
Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum

 , , ,
, , ,  and
and 
Abstract
1. Introduction
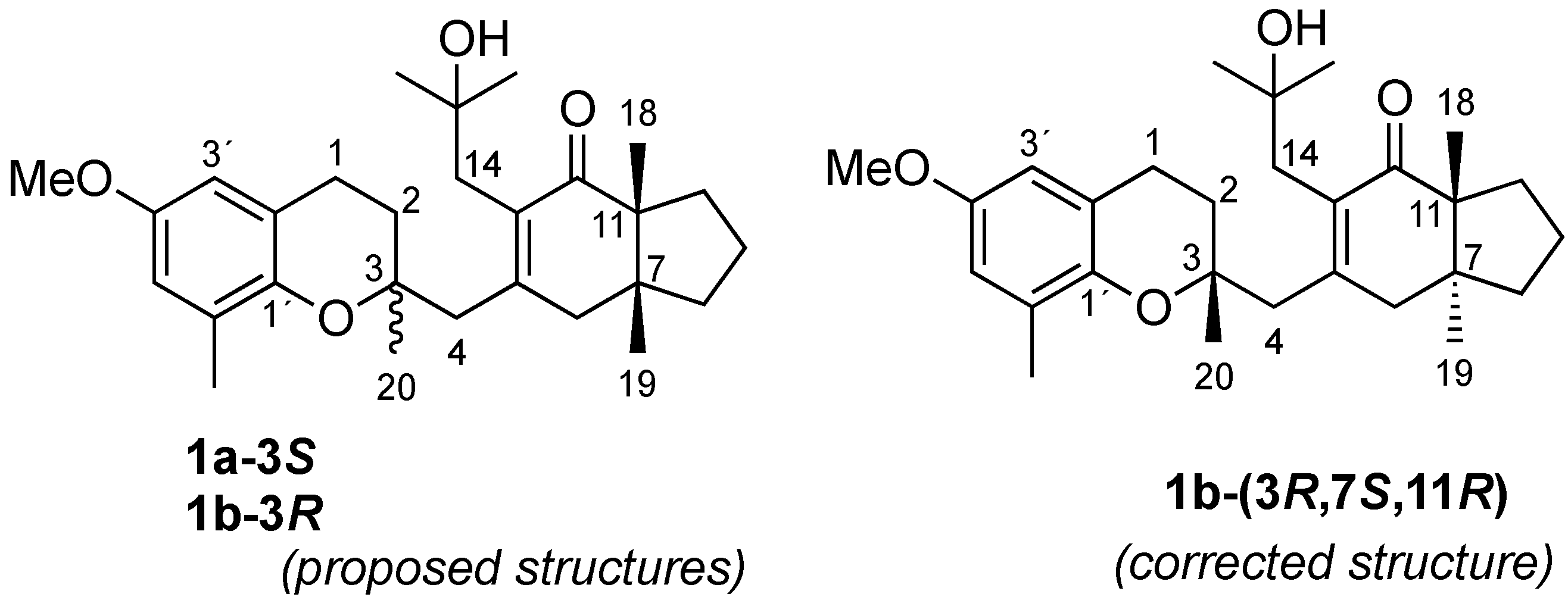
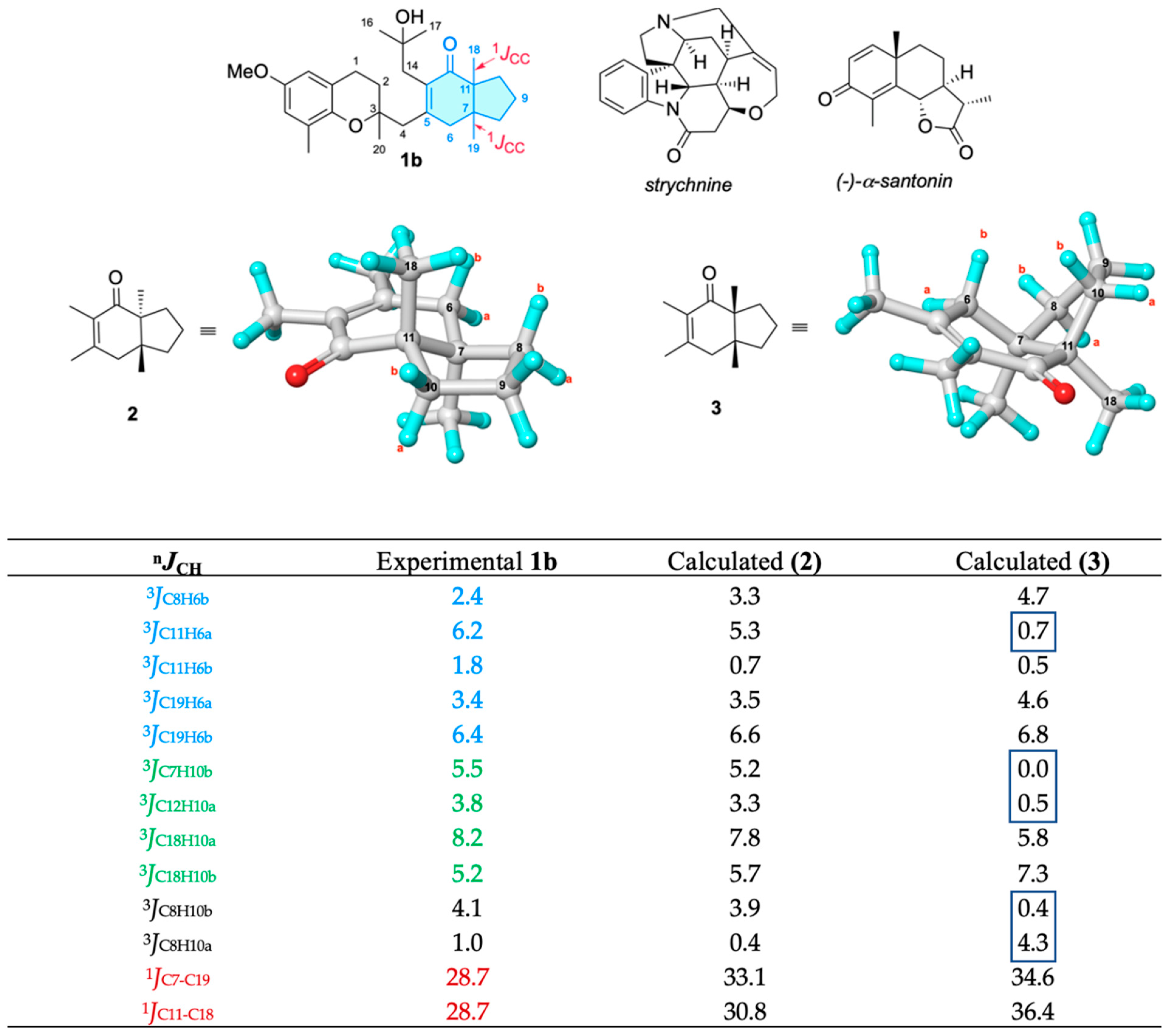
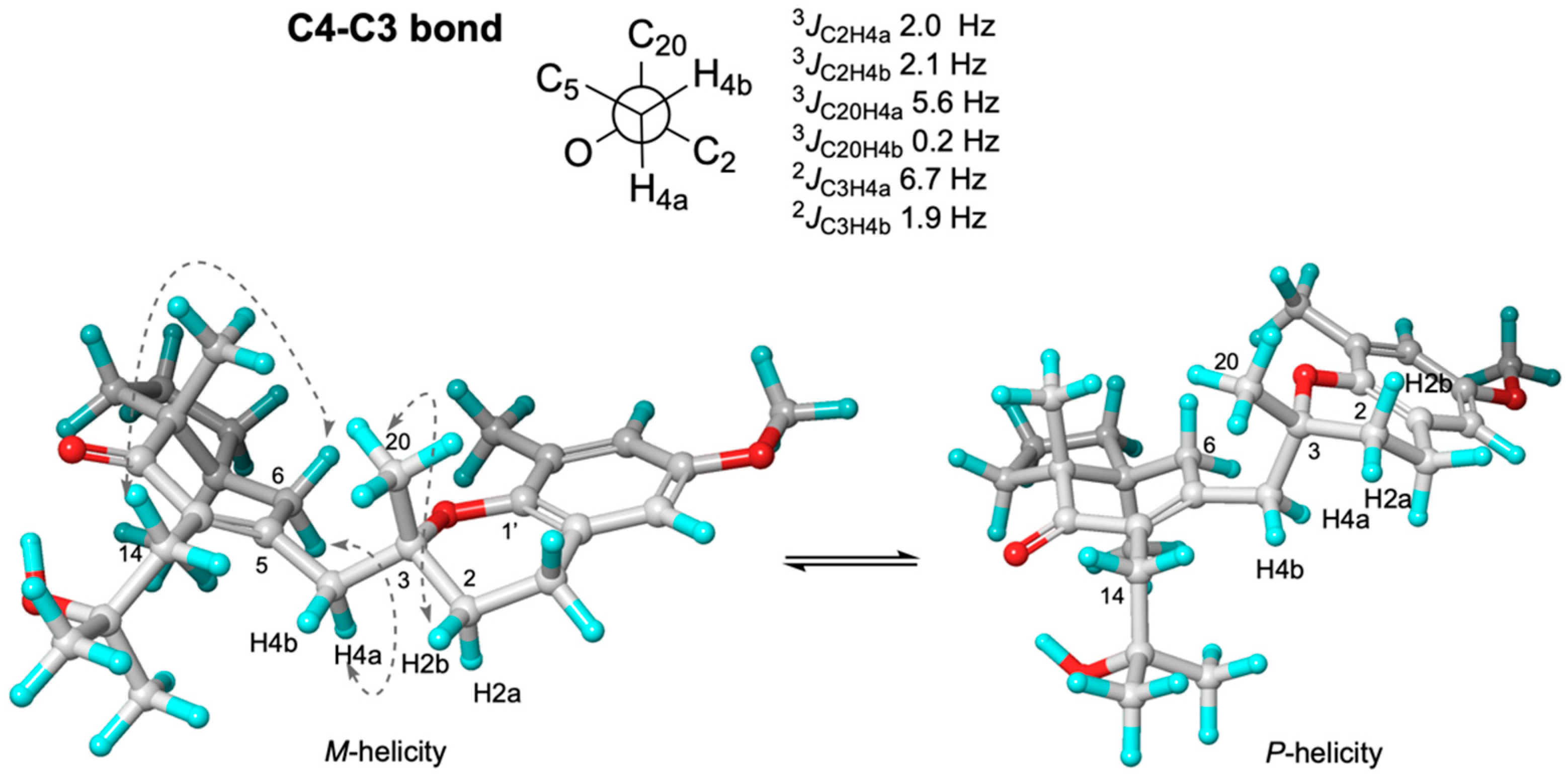
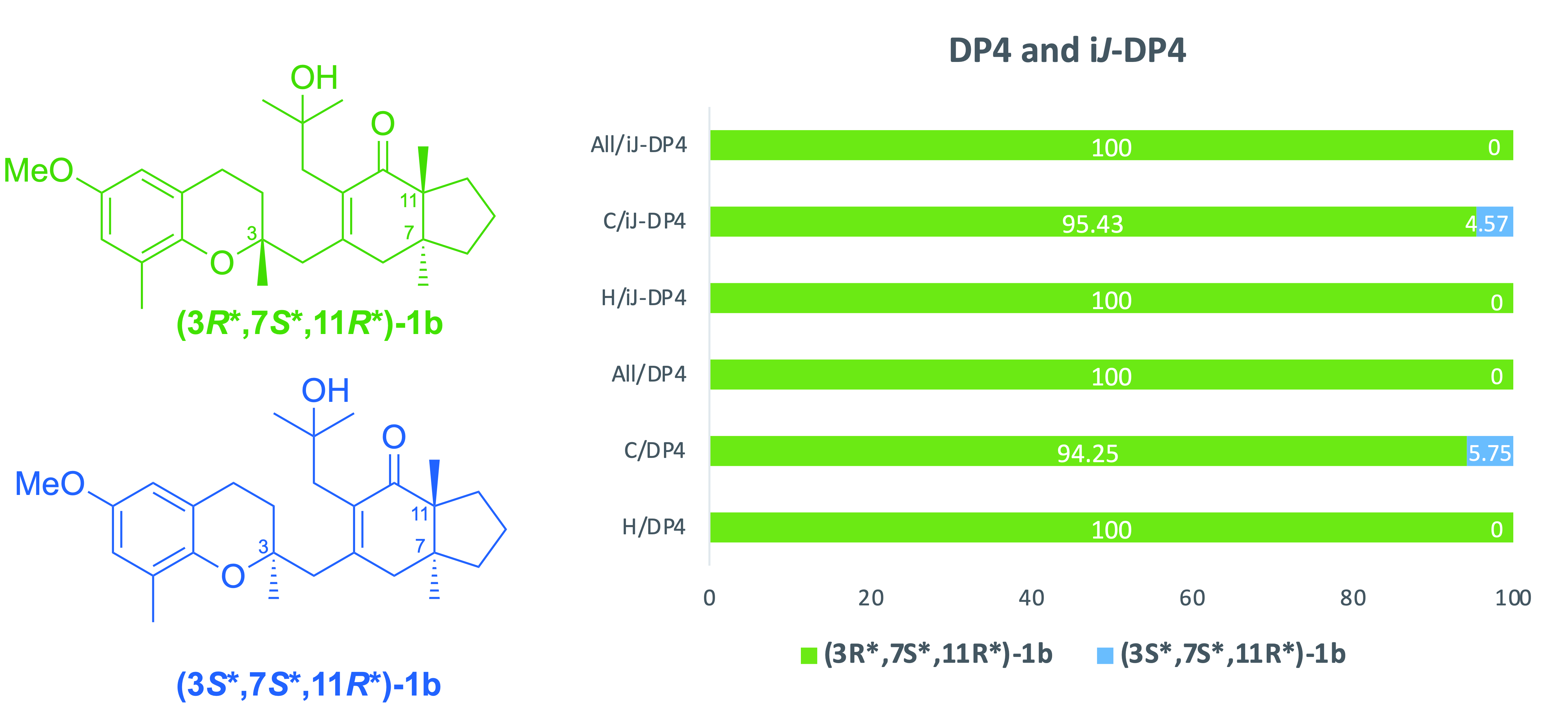
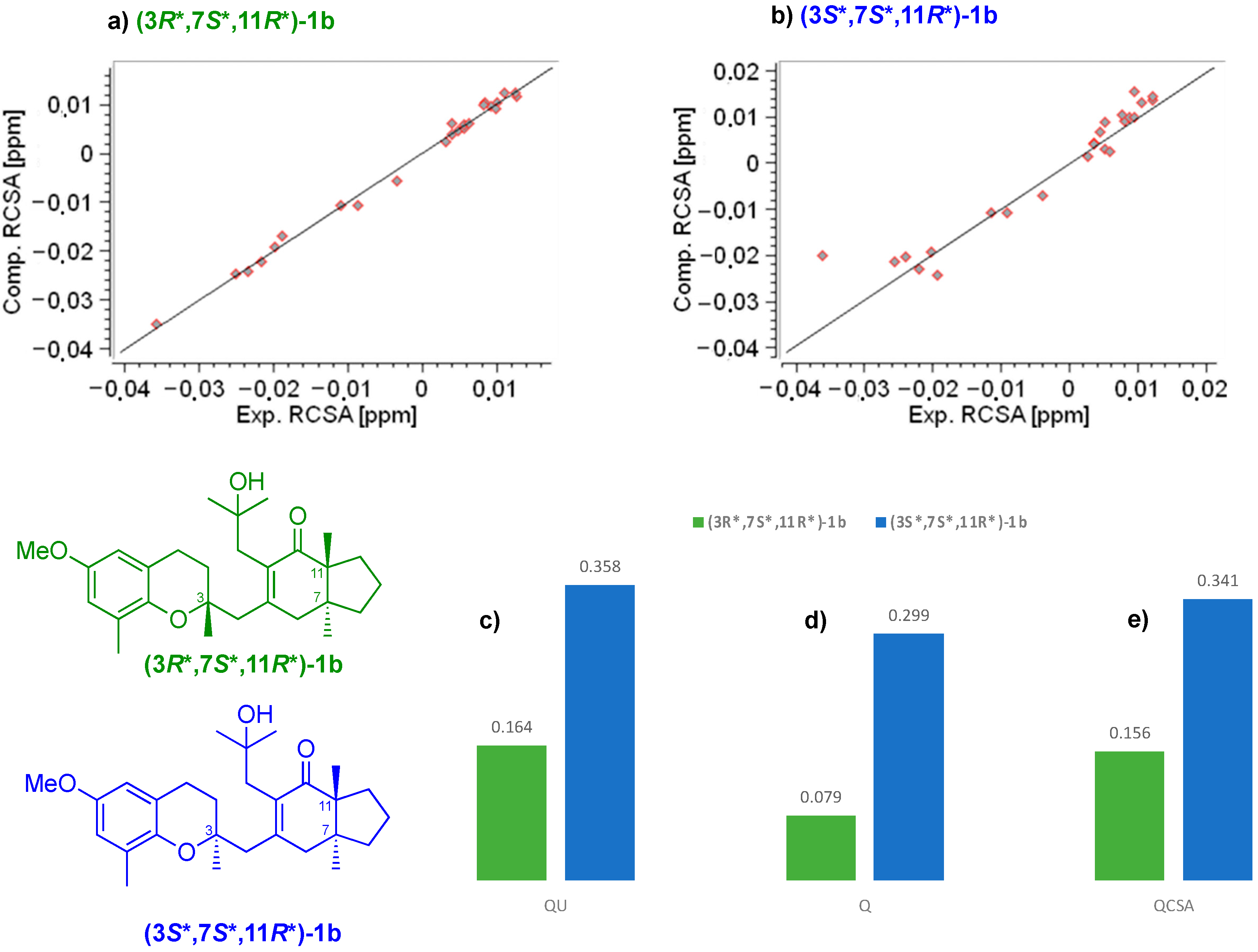
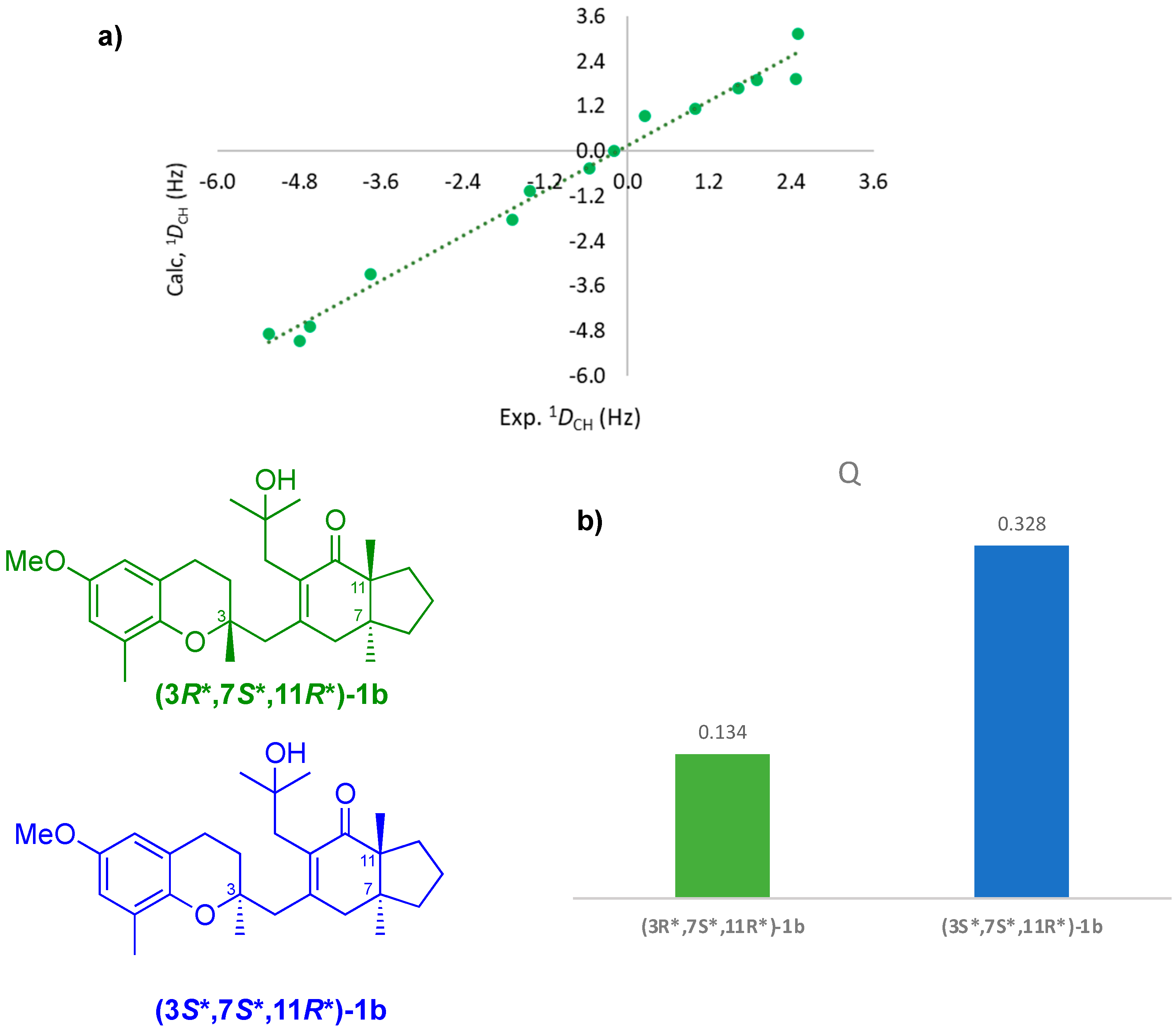
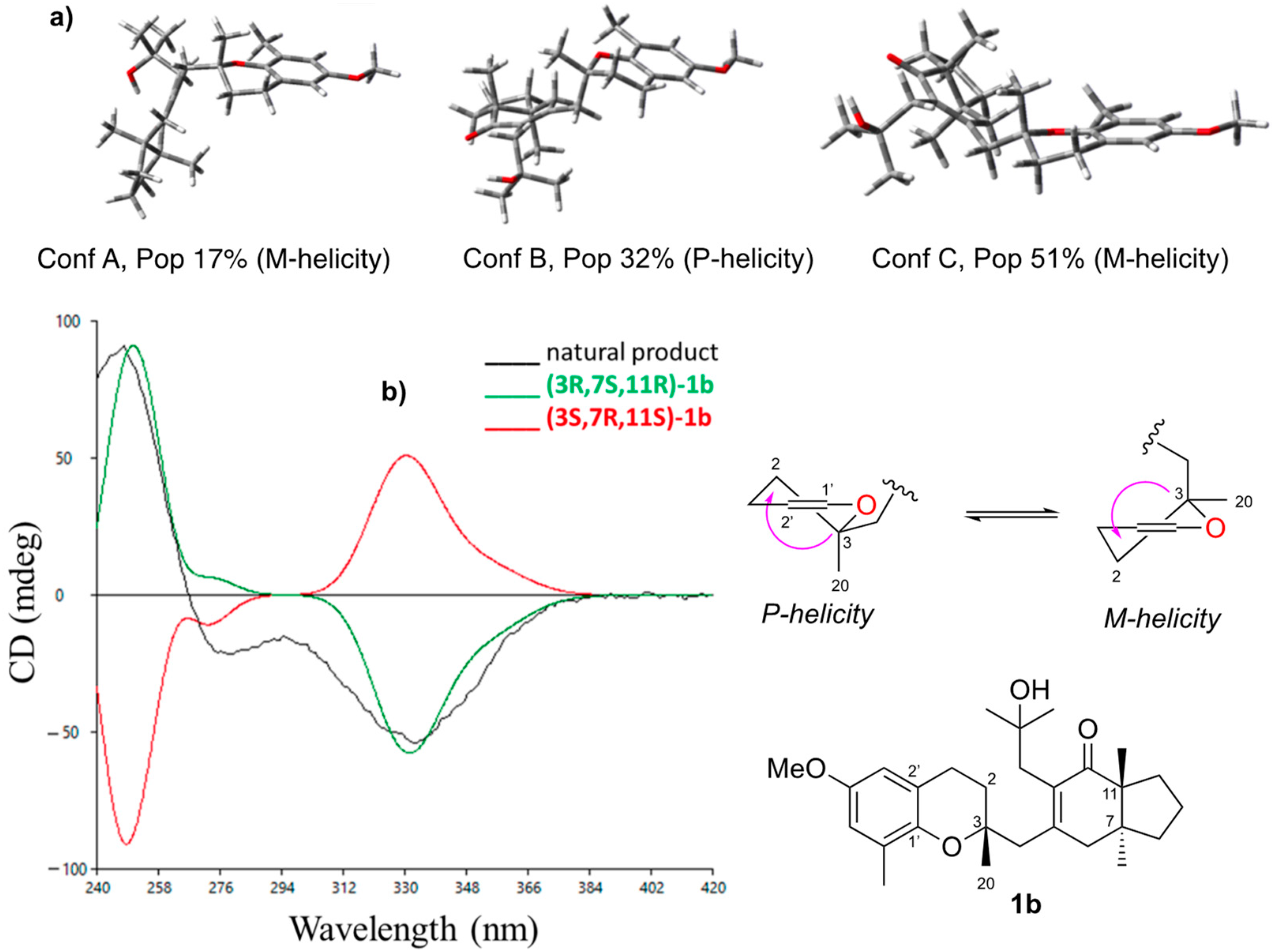
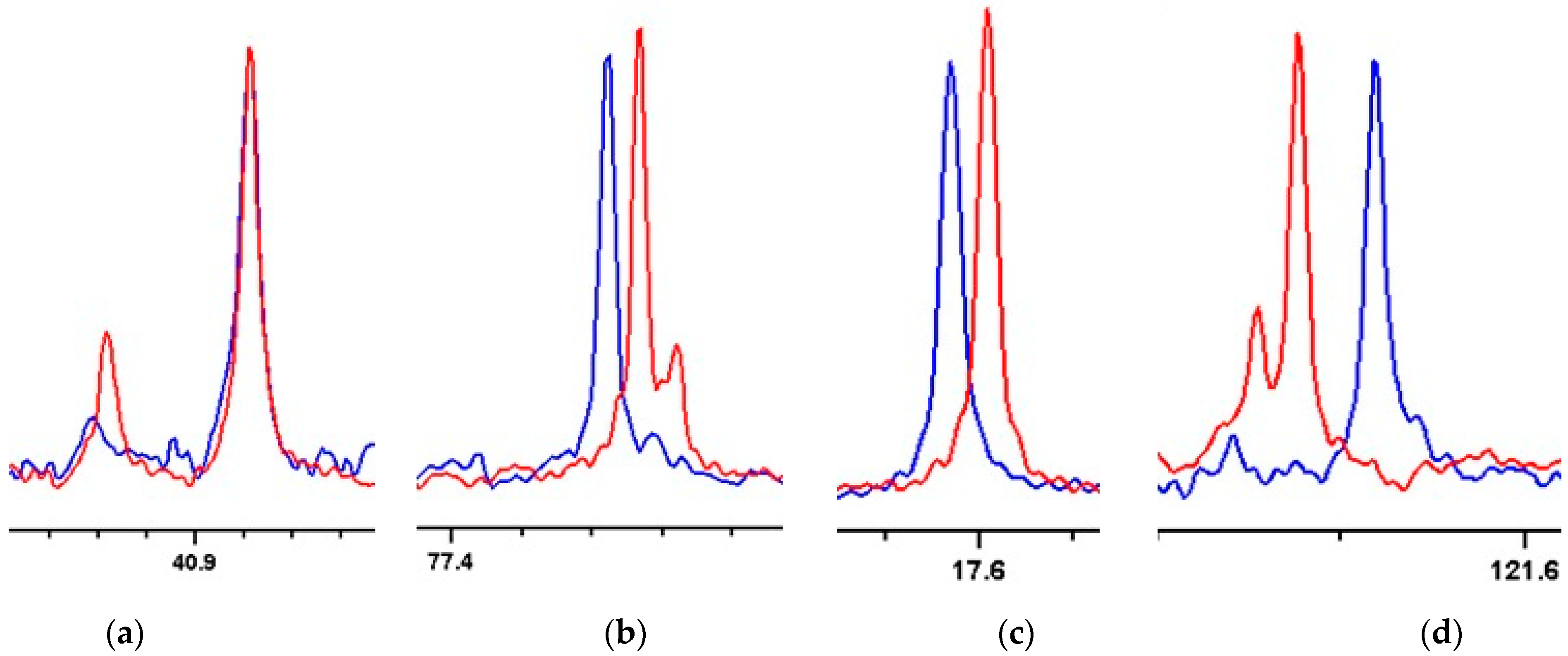
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Data Processing
3.3. Raw Material
3.4. Extraction and Isolation
Purification of 1b
3.5. Computational Section
J-Coupling Calculation
3.6. 13C-RCSA Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norton, T.A. The Growth and Development of Sargassum Muticum (Yendo) Fensholt. J. Exp. Mar. Biol. Ecol. 1977, 26, 41–53. [Google Scholar] [CrossRef]
- Critchley, A.T. Sargassum Muticum: A Taxonomic History Including World-Wide and Western Pacific Distributions. J. Mar. Biol. Assoc. U. K. 1983, 63, 617–625. [Google Scholar] [CrossRef]
- Davison, D.M. Sargassum Muticum in Scotland, 2008: A Review of Information, Issues and Implications; Commissioned Report No. 324; Scottish Natural Heritage: Perth, Scotland, 2009. [Google Scholar]
- Balboa, E.; Conde, E.; Constenla, A.; Falqué, E.; Domínguez, H. Sensory Evaluation and Oxidative Stability of a Suncream Formulated with Thermal Spring Waters from Ourense (NW Spain) and Sargassum Muticum Extracts. Cosmetics 2017, 4, 19. [Google Scholar] [CrossRef]
- Park, S.Y.; Seo, I.S.; Lee, S.J.; Lee, S.P. Study on the Health Benefits of Brown Algae Sargassum Muticum in Volunteers. J. Food Nutr. Res. 2015, 3, 126–130. [Google Scholar] [CrossRef][Green Version]
- Heo, S.J.; Jeon, Y.J. Protective Effect of Fucoxanthin Isolated from Sargassum Siliquastrum on UV-B Induced Cell Damage. J. Photochem. Photobiol. B Biol. 2009, 95, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.; Saleem, M.; Tousif, M.I.; Anwar, M.A.; Surup, F.; Ali, I.; Wang, D.; Mamadalieva, N.Z.; Alshammari, E.; Ashour, M.L.; et al. Meroterpenoids: A Comprehensive Update Insight on Structural Diversity and Biology. Biomolecules 2021, 11, 957. [Google Scholar] [CrossRef]
