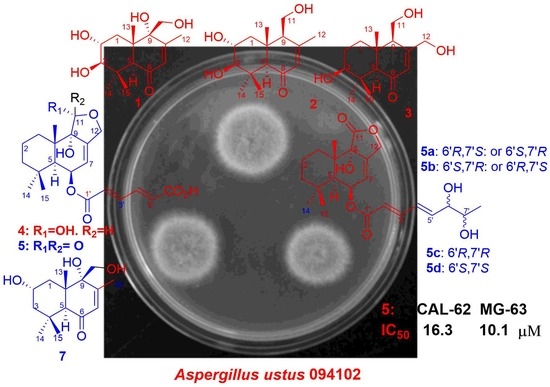
2. Results and Discussion
The bioactive EtOAc extract of the fermentation broth of the mangrove-derived fungus
Aspergillus ustus 094102 was chromatographed on silica gel, Sephadex LH-20, and preparative HPLC columns to give compounds
1–
7 (
Figure 1).
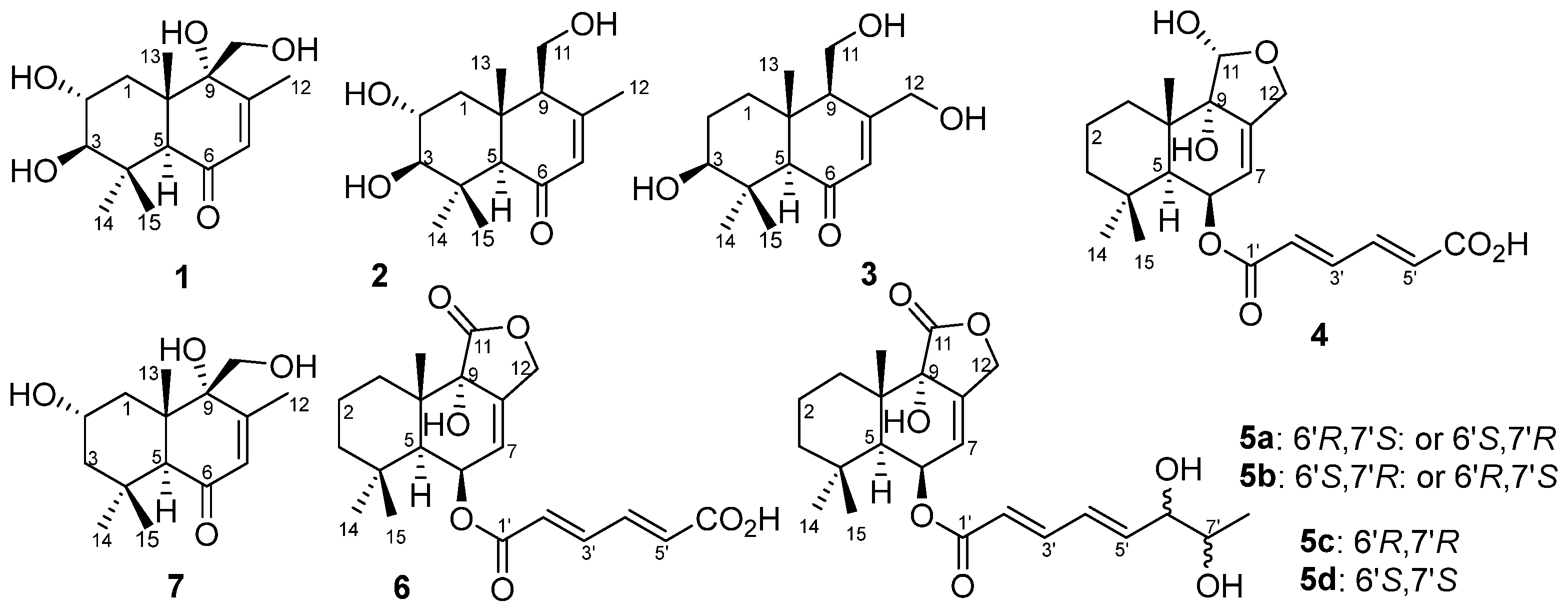
Compound
1 was obtained as a colorless oily solid. Its molecular formula was determined as C
15H
24O
5 based on the high-resolution mass spectrometry (HRMS, ESI-Orbitrap) peak at
m/
z 285.1694 [M+H]
+ or 283.1547 [M–H]
− (
Figure S1), indicating 4 index of hydrogen deficiency (IHD). The IR spectrum at ν
max 3399 and 1663 cm
−1 (
Figure S2), corresponded to a hydroxy and an
α,
β-unsaturated carbonyl group, respectively. The
1H-NMR data (
Table 1,
Figure S3) of
1 revealed four tertiary methyl groups at
δH 1.04 (s, H-13/15), 1.14 (s, H-14) and 1.96 (s, H-12), an oxymethylene signal at
δH 3.53/3.64 (d/d, H-11), a methylene signal at
δH 1.85/1.69 (dd/t, H-1), one olefinic proton signal at
δH 5.61 (d, H-7), three methine signals at
δH 3.47 (dt, H-2), 2.67 (d, H-3) and 2.81 (s, H-5), as well as four exchangeable proton signals at
δH 4.48 (HO-3/2), 4.91 (HO-11) and 5.06 (HO-9). The
13C-NMR and DEPT data (
Table 1,
Figures S4 and S5) of
1 revealed 15 carbon signals, including a ketone carbonyl signal at
δC 199.2 (C-6), two olefinic carbons at
δC 128.2/157.4 (C-7/C-8), four methyl signals at
δC 16.7/19.1/19.2/29.3 (C-15/C-13/C-12/C-14), two methylenes at
δC 38.6/61.9 (C-1/C-11), three methines at
δC 55.0/66.4/81.6 (C-5/C-2/C-3) and three nonhydrogenated carbons at
δC 37.8/45.4/74.6 (C-4/C-10/C-9). These NMR data (
Table 1) were closely related to those of 3
β,9
