
Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents

Abstract
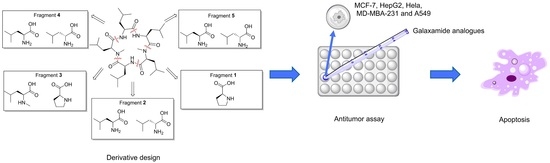
:
1. Introduction
2. Results and Discussion
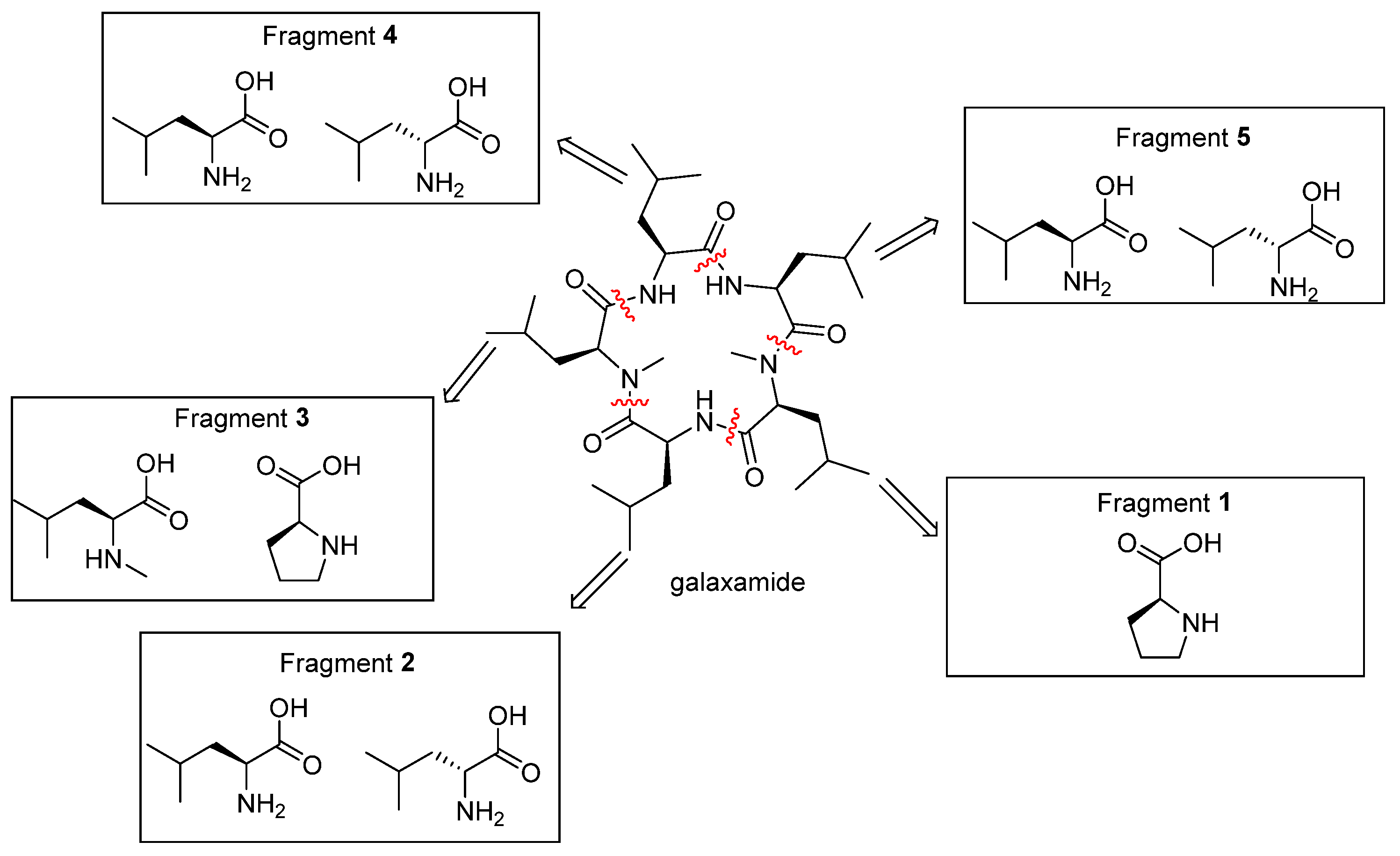
2.1. Chemistry
2.2. Biological Activity
2.2.1. Inhibitory Activities of Galaxamide and Its Analogues
2.2.2. Effects of Analogues on the Cell Cycle of MCF-7
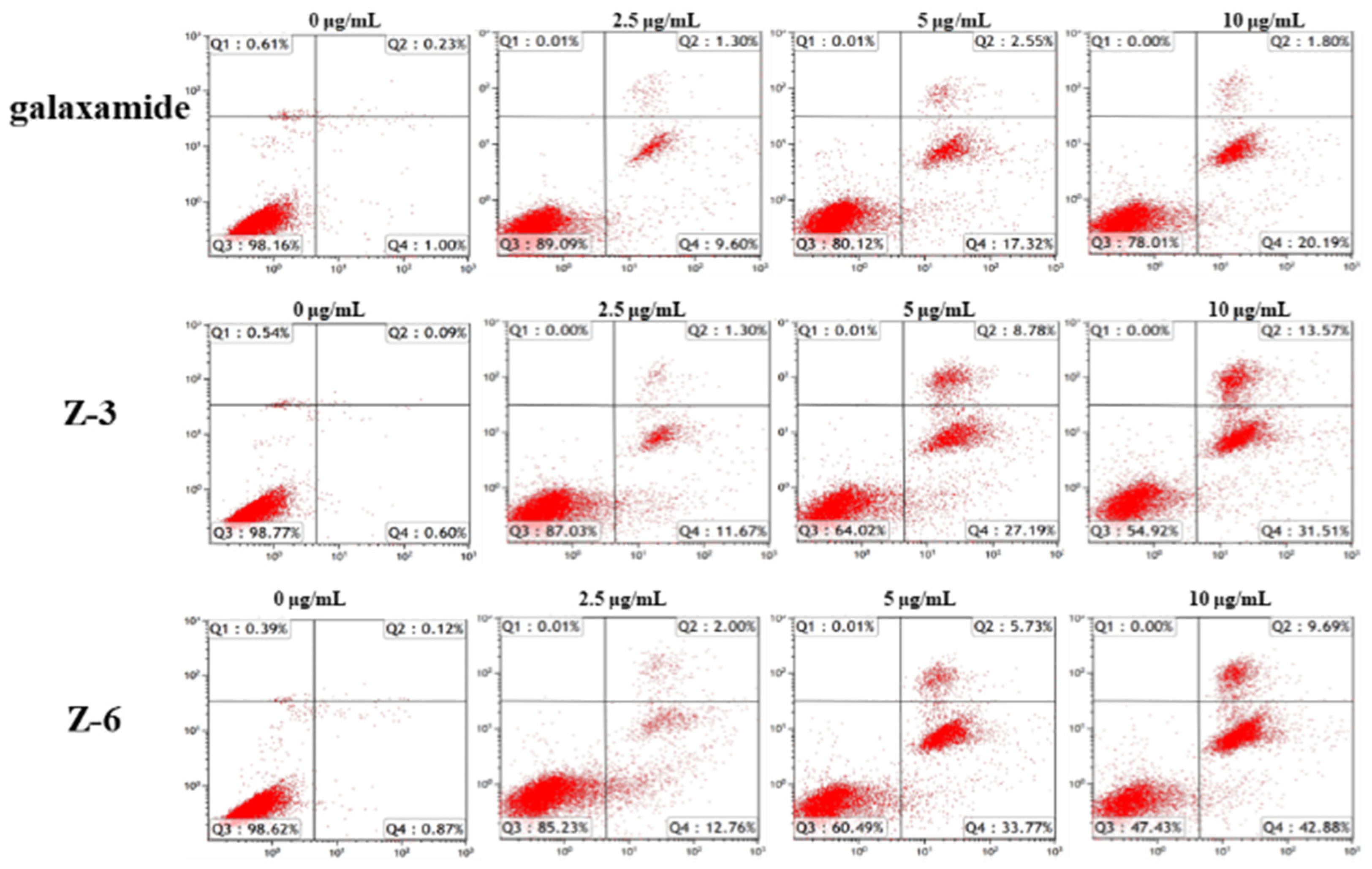
2.2.3. Apoptotic Mechanism Study of Galaxamide and Its Analogues
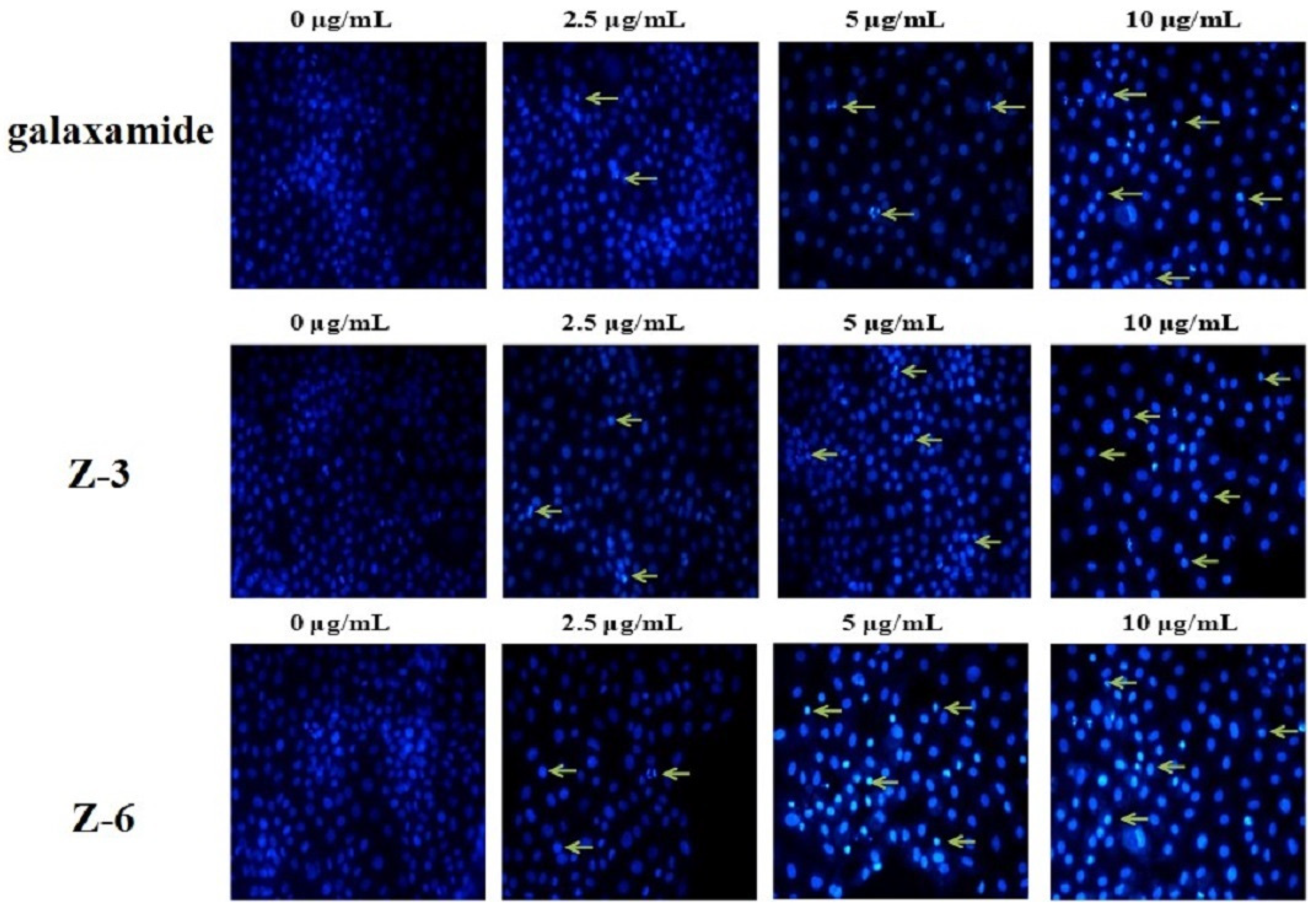
2.2.4. Effects of Galaxamide and Its Analogues on Cell Nuclear Integrity
3. Materials and Methods
3.1. General
3.2. Compounds
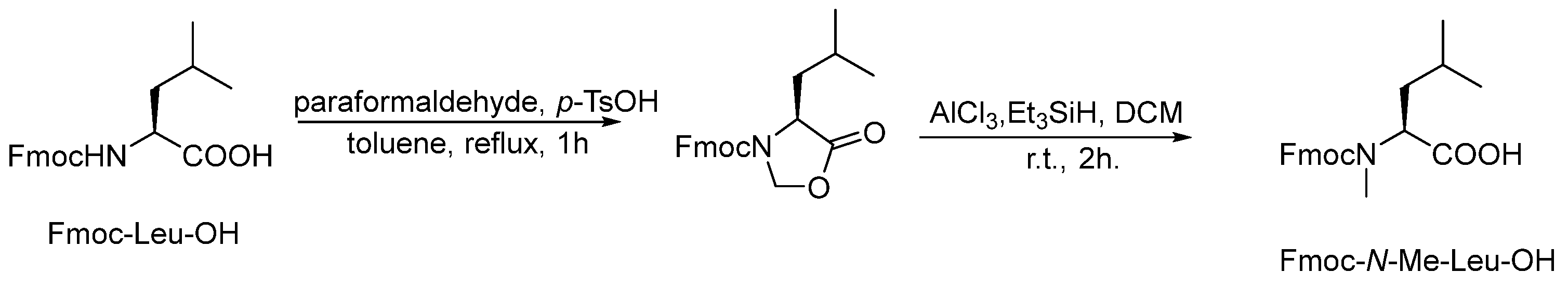
3.2.1. Synthesis of Fmoc-N-Me-Leu-OH
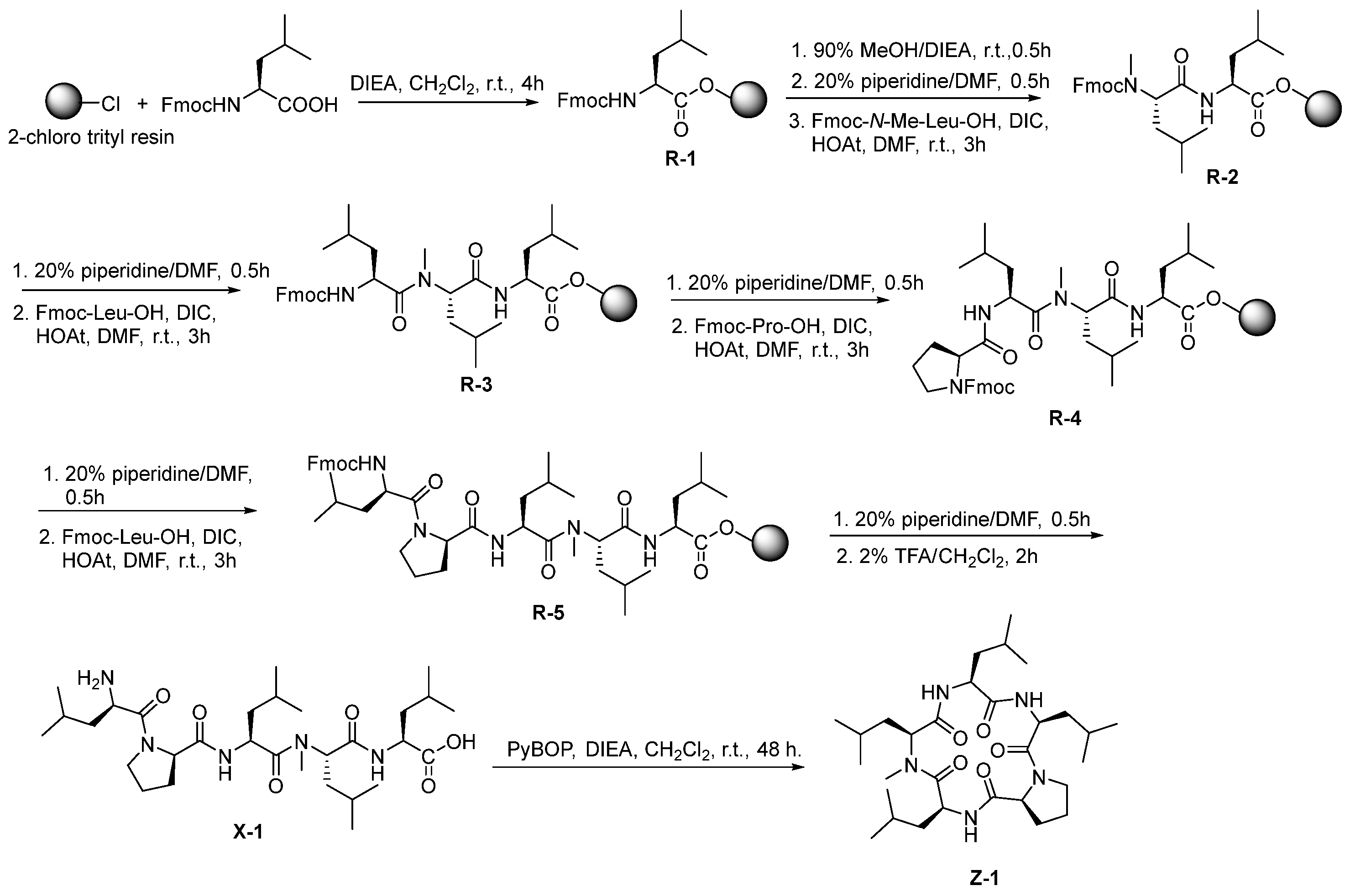
3.2.2. Synthesis of Linear Pentapeptide HO-Leu-N-Me-Leu-Leu-Pro-Leu-H (X-1)
3.2.3. Synthesis of Linear Pentapeptide HO-Leu-Pro-Leu-Pro-Leu-H (X-2)
3.2.4. Synthesis of Linear Pentapeptide HO-D-Leu-Pro-Leu-Pro-Leu-H (X-3)
3.2.5. Synthesis of Linear Pentapeptide HO-Leu-Pro-Leu-Pro-D-Leu-H (X-4)
3.2.6. Synthesis of Linear Pentapeptide HO-Leu-Pro-D-Leu-Pro-Leu-H (X-5)
3.2.7. Synthesis of Linear Pentapeptide HO-D-Leu-Pro-Leu-Pro-D-Leu-H (X-6)