- Kim, J.A.; Ahn, B.N.; Kong, C.S.; Kim, S.K. The Chromene Sargachromanol e Inhibits Ultraviolet A-Induced Ageing of Skin in Human Dermal Fibroblasts. Br. J. Dermatol. 2013, 168, 968–976. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, J.P. New Chromene Derivatives with Radical Scavenging Activities from the Brown Alga Sargassum Siliquastrum. J. Chem. Res. 2017, 41, 116–119. [Google Scholar] [CrossRef]
- Balboa, E.M.; Li, Y.-X.; Ahn, B.-N.; Eom, S.-H.; Domínguez, H.; Jiménez, C.; Rodríguez, J. Photodamage Attenuation Effect by a Tetraprenyltoluquinol Chromane Meroterpenoid Isolated from Sargassum Muticum. J. Photochem. Photobiol. B: Biol. 2015, 148, 51–58. [Google Scholar] [CrossRef]
- Ferdous, U.T.; Yusof, Z.N.B. Algal Terpenoids: A Potential Source of Antioxidants for Cancer Therapy. In Terpenes and Terpenoids; Perveen, S., Al-Taweel, A.M., Eds.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar]
- Fisch, K.M.; Böhm, V.; Wright, A.D.; König, G.M. Antioxidative Meroterpenoids from the Brown Alga Cystoseira Crinita. J. Nat. Prod. 2003, 66, 968–975. [Google Scholar] [CrossRef]
- Valls, R.; Piovetti, L.; Banaigs, B.; Praud, A. Secondary Metabolites from Morocco Brown Algae of the Genus Cystoseira. Phytochemistry 1993, 32, 961–966. [Google Scholar] [CrossRef]
- de Sousa, C.B.; Gangadhar, K.N.; Morais, T.R.; Conserva, G.A.A.; Vizetto-Duarte, C.; Pereira, H.; Laurenti, M.D.; Campino, L.; Levy, D.; Uemi, M.; et al. Antileishmanial Activity of Meroditerpenoids from the Macroalgae Cystoseira Baccata. Exp. Parasitol. 2017, 174, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amico, V.; Cunsolo, F.; Oriente, G.; Piattelli, M.; Ruberto, G. Cystoketal, a New Metabolite From the Brown Alga Cystoseira Balearica. J. Nat. Prod. 1984, 47, 947–952. [Google Scholar] [CrossRef]
