
Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases


 and
and
Abstract
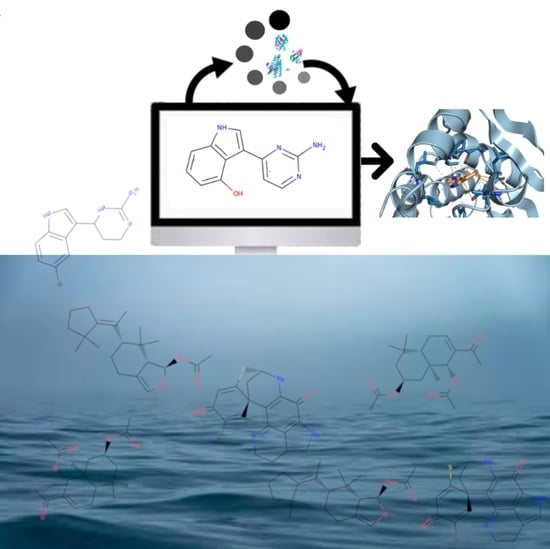
:
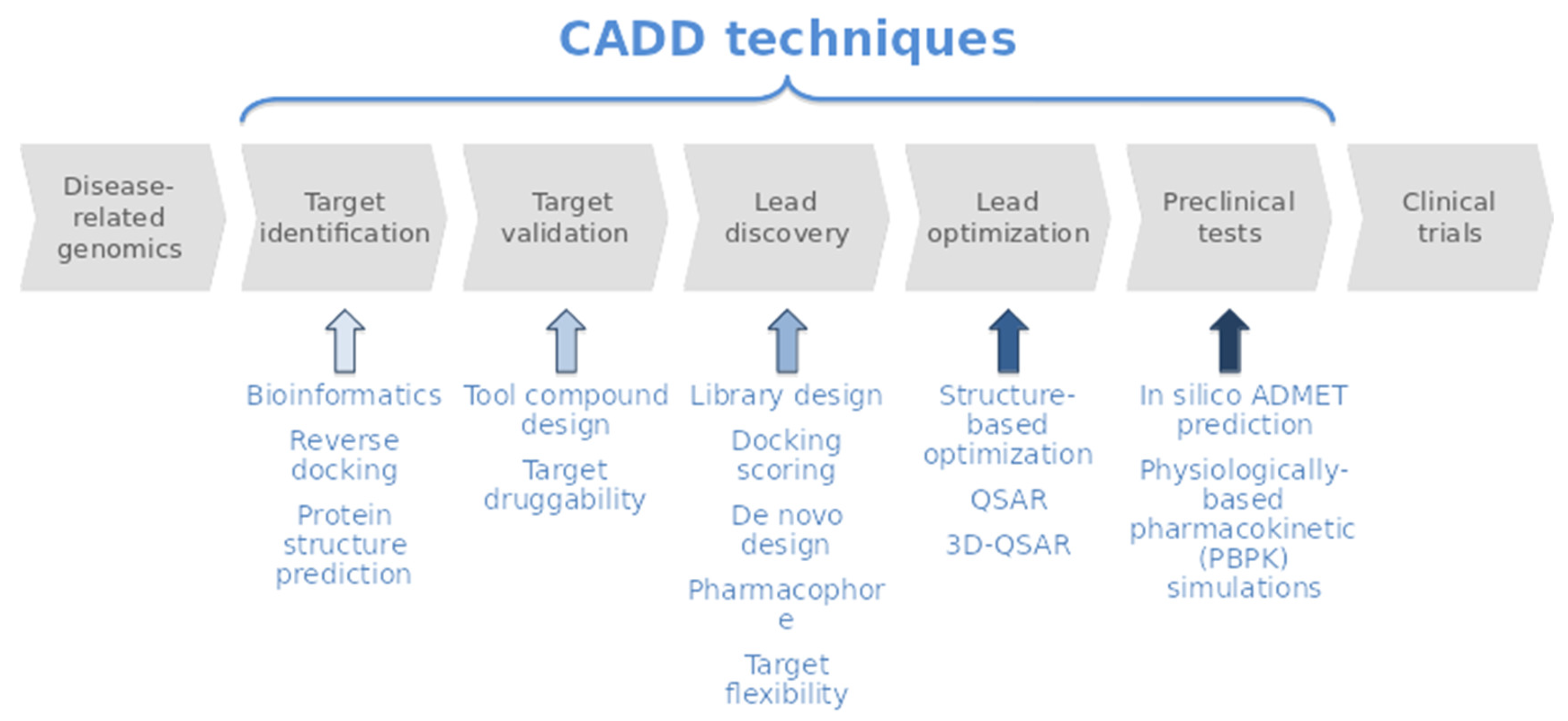