3.2.8. Synthesis of Linear Pentapeptide HO-Leu-Pro-D-Leu-Pro-D-Leu-H (X-7)
3.2.9. Synthesis of Linear Pentapeptide HO-D-Leu-Pro-D-Leu-Pro-D-Leu-H (X-8)
3.2.10. Synthesis of Cyclo (Leu-N-Me-Leu-Leu-Pro-Leu) (Z-1)
3.2.11. Synthesis of Cyclo (Leu-Pro-Leu-Pro-Leu) (Z-2)
3.2.12. Synthesis of Cyclo (Leu-Pro-Leu-Pro-Leu) (Z-3)
3.2.13. Synthesis of Cyclo (Leu-Pro-D-Leu-Pro-Leu) (Z-4)
3.2.14. Synthesis of Cyclo (Leu-Pro-Leu-Pro-D-Leu) (Z-5)
3.2.15. Synthesis of Cyclo (D-Leu-Pro-Leu-Pro-D-Leu) (Z-6)
3.2.16. Synthesis of Cyclo (Leu-Pro-D-Leu-Pro-D-Leu) (Z-7)
3.2.17. Synthesis of Cyclo (D-Leu-Pro-D-Leu-Pro-D-Leu) (Z-8)
3.3. In Vitro Cytotoxic Assays
3.4. MTT Assay
3.5. Flow Cytometry Experiments
3.6. Cell Apoptotic Analysis
3.7. Hoechst 33,342 Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J. Bioactive peptides from a marine mollusk elysiarufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.F.; Cichacz, Z.; Barkoczy, J.; Dorsaz, A.; Herald, D.L.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Tackett, L.P.; Brune, D.C. Isolation and structure of the marine sponge cell growth inhibitory cyclic peptide phakellistin. J. Nat. Prod. 1993, 56, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, A.S.; Bugni, T.S.; Feng, X.D.; Harper, M.K.; Skalicky, J.J.; Mohammed, K.