- Nath, N.; Schmidt, M.; Gil, R.R.; Williamson, R.T.; Martin, G.E.; Navarro-Vázquez, A.; Griesinger, C.; Liu, Y. Determination of Relative Configuration from Residual Chemical Shift Anisotropy. J. Am. Chem. Soc. 2016, 138, 9548–9556. [Google Scholar] [CrossRef]
- Gayathri, C.; Tsarevsky, N.V.; Gil, R.R. Residual Dipolar Couplings (RDCs) Analysis of Small Molecules Made Easy: Fast and Tuneable Alignment by Reversible Compression/Relaxation of Reusable PMMA Gels. Chem. –A Eur. J. 2010, 16, 3622–3626. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical Determination of Acyclic Structures Based on Carbon-Proton Spin-Coupling Constants. A Method of Configuration Analysis for Natural Products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Matsumori, N.; Murata, M.; Tachibana, K. Conformational Analysis of Natural Products Using Long-Range Carbon-Proton Coupling Constants: Three-Dimensional Structure of Okadaic Acid in Solution. Tetrahedron 1995, 51, 12229–12238. [Google Scholar] [CrossRef]
- Pachler, K.G.R. Nuclear Magnetic Resonance Study of Some α-Amino Acids—II. Rotational Isomerism. Spectrochim. Acta 1964, 20, 581–587. [Google Scholar] [CrossRef]
- Pachler, K.G.R. Nuclear Magnetic Resonance Study of Some α-Amino Acids—I. Spectrochim. Acta 1963, 19, 2085–2092. [Google Scholar] [CrossRef]
- Nath, N.; Fuentes-Monteverde, J.C.; Pech-Puch, D.; Rodríguez, J.; Jiménez, C.; Noll, M.; Kreiter, A.; Reggelin, M.; Navarro-Vázquez, A.; Griesinger, C. Relative Configuration of Micrograms of Natural Compounds Using Proton Residual Chemical Shift Anisotropy. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Teles, R.R.; França, J.A.; Navarro-Vázquez, A.; Hallwass, F. Atribuição da Estereoquímica da α-santonina através das medidas do Acoplamento Dipolar Residual. Química Nova 2015, 38, 1345–1350. [Google Scholar]