α,11-trihydroxydrim-7-en-6-one (that is 3
β,9
α,11-trihydroxy-6-oxodrim-7-ene [
7]), indicating the presence of a drimane sesquiterpene skeleton. The key difference was that compound
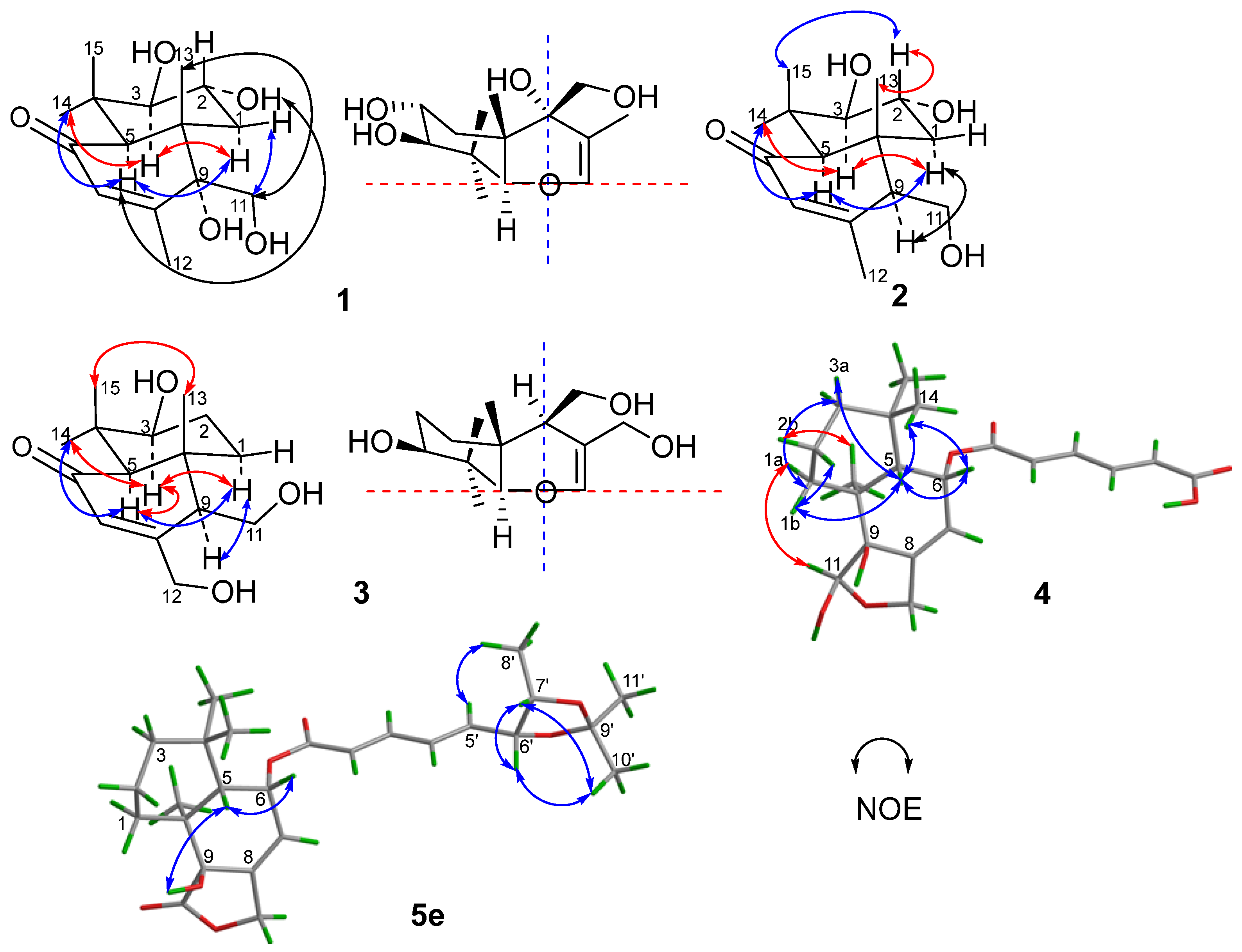
1 possessed an additional hydroxy group that resided at C-2 of ring A. On the basis of correlations in the COSY experiments between HO-3/H-3, H-3/H-2/H-1 and HO-11/H-11, as well as the key HMBC correlations from H-1 to C-5/C-10/C-13, H-3 to C-4/C-14/C-15, H-5 to C-4/C-6/C-9/C-10/C-13/C-14/C-15, H-7 to C-5/C-9/C-12, H-11 to C-8/C-9/C-10, H-12 to C-7/C-8/C-9, H-13 to C-5/C-9/C-10, H-14 to C-3/C-4/C-5/C-15, and H-15 to C-3/C-4/C-14 (
Figure 2 and
Figures S6–S8) further confirmed the constitution of
1 (
Figure 1). The relative configuration was deduced from the NOESY spectrum (
Figure 3 and
Figure S9), which showed correlations of H-1
α to H-3/H-5/HO-9, H-11 to H-1
β/H-2/H-13, and H-2 to H-13 indicated
cis-orientation of HO-2/H-5/HO-9, and H-2/HO-3/CH
3-13/CH
2-11, and a
trans-fused decalin nucleus. The absolute configuration of
1 was determined by its ECD spectrum. On the basis of the octant rule for cyclohexenones [
11,
12,
13], the positive Cotton effect at λ
max 336 nm (∆ε + 8.4) and the negative Cotton effect at λ
max 240 nm (∆ε − 41.3) (
Figure 3 and
Figure S10) indicated the (2
R,3
R,5
S,9
R,10
S)-configuration, consistent with the core configuration of the drimane sesquiterpene, 9
α,11-dihydroxydrim-7-en-6-one (that is 6-oxo-7-drimen-9
α,11-diol [
14]), whose absolute configurations have been established by chemical synthesis. Therefore, compound
1, which we named ustusol F, was determined as (2
R,3
R,5
S,9
R,10
S)-2,3,9,11-tetrahydroxydrim-7-en-6-one.
