, M. The role of n-3 PUFA-derived fatty acid derivatives and their oxygenated metabolites in the modulation of inflammation. Prostaglandins Other Lipid Mediat. 2019, 144, 106351. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, A.I.; Tyrtyshnaia, A.A.; Pislyagin, E.A.; Dyuizen, I.V.; Sultanov, R.M.; Manzhulo, I.V. N-docosahexaenoylethanolamine reduces neuroinflammation and cognitive impairment after mild traumatic brain injury in rats. Sci. Rep. 2021, 11, 756. [Google Scholar] [CrossRef]
- Papa, L.; Brophy, G.M.; Welch, R.D.; Lewis, L.M.; Braga, C.F.; Tan, C.N.; Ameli, N.J.; Lopez, M.A.; Haeussler, C.A.; Giordano, D.I.M.; et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. 2016, 73, 551–560. [Google Scholar] [CrossRef]
- Neselius, S.; Brisby, H.; Granholm, F.; Zetterberg, H.; Blennow, K. Monitoring concussion in a knocked-out boxer by CSF biomarker analysis. Knee Surg. Sports Traumatol. Arthrosc. 2015, 23, 2536–2539. [Google Scholar] [CrossRef]
- Bloomfield, S.M.; McKinney, J.; Smith, L.; Brisman, J. Reliability of S100B in predicting severity of central nervous system injury. Neurocrit. Care 2007, 6, 121–138. [Google Scholar] [CrossRef]
- Kadhim, H.J.; Duchateau, J.; Sébire, G. Cytokines and brain injury: Invited review. J. Intensive Care Med. 2008, 23, 236–249. [Google Scholar] [CrossRef]
- Miñambres, E.; Cemborain, A.; Sánchez-Velasco, P.; Gandarillas, M.; Díaz-Regañón, G.; Sánchez-González, U.; Leyva-Cobián, F. Correlation between transcranial interleukin-6 gradient and outcome in patients with acute brain injury. Crit. Care Med. 2003, 31, 933–938. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Snyder, S.H. Gases as biological messengers: Nitric oxide and carbon monoxide in the brain. J. Neurosci. 1994, 14, 5147–5159. [Google Scholar] [CrossRef] [PubMed]
- Hortobágyi, T.; Görlach, C.; Benyó, Z.; Lacza, Z.; Hortobágyi, S.; Wahl, M.; Harkany, T. Inhibition of neuronal nitric oxide synthase-mediated activation of poly(ADP-ribose) polymerase in traumatic brain injury: Neuroprotection by 3-aminobenzamide. Neuroscience 2003, 121, 983–990. [Google Scholar] [CrossRef]
- Pieper, A.A.; Blackshaw, S.; Clements, E.E.; Brat, D.J.; Krug, D.K.; White, A.J.; Pinto-Garcia, P.; Favit, A.; Conover, J.R.; Snyder, S.H.; et al. Poly(ADP-ribosyl)ation basally activated by DNA strand breaks reflects glutamate-nitric oxide neurotransmission. Proc. Natl. Acad. Sci. USA 2000, 97, 1845–1850. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Moon, H.S.; Cao, D.; Lee, J.; Kevala, K.; Jun, S.; Lovinger, D.; Akbar, M.; Huang, B.X. N-docosahexaenoylethanolamide promotes development of hippocampal neurons. Biochem. J. 2011, 435, 327–336. [Google Scholar] [CrossRef]
- Bottemanne, P.; Muccioli, G.G.; Alhouayek, M. N-acylethanolamine hydrolyzing acid amidase inhibition: Tools and potential therapeutic opportunities. Drug Discov. Today 2018, 23, 1520–1529. [Google Scholar] [CrossRef]
- Tsuboi, K.; Takezaki, N.; Ueda, N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA). Chem. Biodivers. 2007, 4, 1914–1925. [Google Scholar] [CrossRef]
- Vogel, A.; Wilken-Schmitz, A.; Hummel, R.; Lang, M.; Gurke, R.; Schreiber, Y.; Schäfer, M.; Tegeder, I. Low brain endocannabinoids associated with persistent non-goal directed nighttime hyperactivity after traumatic brain injury in mice. Sci. Rep. 2020, 10, 14929. [Google Scholar] [CrossRef]
- Sharma, R.; Laskowitz, D.T. Biomarkers in traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2012, 12, 560–569. [Google Scholar] [CrossRef]
- Strathmann, F.G.; Schulte, S.; Goerl, K.; Petron, D.J. Blood-based biomarkers for traumatic brain injury: Evaluation of research approaches, available methods and potential utility from the clinician and clinical laboratory perspectives. Clin. Biochem. 2014, 47, 876–888. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Cui, Y.; Roberts, C.; Lu, M.; Wilhelmsson, U.; Pekny, M.; Chopp, M. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 2014, 62, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Gorina, R.; Font-Nieves, M.; Marquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Ponomarenko, A.I.; Tyrtyshnaia, A.A.; Ivashkevich, D.O.; Manzhulo, I.V. Mild Traumatic Brain Injury Contributes to the Development of Delayed Neuroinflammation. Neuroimmunomodulation 2022, 29, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fawcett, J. The perineuronal net and the control of CNS plasticity. Cell Tissue Res. 2012, 349, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, D.B.; Cornwall, E.H.; Landar, A.; Song, W. The S100 protein family: History, function, and expression. Brain Res. Bull. 1995, 37, 417–429. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Marini, A.M.; Jiang, X.; Wu, X.; Tain, F.; Zhu, D.; Okagaki, P.; Lipski, R. Role of brain-derived neurotrophic factor and NF-kappaB in neuronal plasticity and survival: From genes to phenotype. Restor. Neurol. Neurosci. 2004, 22, 121–130. [Google Scholar]