- Butts, C.P.; Jones, C.R.; Harvey, J.N. High Precision NOEs as a Probe for Low Level Conformers—A Second Conformation of Strychnine. Chem. Commun. 2011, 47, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The MPW and MPW1PW Models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A.J. Left-Right Correlation Energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Grimblat, N.; Gavín, J.A.; Hernández Daranas, A.; Sarotti, A.M. Combining the Power of J Coupling and DP4 Analysis on Stereochemical Assignments: The J-DP4 Methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, G.; Fernández, R.; Pérez, M.; Millán, R.E.; Jiménez, C.; Rodríguez, J.; Cuevas, C. Enigmazole C: A Cytotoxic Macrocyclic Lactone and Its Ring-Opened Derivatives from a New Species of Homophymia Sponge. J. Nat. Prod. 2022, 85, 1059–1066. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Troche-Pesqueira, E.; Anklin, C.; Gil, R.R.; Navarro-Vázquez, A. Computer-Assisted 3D Structure Elucidation of Natural Products Using Residual Dipolar Couplings. Angew. Chem. Int. Ed. 2017, 56, 3660–3664. [Google Scholar] [CrossRef]
- Navarro-Vázquez, A.; Gil, R.R.; Blinov, K. Computer-Assisted 3D Structure Elucidation (CASE-3D) of Natural Products Combining Isotropic and Anisotropic NMR Parameters. J. Nat. Prod. 2018, 81, 203–210. [Google Scholar] [CrossRef]
- Liu, Y.; Cohen, R.D.; Gustafson, K.R.; Martin, G.E.; Williamson, R.T. Enhanced Measurement of Residual Chemical Shift Anisotropy for Small Molecule Structure Elucidation. Chem. Commun. 2018, 54, 4254–4257. [Google Scholar] [CrossRef]
- Liu, Y.; Saurí, J.; Mevers, E.; Peczuh, M.W.; Hiemstra, H.; Clardy, J.; Martin, G.E.; Williamson, R.T. Unequivocal Determination of Complex Molecular Structures Using Anisotropic NMR Measurements. Science (1979) 2017, 356, eaam5349. [Google Scholar] [CrossRef]