Compound
2 was obtained as a light-yellow oil. Its molecular formula was determined as C
15H
24O
4 based on the HRESIMS peak at
m/
z 269.1751 [M+H]
+ (
Figure S11). The similar IR and UV absorptions to those of
1 implied that they shared the same molecular skeleton (
Figure S12). The 1D NMR data (
Table 1,
Figures S13–S15) were also similar to
1 except for a methine signal at
δC/H 57.1/2.29 which replaced the nonhydrogenated oxycarbon at
δC 74.6, the disappearance of a hydroxy signal at
δH 5.06 (HO-9), and the changes of chemical shifts around C-9. These data combined with the 16 amu less of molecular weight than
1 revealed compound
2 as the 9-deoxy derivative of compound
1. Key COSY of H-9/H-11/HO-11 and HMBC of H-11 to C-8 and C-10 and H-9 to C-10 (
Figure 2,
Figures S16 and S18) supported the inference. The same relative configuration to
1 was deduced from the NOESY correlations of H-2 (
δH 3.45) to H-13 (
δH 0.88), H-15 (
δH 1.03) and H-1
β (
δH 2.09), H-1
α (
δH 1.33) to H-3 (
δH 2.75), H-5 (
δH 2.22) and H-9 (
δH 2.29), and H-3 to H-14 (
δH 1.12) (
Figure 3 and
Figure S19). The absolute configuration of the
threo-2,3-diol in
2 was assigned by a dimolybdenum-induced ECD method [
15,
16]. Upon addition of Mo
2(OAc)
4 to a DMSO solution of compound
2, a chiral dimolybdenum complex was generated in situ as an auxiliary chromophore. Because the contribution from the inherent ECD was subtracted to give the induced ECD of the complex, the observed sign of the Cotton effect in the induced spectrum originates solely from the chirality of the
ortho-diol moiety expressed by the sign of the O–C–C–O torsion angle. The positive Cotton effect at λ
max 332 (∆
ε + 6.8) nm (
Figure S20) permitted us to assign the (2
R,3
R)-configuration on the basis of Snatzke’s empirical rule [
15]. In addition, compounds
1 and
2 also showed a similar ECD Cotton effect, indicating the same absolute configuration. Thus, compound
2, which we named 9-deoxyustusol F, was determined as (2
R,3
R,5
R,9
S,10
R)-2,3,11-trihydroxydrim-7-en-6-one.
Compound
3 was obtained as a colorless oily solid. Its molecular formula was determined as C
15H
24O
4 based on the HRESIMS peak at
m/
z 269.1750 [M+H]
+ (
Figure S21), indicating an isomer of
2. Similar 1D NMR data (
Table 1,
Figures S23–S25) with
2 were observed. In addition, a methylene signal (
δH/C 1.47/26.7) and an oxymethylene signal (
δH/C 4.19/4.26/61.3) replaced the methyl signal (
δH/C 1.98/21.5) and oxymethine signal (
δH/C 3.45/66.1). Key COSY of H-1/H
2-2/H-3 as well as the HMBC of H
2-12 (
δH 4.19/4.26) to C-8 (
δC 162.3), H-7 (
δH 5.96) to C-12 (
δC 61.3) and H
2-2 (
δH 1.47) to C-4 (
δC 37.5) and C-10 (
δC 41.7) revealed that 2-OH was moved to C-12 to form 2-CH
2 and 12-CH
2OH, respectively (
Figure 2,
Figures S26 and S28). The relative configuration of compound
3 was deduced from the NOE difference (NOEdiff) experiment. NOEdiff of
3 showed that H-5 (
δH 2.15) and H-1a (
δH 1.44) were enhanced after the irradiation of H-9 (
δH 2.31), while H-3 (
δH 3.02) and H-9 (
δH 2.31) were enhanced after the irradiation of H-5. The NOE enhancements of H-3 (
δH 3.02) and H-5 (
δH 2.15) were also observed after H-14 (
δH 1.10) was irradiated, while H-13 (
δH 0.80) was enhanced after the irradiation of H-15 (
δH 0.99). H-1b (
δH 1.88) and H-15 was enhanced after the irradiation of H-13 (
Figure S29). These NOE data indicated the
cis-orientation of H-3, H-5, H-9 and H-14 as well as H-13 and H-15, indicating the same relative configuration of
3 to
2 in the chiral centers of C-3, C-5, C-9, and C-10. The similar ECD spectrum to that of
2 implied the same absolute configuration, which was confirmed by octant rule for cyclohexanone [
11,
12,
13], the positive Cotton effect at λ
max 335 nm (∆ε + 10.6) and the negative Cotton effect at λ
max 241 nm (∆ε – 19.1) (
Figure 4 and
Figure S30). Accordingly, compound
3, which we named ustusol G, was elucidated as (3
S,5
R,9
R,10
R)-3,11,12-trihydroxydrim-7-en-6-one.