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Theopapuamide, a cyclic depsipeptide from a papua new guinea lithistid sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 1582–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampella, A.; D’Auria, M.V.; Paloma, L.G.; Casapullo, A.; Minale, L.; Debitus, C.; Henin, Y.; Callipeltin, A. An anti-HIV cyclic depsipeptide from the new caledonian lithistida sponge Callipelta sp. J. Am. Chem. Soc. 1996, 118, 6202–6209. [Google Scholar] [CrossRef]
- Kashinath, K.; Vasudevan, N.; Reddy, D.S. Studies toward the synthesis of potent anti-inflammatory peptides solomonamides A and B: Synthesis of a macrocyclic skeleton and key fragment 4-amino-6-(2′-amino-4′-hydroxyphenyl)-3-hydroxy-2-methyl-6-oxohexanoic acid (AHMOA). Org. Lett. 2012, 14, 6222–6225. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; De Marino, S.; Sepe, V.; Monti, M.C.; Luciano, P.; Valeria D’Auria, M.; Debitus, C.; Bucci, M.; Vellecco, V.; Zampella, A. Perthamides C and D, two new potent anti-inflammatory cyclopeptides from a solomon lithistid sponge Theonella swinhoei. Tetrahedron 2009, 65, 10424–10429. [Google Scholar] [CrossRef]