- Schmidt, M.; Sun, H.; Rogne, P.; Scriba, G.K.E.; Griesinger, C.; Kuhn, L.T.; Reinscheid, U.M. Determining the Absolute Configuration of (+)−Mefloquine HCl, the Side-Effect-Reducing Enantiomer of the Antimalaria Drug Lariam. J. Am. Chem. Soc. 2012, 134, 3080–3083. [Google Scholar] [CrossRef] [PubMed]
- Tukey, J.W. Abstracts of Papers. Ann. Math. Stat. 1958, 29, 614–623. [Google Scholar] [CrossRef]
- Thiele, C.M.; Bermel, W. Speeding up the Measurement of One-Bond Scalar (1J) and Residual Dipolar Couplings (1D) by Using Non-Uniform Sampling (NUS). J. Magn. Reson. 2012, 216, 134–143. [Google Scholar] [CrossRef]
- Furrer, J.; John, M.; Kessler, H.; Luy, B. J-Spectroscopy in the Presence of Residual Dipolar Couplings: Determination of One-Bond Coupling Constants and Scalable Resolution. J. Biomol. NMR 2007, 37, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Antus, S.; Snatzke, G.; Steinke, I. Circulardichroismus, LXXXI. Synthese Und Circulardichroismus von Steroiden Mit Isochromanon-Chromophor. Liebigs Ann. Der Chem. 1983, 1983, 2247–2261. [Google Scholar] [CrossRef]
- Nozoe, S.; Hirai, K.; Snatzke, F.; Snaizke, G. Circular Dichroism-LXIV, On The Chiroptical Properties Of Siccamn Derivatives And The Absolute Configuration Of Siccanochromene-A. Tetrahedron 1974, 30, 2773–2776. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Chakraborty, D.K. Absolute Stereochemistry of Chiral Molecules from Ab Initio Theoretical and Experimental Molecular Optical Rotations. J. Am. Chem. Soc. 1998, 120, 6160–6164. [Google Scholar] [CrossRef]
- Stephens, P.J.; McCann, D.M.; Cheeseman, J.R.; Frisch, M.J. Determination of Absolute Configurations of Chiral Molecules Using Ab Initio Time-Dependent Density Functional Theory Calculations of Optical Rotation: How Reliable Are Absolute Configurations Obtained for Molecules with Small Rotations? Chirality 2005, 17, S52–S64. [Google Scholar] [CrossRef]
- Polavarapu, P.L. Optical Rotation: Recent Advances in Determining the Absolute Configuration. Chirality 2002, 14, 768–781. [Google Scholar] [CrossRef]