Compound
4 was obtained as a colorless solid. Its molecular formula was determined as C
21H
28O
7 based on the HRESIMS peak at
m/
z 391.1762 [M–H]
–, indicating 8 HIDs (
Figure S32). The IR spectrum showed absorption bands of hydroxyl and conjugated carbonyl at ν
max 3434 and 1696 cm
−1 (
Figure S33), respectively. The 1D NMR spectra of
4 (
Table 2,
Figures S34–S36) were very similar to those of (2
E,4
E)-(strobilactone A-6-yl)-5-carboxypenta-2,4-dienoate (that is mono(6-strobilactone B) ester of (
E,
E)-2,4-hexadienedioic acid [
7]), which we named ustusolate J (
6) for convenience, suggesting that they shared the same molecular scaffold. The only difference was a replacement of the lactone carbonyl signal (
δC 174.6 in
6) by the hemiacetal methine group (
δC/H 97.4/5.20 in
4). In addition, the chemical shifts for C-9 and C-7 have a great increase and decrease, respectively (
Table 2 and
Figure S35). These data combined with a 2 amu more than
6 suggested that the γ-lactone of
6 was reduced to the corresponding hemiacetal in
4. The key HMBC correlations from hemiacetal proton (
δH-11 5.20) to C-9 (
δC 76.4)/C-10 (
δC 38.0)/C-12 (
δC 65.8), from H-12 (
δH 4.08/4.38) to C-7 (
δC 117.0)/C-8 (
δC 143.2)/C-9/C-11 (
δC 97.3), and from H-7 (
δH 5.49) to C-5 (
δC 45.1)/C-9 verified the deduction (
Figure 2 and
Figure S39). Compound
4 displayed the key NOESY correlations of H-6 (
δH 5.58) with H-5 (
δH 2.07) and H-14 (
δH 0.91), H-5 with H-1b (
δH 1.86) and H-2a (
δH 1.42), H-11 (
δH 5.20) with H-1a (
δH 1.22), as well as H-13 (
δH 1.12) with H-2b (
δH 1.58) (
Figure 3 and
Figure S40), indicating
cis-orientation of H-5 with H-6 and
trans-orientation of H-5 with H-11 and H-13 which is the same relative configuration of
4 to
1 and
6 in the decalin (decahydronaphthalene) nucleus. The same relative configuration of HO-9 was deduced from the same biosynthetic pathway to those of compounds
1 and
5–
7. Subsequently, the same ECD pattern of
4–
6 (
Figure S78) implied the same absolute configuration of the drimane nucleus. Compound
4, which we named ustusolate H, was thus elucidated as (5
S,6
R,9
S,10
S,11
R,2′
E,4′
E)-(11-deoxy-11-hydroxystrobilactone A-6-yl)-5-carboxypenta-2,4-dienoate.
Compound
5 was obtained as a yellow oil. Its molecular formula was determined as C
21H
28O
7 based on the ESIMS peak at
m/
z 419.1 for [M–H]
– and
m/
z 464.9 for [M + HCO
2]
–(
Figure S42), indicating 8 HIDs. A literature search verified that the constitution (planar structure) of compound
5 was the same as the (strobilactone A-6-yl) (2
E,4
E)-6,7-dihydroxyocta-2,4-dienoate (that is (6-strobilactone-B) esters of (
E,
E)-6,7-dihydroxy-2,4-octadienoic acid [
7]), for almost the same NMR data. However, four sets of
13C NMR signals of compound
5 (
Figure S40) for the side chain at
δC 165.51/165.50/165.49/165.47 (C-1′), 120.03/120.99/119.95/119.90 (C-2′), 145.41/145.37/145.34/145.26 (C-3′), 127.54/127.35/127.16/126.98 (C-4′), 146.18/146.12/145.48/145.45 (C-5′), 75.16/75.00/74.64/74.46 (C-6′), 69.64/69.62/69.33/69.32 (C-7′), and 19.34/19.26/18.26/18.24 (C-8′) were observed, indicating four stereoisomers of
5 resulted from the
ortho-diol chiral centers of the side chain. With the help of HPLC, compound
5, which we named ustusolate I for convenience, was confirmed to have four baseline-separated peaks, then purified
5a,
5b,
5c, and
5d were obtained (
Figure S83). The NMR differences of
5a–
5d were concentrated in the side chains (
Table 3 and
Table 4,
Figures S45–S60), and indicated that compounds
5a and
5b,
5c, and
5d were two pairs of enantiomers of the
ortho-diol in the side chain. To elucidate the relative configuration of 6′,7′-diol moiety, the acetonide (
5e) was prepared from
5a (
Figure 4). The 1D and 2D NMR spectra (
Table 3 and
Table 4,
Figures S66–S70), as well as the NOESY correlations of H-5′ (
δH 6.24)/H
3-8′ (
δH 1.01) and H
3-11′ (
δH 1.40), H-6′ (
δH 4.62)/H-7′ (
δH 4.34) and H
3-10′ (
δH 1.28) in
5e (
Figure 3 and
Figure S71) clearly suggested an
erythro-6′,7′-diol in
5a and
5b, and a
threo-6′,7′-diol in
5c and
5d was accordingly elucidated. This conclusion is consistent with the chemical shift rule of methyl carbon (
δCH3) for 1-methyl-1,2-diol by chemical synthesis, that is 18.1–18.6 and 19.1–19.6 ppm for
threo- and
erythro-1,2-diol, respectively [
17]. The absolute configuration of the
threo-6′,7′-diol in
5c and
5d was assigned by a dimolybdenum-induced ECD method [
15,
16] in the same manner as that of compound
2. Upon addition of Mo
2(OAc)
4 to a solution of compounds
5c and
5d in DMSO, a chiral dimolybdenum complex was generated in situ as an auxiliary chromophore. According to the negative ECD Cotton effects of
5c at λ
max 303 (Δ
ε – 7.9) nm and the positive ECD Cotton effects of compound
5d at λ
max 305 (Δ
ε +2.37) nm (
Figures S73 and S74), the absolute configuration of
threo-6′,7′-diol in
5c and
5d were determined to be (6′
R,7′
R) and (6′
S,7′
S), respectively. Thus, the structure of
5c and
5d was unambiguously determined as (2′
E,4′
E,6′
R,7′
R)-ustusolate I (
5c) and (2′
E,4′
E,6′
S,7′
S)-ustusolate I (
5d), respectively. Unfortunately, the absolute configuration of compounds
5a and
5b were not determined yet in this paper, which we tentatively named (2′
E,4′
E;6′,7′-
erythro)-ustusolate I (
5a) and (2′
E,4′
E;
ent-6′,7′-
erythro)-ustusolate I (
5b), respectively.