- Bewley, C.A.; Detritus, C.; Faulkner, D.J. Microsclerodermins A and B. antifungal cyclic peptides from the lithistid sponge Microscleroderma sp. J. Am. Chem. Soc. 1994, 116, 7631–7636. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Faulkner, D.J. Microsclerodermins C-E, sntifungal cyclic peptides from the lithistid marine sponges Theonella sp. and Microscleroderma sp. Tetrahedron 1998, 54, 3043–3056. [Google Scholar] [CrossRef]
- Qureshi, A.; Colin, P.L.; Faulkner, D.J. Microsclerodermins F-I, antitumor and antifungal cyclic peptides from the lithistid sponge Microscleroderma sp. Tetrahedron 2000, 56, 3679–3685. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Harper, J.B.; Hunter, L. Incrementally increasing the length of a peptide backbone: Effect on macrocyclization efficiency. Org. Biomol. Chem. 2014, 12, 4598–4601. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Jimenez, G.M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.M. Bioactivep peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Y.; Dahiya, R.; Qin, H.L.; Mourya, R.; Maharaj, S. Natural proline-rich cyclopolypeptides from marine organisms: Chemistry, synthetic methodologies and biological status. Mar. Drugs 2016, 14, 194–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.J.; Liao, X.J.; Xu, S.H.; Diao, J.Z.; Du, B.; Zhou, X.L.; Pan, S.S. Isolation, structure determination, and synthesis of galaxamide, a rare cytotoxic cyclic pentapeptide from a marine algae Galaxaura filamentosa. Org. Lett. 2008, 10, 4569–4572. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liao, X.J.; Qiu, S.L.; Liu, Z.H.; Du, B.; Xu, S.H. Paper synthesis, cytotoxicity and apoptosis induction in human tumor cells by galaxamide and its analogues. Mar. Drugs 2014, 12, 4521–4538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunagariya, J.; Zhong, S.H.; Chen, J.; Bai, D.; Bhadja, P.; Long, W.; Liao, X.; Tang, X.; Xu, S. Design and aynthesis of analogues of marine natural product galaxamide, an N-methylated cyclic pentapeptide, as potential anti-tumor agent in vitro. Mar. Drugs 2016, 14, 161–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.F.; Yu, S.M.; Zhong, S.H.; Zhao, B.X.; Qiu, S.L.; Chen, J.W.; Lunagariya, J.; Liao, X.J.; Xu, S.H. D-amino