- Kairisalo, M.; Korhonen, L.; Sepp, M.; Pruunsild, P.; Kukkonen, J.P.; Kivinen, J.; Timmusk, T.; Blomgren, K.; Lindholm, D. NF-κB-dependent regulation of brain-derived neurotrophic factor in hippocampal neurons by X-linked inhibitor of apoptosis protein. Eur. J. Neurosci. 2009, 30, 958–966. [Google Scholar] [CrossRef]
- Sniderhan, L.F.; Stout, A.; Lu, Y.; Chao, M.V.; Maggirwar, S.B. Ankyrin-rich membrane spanning protein plays a critical role in nuclear factor-κb signaling. Mol. Cell. Neurosci. 2008, 38, 404–416. [Google Scholar] [CrossRef][Green Version]
- Chao, C.C.; Ma, Y.L.; Lee, E.H.Y. Brain-Derived Neurotrophic Factor Enhances Bcl-xL Expression Through Protein Kinase Casein Kinase 2-Activated and Nuclear Factor Kappa B-Mediated Path-way in Rat Hippocampus. Brain Pathol. 2011, 21, 150–162. [Google Scholar] [CrossRef]
- Maqbool, A.; Lattke, M.; Wirth, T.; Baumann, B. Sustained, neuron-specific IKK/NF-κB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener. 2013, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Chen, H.; Kim, H.Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflamm. 2019, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Liu, N.-K.; Wu, X.; Wang, Y.; Xia, Y.; Sun, Y.; Lai, Y.; Li, R.; Shekhar, A.; Xu, X.-M. Disrupting nNOS-PSD95 Interaction Improves Neurological and Cognitive Recoveries after Traumatic Brain Injury. Cereb. Cortex 2020, 30, 3859–3871. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Matsuda, F.; Annamalai, B.; Dhindsa, T.S.; Singh, I.; Singh, A.K. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016, 1630, 159–170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Yun, X.; Maximov, V.D.; Yu, J.; Zhu, H.; Vertegel, A.A.; Kindy, M.S. Nanoparticles for targeted delivery of antioxidant enzymes to the brain after cerebral ischemia and reperfusion injury. J. Cereb. Blood Flow Metab. 2013, 33, 583–592. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox regulation of inflammation: Old elements, a new story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef]
- Starinets, A.; Tyrtyshnaia, A.; Kipryushina, Y.; Manzhulo, I. Analgesic Activity of Synaptamide in a Rat Sciatic Nerve Chronic Constriction Injury Model. Cells Tissues Organs 2022, 211, 73–84. [Google Scholar] [CrossRef]
- Latyshev, N.A.; Ermolenko, E.V.; Kasyanov, S.P. Concentration and purification of polyunsaturated fatty acids from squid liver processing wastes. Eur. J. Lipid Sci. Technol. 2014, 116, 1608–1613. [Google Scholar] [CrossRef]
- Statler, K.D.; Alexander, H.; Vagni, V.; Holubkov, R.; Dixon, C.E.; Clark, R.S.; Jenkins, L.; Kochanek, P.M. Isoflurane exerts neuroprotective actions at or near the time of severe traumatic brain injury. Brain Res. 2006, 1076, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Schildge, S.; Bohrer, C.; Beck, K.; Schachtrup, C. Isolation and culture of mouse cortical astrocytes. J. Vis. Exp. 2013, 71, 50079. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarenko, A.; Tyrtyshnaia, A.; Ivashkevich, D.; Ermolenko, E.; Dyuizen, I.; Manzhulo, I. Synaptamide Modulates Astroglial Activity in Mild Traumatic Brain Injury. Mar. Drugs 2022, 20, 538. https://doi.org/10.3390/md20080538
Ponomarenko A, Tyrtyshnaia A, Ivashkevich D, Ermolenko E, Dyuizen I, Manzhulo I. Synaptamide Modulates Astroglial Activity in Mild Traumatic Brain Injury. Marine Drugs. 2022; 20(8):538. https://doi.org/10.3390/md20080538
Chicago/Turabian StylePonomarenko, Arina, Anna Tyrtyshnaia, Darya Ivashkevich, Ekaterina Ermolenko, Inessa Dyuizen, and Igor Manzhulo. 2022. "Synaptamide Modulates Astroglial Activity in Mild Traumatic Brain Injury" Marine Drugs 20, no. 8: 538. https://doi.org/10.3390/md20080538
APA StylePonomarenko, A., Tyrtyshnaia, A., Ivashkevich, D., Ermolenko, E., Dyuizen, I., & Manzhulo, I. (2022). Synaptamide Modulates Astroglial Activity in Mild Traumatic Brain Injury. Marine Drugs, 20(8), 538. https://doi.org/10.3390/md20080538