Compounds
6 and
7 which could be a 3-deoxy derivative of ustusol F (
1) were identified by respective comparison of NMR data with those of mono(6-strobilactone-B) ester of (
E,
E)-2,4-hexadienedioic acid [
7] and 2
α,9
α,11-trihydroxy-6-oxodrim-7-ene [
7]. The same ECD pattern of compound
6 with
5 (
Figure S78) and compound
7 with
1 (
Figure S31) indicated they shared the same absolute configuration. Thus, compounds
6 and
7 were respectively identified as (5
S,6
R,9
S,10
S,2′
E,4′
E)-(strobilactone A-6-yl)-5-carboxypenta-2,4-dienoate (ustusolate J,
6) and (2
S,5
S,9
R,10
S)-2,9,11-trihydroxydrim-7-en-6-one (ustusol B,
7) in this paper. In addition, compound 7 showed almost the same NMR data as our previously reported ustusol B [
4] (
Table S1) and displayed the same retention times in the co-HPLC (
Figure S91). Thus, the structure of ustusol B was revised as structure
7, which was named ustusol B.
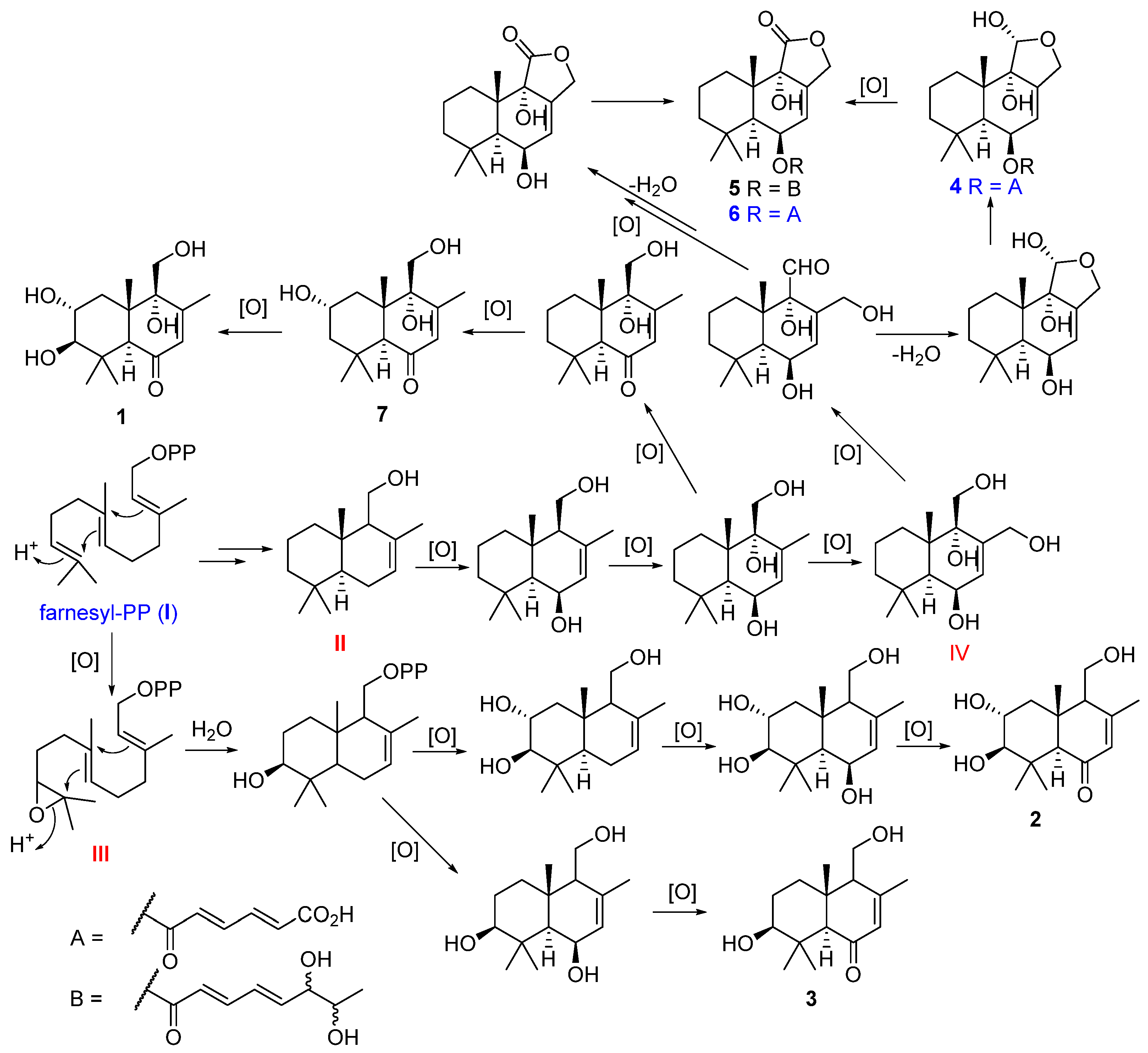
The drimane sesquiterpenoids
1–
7 were postulated to be biosynthesized from farnesyl-PP (
I) which generated intermediate
II,
III and
IV after cyclization and oxidation. The intermediates
II and
III were subjected to further oxidation to form compounds
1,
2,
3, and
7. The intermediate
II was further oxidized to intermediate
IV, and the latter was subjected to oxidation, hemi acetalization, and esterification to form compounds
4,
5, and
6 (
Figure 5).