acid position influences the antiCancer activity of galaxamide analogs: An apoptotic mechanism study. Int. J. Mol. Sci. 2017, 18, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redman, J.E.; Ghadiri, M.R. Synthesis of photoactive p-azidotetrafluorophenylalanine containing peptide by solid-phase fmoc methodology. Org. Lett. 2002, 4, 4467–4469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Govender, T.; Norstrom, T.; Arvidsson, P.I. An improved synthesis of fmoc-N-methyl-α-amino acids. J. Org. Chem. 2005, 70, 6918–6920. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 (μg/mL) | |||||
|---|---|---|---|---|---|---|
| MCF-7 | HepG2 | MD-MBA-231 | Hela | A549 | HUVEC | |
| galaxamide | 11.33 ± 2.95 | 5.20 ± 0.52 | 8.73 ± 0.29 | 8.53 ± 0.73 | 6.99 ± 0.63 | >40 |
| Z-1 | 5.85 ± 1.28 | 7.57 ± 0.17 | 17.81 ± 0.60 | 11.56 ± 0.65 | 4.92 ± 0.84 | >40 |
| Z-2 | 4.68 ± 1.22 | 11.95 ± 0.64 | 14.45 ± 1.10 | 14.50 ± 0.36 | 7.97 ± 2.06 | >40 |
| Z-3 | 2.25 ± 0.42 | 5.05 ± 0.45 | 6.34 ± 0.60 | 5.57 ± 0.45 | 6.43 ± 0.43 | >40 |
| Z-4 | 4.88 ± 0.58 | 15.23 ± 1.14 | 22.09 ± 1.12 | 7.38 ± 0.83 | 9.85 ± 0.95 | >40 |