- Górecki, M.; Suszczyńska, A.; Woźnica, M.; Baj, A.; Wolniak, M.; Cyrański, M.K.; Witkowski, S.; Frelek, J. Chromane Helicity Rule-Scope and Challenges Based on an ECD Study of Various Trolox Derivatives. Org. Biomol. Chem. 2014, 12, 2235–2254. [Google Scholar] [CrossRef]
- Batista, J.M.; Batista, A.N.L.; Rinaldo, D.; Vilegas, W.; Cass, Q.B.; Bolzani, V.S.; Kato, M.J.; López, S.N.; Furlan, M.; Nafie, L.A. Absolute Configuration Reassignment of Two Chromanes from Peperomia Obtusifolia (Piperaceae) Using VCD and DFT Calculations. Tetrahedron Asymmetry 2010, 21, 2402–2407. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Melo Sousa, C.M.; Giordani, R.B.; Almeida, W.A.M.; Griesinger, C.; Gil, R.R.; Navarro-Vázquez, A.; Hallwass, F. Effect of the Solvent on the Conformation of Monocrotaline as Determined by Isotropic and Anisotropic NMR Parameters. Magn. Reson. Chem. 2021, 59, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kolmer, A.; Edwards, L.J.; Kuprov, I.; Thiele, C.M. Conformational Analysis of Small Organic Molecules Using NOE and RDC Data: A Discussion of Strychnine and α-Methylene-γ-Butyrolactone. J. Magn. Reson. 2015, 261, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Calculation of Optical Rotation Using Density Functional Theory. J. Phys. Chem. A 2001, 105, 5356–5371. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Anta, C.; González, N.; Rodríguez, J.; Jiménez, C. A New Secosterol from the Indonesian Octocoral Pachyclavularia Violacea. J. Nat. Prod. 2002, 65, 1357–1359. [Google Scholar] [CrossRef]
- Palermo, G.; Riccio, R.; Bifulco, G. Effect of Electronegative Substituents and Angular Dependence on the Heteronuclear Spin−Spin Coupling Constant 3 J C−H: An Empirical Prediction Equation Derived by Density Functional Theory Calculations. J. Org. Chem. 2010, 75, 1982–1991. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C. The Relationship between Proton-Proton NMR Coupling Constants and Substituent Electronegativities—I. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Hallwass, F.; Schmidt, M.; Sun, H.; Mazur, A.; Kummerlöwe, G.; Luy, B.; Navarro-Vázquez, A.; Griesinger, C.; Reinscheid, U.M. Residual Chemical Shift Anisotropy (RCSA): A Tool for the Analysis of the Configuration of Small Molecules. Angew. Chem. Int. Ed. 2011, 50, 9487–9490. [Google Scholar] [CrossRef] [PubMed]
- Hallwass, F.; Teles, R.R.; Hellemann, E.; Griesinger, C.; Gil, R.R.; Navarro-Vázquez, A. Measurement of Residual Chemical Shift Anisotropies in Compressed Polymethylmethacrylate Gels. Automatic Compensation of Gel Isotropic Shift Contribution. Magn. Reson. Chem. 2018, 56, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Cornilescu, G.; Marquardt, J.L.; Ottiger, M.; Bax, A. Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J. Am. Chem. Soc. 1998, 120, 6836–6837. [Google Scholar] [CrossRef]
- Losonczi, J.A.; Andrec, M.; Fischer, M.W.F.; Prestegard, J.H. Order Matrix Analysis of Residual Dipolar Couplings Using Singular Value Decomposition. J. Magn. Reson. 1999, 138, 334–342. [Google Scholar] [CrossRef]
- Navarro-Vázquez, A. MSpin-RDC. A Program for the Use of Residual Dipolar Couplings for Structure Elucidation of Small Molecules. Magn. Reson. Chem. 2012, 50, S73–S79. [Google Scholar] [CrossRef]
- Hellemann, E.; Teles, R.R.; Hallwass, F.; Barros, W.; Navarro-Vázquez, A.; Gil, R.R. Mechanical Behavior of Polymer Gels for RDCs and RCSAs Collection: NMR Imaging Study of Buckling Phenomena. Chem. –A Eur. J. 2016, 22, 16632–16635. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1b | ||
|---|---|---|
| Position | δC, mult. a,b | δH, mult., J (in Hz) c |
| 1 | 23.06 CH2 | 2.774 (t, 6.9) |
| 2 | 34.04 CH2 | H2a: 1.859 (dt, 13.5, 6.9) |
| H2b: 1.805 (dt, 13.5, 6.9) | ||
| 3 | 76.80 qC | - |
| 4 | 45.15 CH2 | H4b: 2.705 (d, 13.7) |
| H4a: 2.514 (d, 13.7) | ||
| 5 | 155.10 qC | - |
| 6 | 44.77 CH2 | H6b: 3.028 (d, 18.7) |