The antiproliferations of compounds
1–
7 were evaluated against 29 human cancer cell lines and a normal cell line (the names of cell lines are listed in the
Supplementary Files) by the cell counting Kit-8 (CCK-8) methods [
18,
19]. Only compound
5, the mixture of
5a/
5b/
5c/
5d, showed antiproliferative activity against the human thyroid cancer cells (CAL-62) and human osteosarcoma cells (MG-63) with the IC
50 values of 16.28 ± 1.01 and 10.08 ± 0.04 µM, respectively, while the pure compounds
5a–
5d were inactive (IC
50 ≥ 50 µM). The IC
50 values of doxorubicin (positive control) against CAL-62 and MG-63 were 0.062 ± 0.022 and 0.096 ± 0.012 µM, respectively. The bacteriostatic activities of compounds
1–
7 against 6 human pathogenic bacteria and 6 aquatic pathogenic bacteria (the names are listed in the
Supplementary Files) were tested by the diffusion method of filter paper, but no inhibition zone was observed at the concentration of 100 μg/mL for compounds
1–
7.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured with a JASCO P-1020 digital polarimeter. UV data were recorded with a Beckman DU 640 spectrophotometer, and ECD data were collected using a JASCO J-715 spectropolarimeter. IR spectra were taken on a Nicolet NEXUS 470 spectrophotometer as KBr disks. 1H, 13C, DEPT, HMQC, HMBC, COSY, and NOESY NMR spectra were recorded on a JEOL JNM-ECP 600 spectrometer or a Bruker Avance 500 spectrometer in DMSO-d6 solution and were referenced to the corresponding residual solvent signals (δH/C 2.50/39.52 for DMSO-d6). HRESIMS spectra were collected using a Q-TOF Ultima Global GAA076 LC mass spectrometer. ESIMS data were measured using a Waters ACQUITY SQD 2 UPLC/MS system with a reversed-phase C18 column (ACQUITY UPLC BEH C18, 2.1 × 50 mm, 1.7 μm) at a flow rate of 0.4 mL/min. Semipreparative HPLC was performed using an ODS column (YMC- pack ODS-A, 10 × 250 mm, 5 μm, 4 mL/min) and a phenyl column (YMC-pack Ph, 10 × 250 mm, 5 μm, 4 mL/min). Vacuum–liquid chromatography (VLC) utilized silica gel H (Qingdao Marine Chemical Factory, Qingdao, China). TLC were carried out by plates precoated with silica gel GF254 (10–40 μm, Qingdao Marine Chemical Factory) and Sephadex LH-20 (Amersham Pharmacia Biotech, Buckinghamshire, UK) were used for column chromatography (CC).
3.2. Fungal Material
The mangrove fungal strain
A. ustus 094102 was isolated from the rhizosphere soil of the mangrove plant
Bruguiera gymnorrhiza grown in Wenchang, Hainan Province of China. It was identified according to the morphological characteristics and the ITS sequences [
4,
5].
3.3. Cultivation and Extraction
The fungus A. ustus 094102 was statically cultured at 25 °C for 28 days in one hundred 1000 mL conical flasks, each containing 300 mL of the liquid medium that was prepared by dissolving maltose (20 g), mannitol (20 g), glucose (10 g), monosodium glutamate (10 g), yeast extract (3 g), corn steep liquor (1 g), CaCO3 (2 g), KH2PO4 (0.5 g), MgSO4·7H2O (0.3 g), and sea salt (33 g) in 1 L of tap water (pH 7.0). The whole fermentation broth (30 L) was filtered by cheesecloth to separate the mycelia from the filtrate. The mycelia were extracted three times with an 80% volume of aqueous acetone. The acetone solution was concentrated under reduced pressure to give an aqueous solution. The aqueous solution was extracted three times with an equivalent volume of ethyl acetate (EtOAc), while the filtrate was extracted three times with an equivalent volume of EtOAc. All EtOAc extracts were combined and concentrated under vacuum to give 240 g of crude gum.
3.4. Purification
The crude gum (240 g) was separated into ten fractions (Fr1–Fr10) on a silica gel VLC column using a stepwise gradient elution of petroleum ether (PE), PE-CH2Cl2 (1:1–0:1) followed by CH2Cl2-MeOH (1:0–1:1). Fr9 (26 g) was fractionated on Sephadex LH-20, eluted with CH2Cl2-MeOH (1:1), to obtain three subfractions (Fr9.1–Fr9.3). Fr9.2 (8 g) was further separated into five subfractions (Fr9.2.1–Fr9.2.5) by VLC on the RP-18 column using a stepwise gradient elution of MeOH-H2O (9:1–1:1), among which the elution of 40% MeOH–H2O gave compound 7 (9.2 mg). Compounds 1 (6.2 mg, tR 11.8 min) and 2 (32 mg, tR 18.7 min) were obtained from Fr9.2.2 (1.7 g) by semipreparative HPLC over an ODS column eluting with 15% MeCN-H2O containing 0.5‰ Et3N. Fr7 (12 g) was fractionated on Sephadex LH-20, eluted with MeOH-CH2Cl2 (1:1), to obtain four subfractions (Fr7.1–Fr7.4). Fr7.4 (3.3 g) was further purified by semipreparative HPLC over an ODS column eluting with 40% MeCN-H2O containing 0.5‰ TFA (trifluoroacetic acid) to yield compound 4 (7.6 mg, tR 16.5 min). Fr7.3 (1.3 g) was fractionated into four subfractions (Fr7.3.1–Fr7.3.5) on a RP-18 column using a stepwise gradient elution of MeOH-H2O (1:9–2:3). Fr7.3.2 (300 mg) was further separated by semipreparative HPLC on an ODS column eluted with 20% MeCN-H2O to yield compound 3 (3.1 mg, tR 7.8 min). Fr6 (17.6 g) was further fractionated on Sephadex LH-20 eluted with MeOH-CH2Cl2 (1:1) to afford four subfractions (Fr6.1–Fr6.4). Fr6.2 (1.1 g) was further separated by semipreparative HPLC on an ODS column eluted with 40% MeCN-H2O containing 0.5‰ TFA to yield compound 6 (16.3 mg, tR 15 min), while compound 5 (860 mg, tR 17.0 min) was purified from Fr6.4 (9 g) by semipreparative HPLC on an ODS column eluted with 65% MeCN-H2O. Pure compounds 5a (8.8 mg, tR 39 min), 5b (5.4 mg, tR 42 min), 5c (7.3 mg, tR 44 min) and 5d (6.8 mg, tR 46 min) were obtained from compound 5 by a careful separation on an ODS column eluted with 50% MeOH-H2O.