| Z-5 | 13.43 ± 1.96 | 14.98 ± 0.93 | 8.48 ± 1.12 | 12.12 ± 0.95 | 9.50 ± 1.13 | >40 |
| Z-6 | 1.65 ± 0.30 | 2.91 ± 0.17 | 4.59 ± 0.27 | 5.69 ± 0.37 | 5.96 ± 0.41 | 38.72 ± 0.67 |
| Z-7 | 13.28 ± 3.28 | 13.85 ± 1.99 | 16.52 ± 1.70 | 18.21 ± 1.35 | 9.97 ± 1.00 | >40 |
| Z-8 | 7.82 ± 1.18 | 9.00 ± 0.83 | 20.16 ± 2.24 | 7.97 ± 0.61 | 15.88 ± 1.42 | >40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Liao, X.; Zhong, S.; Zhao, B.; Xu, S. Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents. Mar. Drugs 2022, 20, 158. https://doi.org/10.3390/md20030158
Li D, Liao X, Zhong S, Zhao B, Xu S. Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents. Marine Drugs. 2022; 20(3):158. https://doi.org/10.3390/md20030158
Chicago/Turabian StyleLi, Daichun, Xiaojian Liao, Shenghui Zhong, Bingxin Zhao, and Shihai Xu. 2022. "Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents" Marine Drugs 20, no. 3: 158. https://doi.org/10.3390/md20030158
APA StyleLi, D., Liao, X., Zhong, S., Zhao, B., & Xu, S. (2022). Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents. Marine Drugs, 20(3), 158. https://doi.org/10.3390/md20030158