| H6a: 2.237 (d, 18.7) | ||
| 7 | 45.25 qC | - |
| 8 | 35.38 CH2 | H8b: 1.754 (m) |
| H8a: 1.520 (m) | ||
| 9 | 19.32 CH2 | 1.744 (m) |
| 10 | 30.03 CH2 | H10a: 1.944 (td, 12.0, 11.9, 6.8) |
| H10b: 1.441 (ddd, 13.1, 8.4, 3.1) | ||
| 11 | 55.42 qC | - |
| 12 | 209.32 qC | - |
| 13 | 133.45 qC | - |
| 14 | 40.37 CH2 | H14b: 2.570 (d, 14.3) |
| H14a: 2.506 (d, 14.3) | ||
| 15 | 71.17 qC | - |
| Me16- | 31.88 CH3 | 1.235 (s) |
| Me17- | 29.08 CH3 | 1.041 (s) |
| Me18- | 21.51 CH3 | 1.105 (s) |
| Me19- | 22.71 CH3 | 0.804 (s) |
| Me20- | 24.28 CH3 | 1.225 (s) |
| 1′ | 145.77 qC | - |
| 2′ | 121.16 qC | - |
| 3′ | 111.58 CH | 6.450 (d, 3.0) |
| 4′ | 153.13 qC | - |
| 5′ | 115.64 CH | 6.563 (d, 3.0) |
| 6′ | 127.43 qC | - |
| MeO-4′ | 55.97 CH3 | 3.701 (s) |
| Me-6′ | 17.08 CH3 | 2.165 (s) |
| OH | - | 3.983 (br s) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes-Monteverde, J.C.C.; Nath, N.; Forero, A.M.; Balboa, E.M.; Navarro-Vázquez, A.; Griesinger, C.; Jiménez, C.; Rodríguez, J. Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Mar. Drugs 2022, 20, 462. https://doi.org/10.3390/md20070462
Fuentes-Monteverde JCC, Nath N, Forero AM, Balboa EM, Navarro-Vázquez A, Griesinger C, Jiménez C, Rodríguez J. Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Marine Drugs. 2022; 20(7):462. https://doi.org/10.3390/md20070462
Chicago/Turabian StyleFuentes-Monteverde, Juan Carlos C., Nilamoni Nath, Abel M. Forero, Elena M. Balboa, Armando Navarro-Vázquez, Christian Griesinger, Carlos Jiménez, and Jaime Rodríguez. 2022. "Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum" Marine Drugs 20, no. 7: 462. https://doi.org/10.3390/md20070462
APA StyleFuentes-Monteverde, J. C. C., Nath, N., Forero, A. M., Balboa, E. M., Navarro-Vázquez, A., Griesinger, C., Jiménez, C., & Rodríguez, J. (2022). Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Marine Drugs, 20(7), 462. https://doi.org/10.3390/md20070462