3.5. The Preparation of Acetonide (5e) for Relative Configuration
According to our procedure [
16], compound
5a (5 mg) in acetone (3 mL) was added to the mixture of 2,2-dimethoxypropane (1 mL), pyridinium
p-toluenesulfonate (PPTS, 26 mg) and
N,
N-dimethylformamide (DMF, 1 mL). The resulting solution was stirred at room temperature (rt) for 12 h, and then 5 mL of H
2O was added. The reaction solution was extracted with 15mL of CH
2Cl
2, and the organic phase was concentrated under reduced pressure. The residue was purified by semipreparative HPLC (95% MeOH-H
2O) to yield the acetonide
5e (3.4 mg, t
R 5.7 min). Its structure was identified by ESIMS (
Figure S65) and NMR data (
Table 3 and
Table 4,
Figure 4 and
Figures S66–S71).
3.6. The Induced ECD Spectra of Compounds 2, 5c, and 5d for Absolute Configuration
According to a published procedure [
16,
17], analytical pure DMSO was dried with 4 Å molecular sieves and was used to prepare 0.6 mg/mL of Mo
2(OAc)
4 solution. To three pieces of this solution (each 1 mL, 1.40 μmol), compounds
2 (0.5 mg, 1.86 μmol),
5c (0.8 mg, 1.90 μmol), and
5d (0.8 mg, 1.90 μmol) were respectively added and the first ECD spectra of the mixtures were recorded immediately. Then, ECD spectra were continuously recorded every 10 min until stationary. The inherent ECD spectrum was subtracted. The observed signs of the diagnostic bands in the region of λ
max 300–400 nm in the induced ECD spectra were correlated to the absolute configuration of the
ortho-diol moiety.
(2
R,3
R,5
S,9
R,10
S)-2,3,9,11-Tetrahydroxydrim-7-en-6-one (ustusol F,
1): colorless oil; [α]
23D −56.0 (
c 0.11, MeOH); UV (MeOH) λ
max (log
ε) 232 (0.82) nm; ECD (1.76 mM, MeOH) λ
max (Δ
ε) 336 (+8.4), 271 (−3.2), 240 (−41.3), 215 (−12.8) nm; IR (KBr)
νmax 3399, 2959, 1663, 1439, 1384, 1243, 1062, 1027 cm
−1;
1H and
13C NMR see
Table 1; HRESIMS
m/
z 285.1694 [M+H]
+ (calcd for C
15H
24O
5, 285.1697), or 283.1547 [M–H]
− (calcd for C
15H
23O
5, 283.1551).
(2
R,3
R,5
R,9
S,10
R)-2,3,11-Trihydroxydrim-7-en-6-one (9-deoxyustusol F,
2): yellow oil; [α]
23D −56 (
c 0.06, MeOH); UV (MeOH) λ
max (log
ε) 238 (1.65) nm; ECD (1.87 mM, MeOH) λ
max (Δ
ε) 334 (+6.8), 264 (−1.3), 240 (−18.3), 220 (−14.3) nm; IR (KBr)
νmax 3398, 2942, 1659, 1440, 1382, 1237, 1152, 1060, 983 cm
−1;
1H and
13C NMR see
Table 1; HRESIMS
m/
z 269.1751 [M+H]
+ (calcd for C
15H
24O
4, 269.1747).
(3
S,5
R,9
R,10
R)-3,11,12-Trihydroxydrim-7-en-6-one (ustusol G,
3): colorless oil; [α]
23D −71 (
c 0.04, MeOH); UV (MeOH) λ
max (log
ε) 240 (1.60) nm; ECD (1.87 mM, MeOH) λ
max (Δ
ε) 335 (+10.6), 265 (−2.6), 241 (−19.1), 205 (−71.7) nm;
1H and
13C NMR see
Table 1; HRESIMS
m/
z 269.1750 [M+H]
+ (calcd for C
15H
24O
4, 269.1747).
(5
S,6
R,9
S,10
S,11
R,2′
E,4′
E)-6-(11-Deoxy-11-hydroxystrobilactone A-6-yl)-5-carboxypenta-2,4-dienoate (ustusolate H,
4): colorless solid; [α]
25D −96 (
c 0.2, MeOH); UV (MeOH) λ
max (log
ε) 264 (1.54) nm; ECD (0.64 mM, MeOH) λ
max (Δ
ε) 264 (−6.2), 232 (−3.3), 205 (−11.1) nm; IR (KBr)
νmax 3434, 2953, 2926, 2856, 1684, 1640, 1460, 1398, 1310, 1260, 1208, 1136, 1028, 913 cm
−1;
1H and
13C NMR see
Table 2; HRESIMS
m/
z 391.1762 [M–H]
– (calcd for C
21H
27O
7, 391.1762).
((5S,6R,9S,10S)-Strobilactone A-6-yl) (2E,4E)-6,7-dihydroxyocta-2,4-dienoate (ustusolate I, 5): light yellow oil; UV (MeOH) λmax (log ε) 265 (4.15) nm. 1H NMR (DMSO-d6, 500 MHz) δH 1.83 (d, J = 13.6 Hz, 1H, H-1α), 1.95 (dd, J = 4.4, 13.6 Hz, 1H, H-1β); 1.59 (m, 1H, H-2α), 1.47 (m, 1H, H-2β); 1.20 (td, J = 3.2, 13.1 Hz, 1H, H-3α), 1.34 (d, J = 12.3 Hz, 1H, H-3β); 2.00 (d, J = 5.0 Hz, 1H, H-5); 5.59 (brs, 1H, H-6); 5.79 (brs, 1H, H-7); 4.88 (dt, J = 2.3, 12.6 Hz, 1H, H-12α), 4.78 (d, J = 12.6 Hz, 1H, H-12β); 1.06 (s, 3H, H-13); 0.92 (s, 3H, H-14); 1.07 (s, 3H, H-15); 5.94 (d, J = 15.3 Hz, 1H, H-2′); 7.20/7.23 (m, 1H, H-3′); 6.40/6.44 (m, 1H, H-4′); 6.30/6.34 (m, 1H, H-5′); 3.85/3.97 (m, 1H, H-6′); 3.49/3.56 (m, 1H, H-7′); 0.94/1.02 (d, J = 6.2 Hz, 3H, H-8′); 5.02 (brs, 1H, HO-6′); 4.61/4.66 (brs, 1H, HO-7′); 13C NMR (DMSO-d6,125 MHz) δC 29.6 (CH2, C-1), 17.5 (CH2, C-2), 44.5 (CH2, C-3), 33.4 (C, C-4), 44.2 (CH, C-5), 65.8 (CH, C-6), 121.4 (CH, C-7), 136.6 (C, C-8), 73.2 (C, C-9), 37.3 (C, C-10), 174.4 (C, C-11), 68.3 (CH2, C-12), 18.3 (CH3, C-13), 32.2 (CH3, C-14), 24.4 (CH3, C-15), 165.51/165.50/165.49/165.47 (C, C-1′), 120.03/120.99/119.95/119.90 (CH, C-2′), 145.41/145.37/145.34/145.26 (CH, C-3′), 127.54/127.35/127.16/126.98 (CH, C-4′), 146.18/146.12/145.48/145.45 (CH, C-5′), 75.16/75.00/74.64/74.46 (CH, C-6′), 69.64/69.62/69.33/69.32 (CH, C-7′), and 19.34/19.26/18.26/18.24 (CH3, C-8′); ESIMS peak at m/z 419.1 for [M–H]– and m/z 464.9 for [M + HCO2]– (C23H32O7).
(2′
E,4′
E;6′,7′-
erythro)-Ustusolate I (
5a): light yellow oil; [α]
23D −35 (
c 0.30, MeOH); UV (MeOH) λ
max (log
ε) 268 (4.15) nm; ECD (0.60 mM, MeOH) λ
max (Δ
ε) 255 (−8.3), 236 (−8.9), 208 (−21.2) nm;
1H and
13C NMR see
Table 3 and
Table 4; ESIMS
m/
z 421.2 [M+H]
+ (C
23H
32O
7).
(2′
E,4′
E;
ent-6′,7′-
erythro)-Ustusolate I (
5b): light yellow oil; [α]
23D −42 (
c 0.30, MeOH); UV (MeOH) λ
max (log
ε) 261 (4.39) nm; ECD (0.60 mM, MeOH) λ
max (Δ
ε) 256 (−11.0), 234 (−8.9), 209 (−21.4) nm;
1H and
13C NMR see
Table 3 and
Table 4; ESIMS
m/
z 421.2 [M+H]
+ (C
23H
32O
7).
(2′
E,4′
E,6′
R,7′
R)-Ustusolate I (
5c): light yellow oil; [α]
23D −105 (
c 0.30, MeOH; UV (MeOH) λ
max (log
ε) 261 (4.42) nm; ECD (0.60 mM, MeOH) λ
max (Δ
ε) 256 (−8.6), 234 (−8.5), 208 (−22.9) nm;
1H and
13C NMR see
Table 3 and
Table 4; ESIMS
m/
z 421.2 [M+H]
+ (C
23H
32O
7).
(2′
E,4′
E,6′
S,7′
S)-Ustusolate I (
5d): light yellow oil; [α]
23D −79 (
c 0.29, MeO; UV (MeOH) λ
max (log
ε) 262 (4.36) nm; ECD (0.60 mM, MeOH) λ
max (Δ
ε) 259 (−10.4), 234 (−8.2), 208 (−18.9) nm;
1H and
13C NMR see
Table 3 and
Table 4; ESIMS
m/
z 421.2 [M+H]
+ (C
23H
32O
7).
(2′
E,4′
E;6′,7′-
erythro)-Ustusolate I-6′,7′-acetonide (
5e): light yellow oil; [α]
22D −102 (
c 0.16, MeOH); UV (MeOH) λ
max (log
ε) 268 (4.15) nm; ECD (0.60 mM, MeOH) λ
max (Δ
ε) 256 (−15.1), 232 (−13.8), 210 (−39.4) nm;
1H and
13C NMR see
Table 3 and
Table 4; ESIMS
m/
z 421.2 [M+H]
+ (C
26H
36O
7).
(5
S,6
R,9
S,10
S,2′
E,4′
E)-(Strobilactone A-6-yl)-5-carboxypenta-2,4-dienoate (ustusolate J,
6): colorless solid; [α]
20D −280 (
c 0.65, MeOH); UV (MeOH) λ
max (log
ε) 265 (1.84) nm; ECD (0.64 mM, MeOH) λ
max (Δ
ε) 261 (−11.4), 232 (−8.9), 207 (−23.2) nm; IR (KBr)
νmax 3400, 3320, 2950, 2928, 1658, 1615, 1460, 1385, 1290,1208, 1155, 1078, 970 cm
−1;
1H and
13C NMR see
Table 2; ESIMS
m/
z 459.5 [M–H]
– (C
21H
26O
7).
(2
S,5
S,9
R,10
S)-2,9,11-Trihydroxydrim-7-en-6-one (ustusol B,
7): light yellow solid; [α]
23D −140 (
c 0.1, MeOH); UV (MeOH) λ
max (log
ε) 252 (1.33) nm; ECD (1.87 mM, MeOH) λ
max (Δ
ε) 335 (+11.9), 268 (−3.3), 241 (−66.9), 214 (−18.7) nm; IR (KBr)
νmax 3400, 3320, 2950, 2928, 1658, 1615, 1460, 1385, 1290, 1208, 1155, 1078, 970 cm
−1;
1H and
13C NMR see
Table 1; ESIMS
m/
z 269.2 [M+H]
+, 291.2 [M+Na]
+ (C
15H
24O
4).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}