1. Introduction
2. Results and Discussion
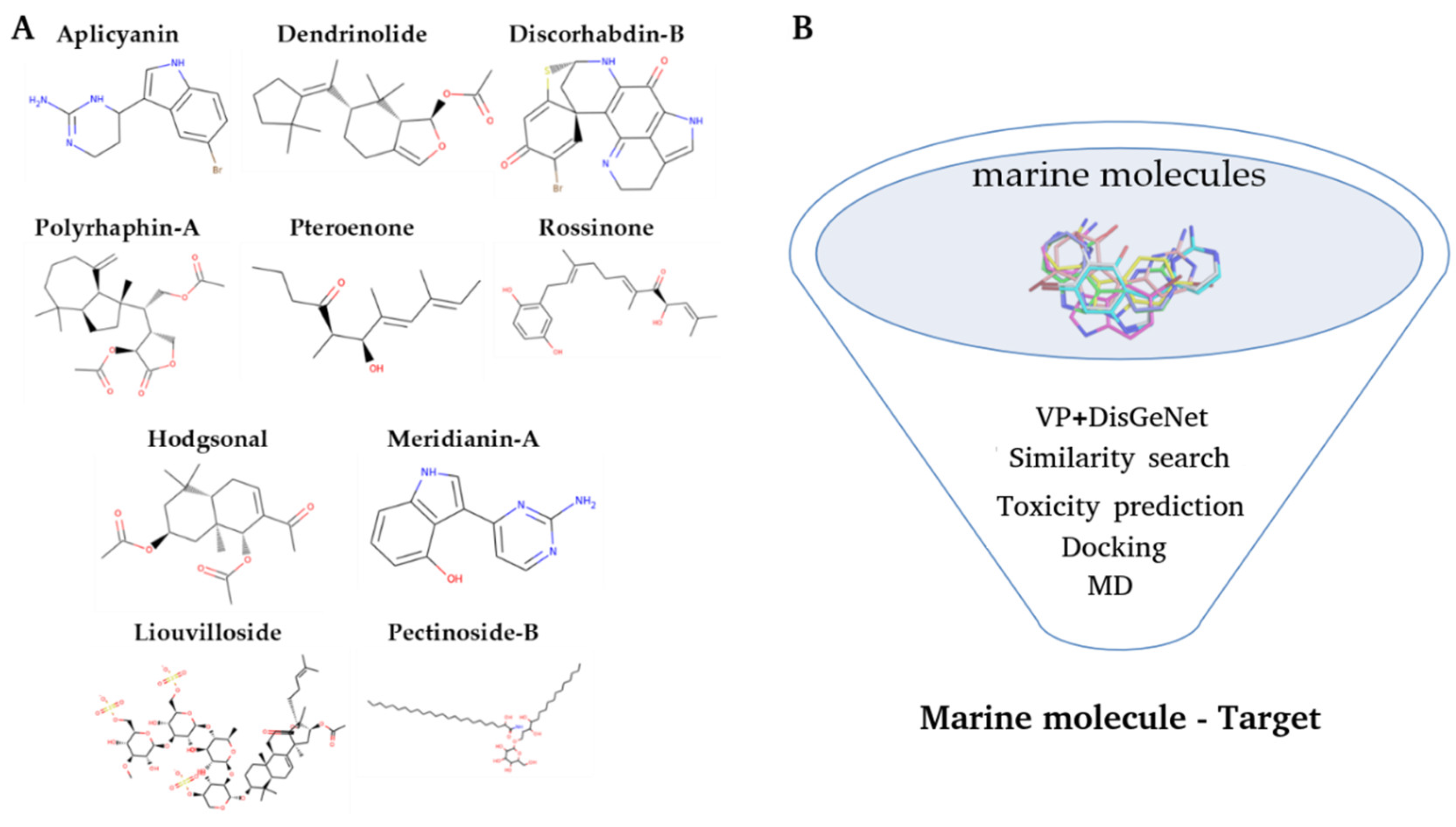
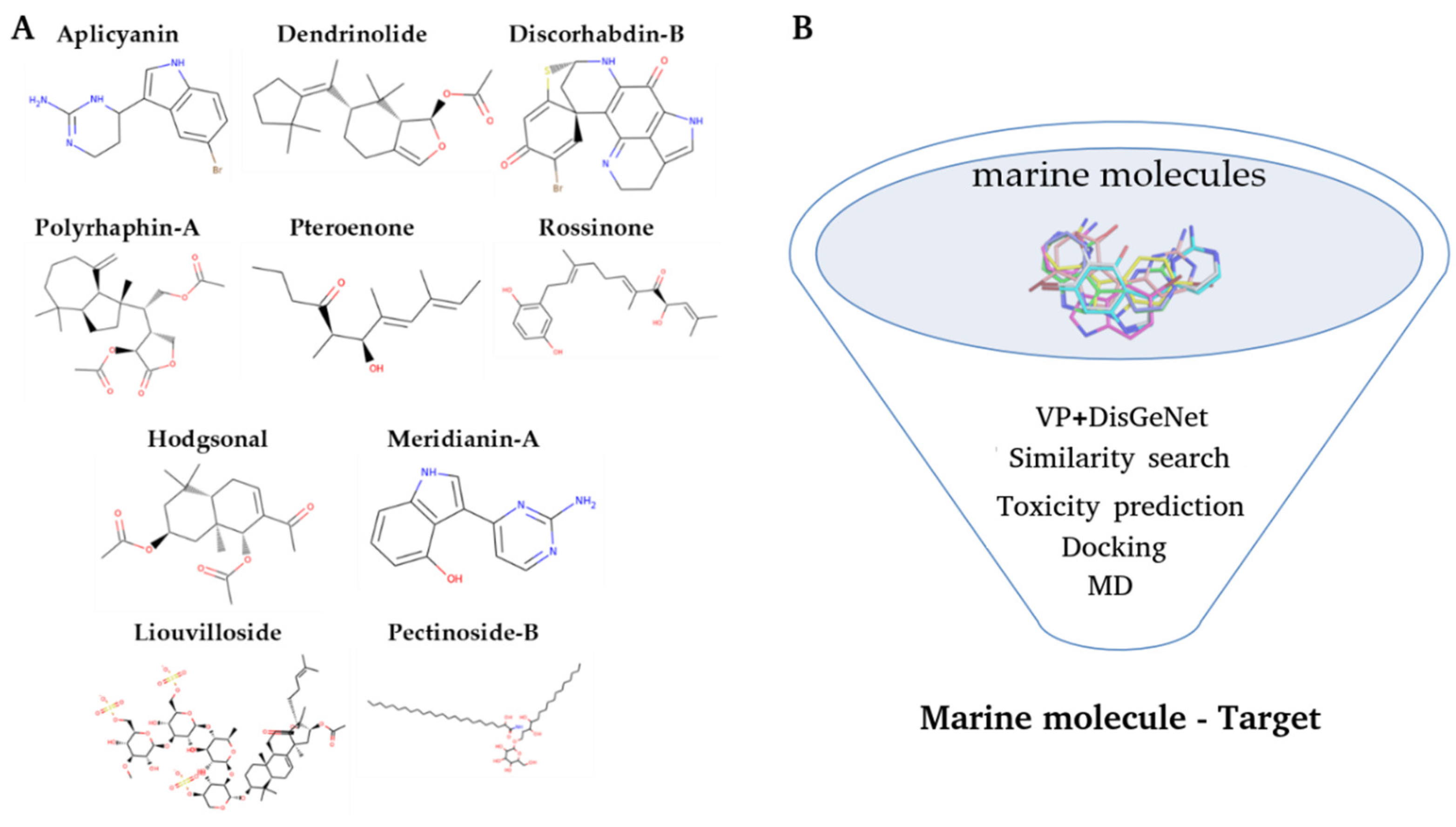
2.1. Virtual Profiling
2.2. Toxicity Prediction
2.3. Virtual Profiling Validation. In Silico Binding Studies
2.3.1. Docking and Molecular Dynamics Simulations
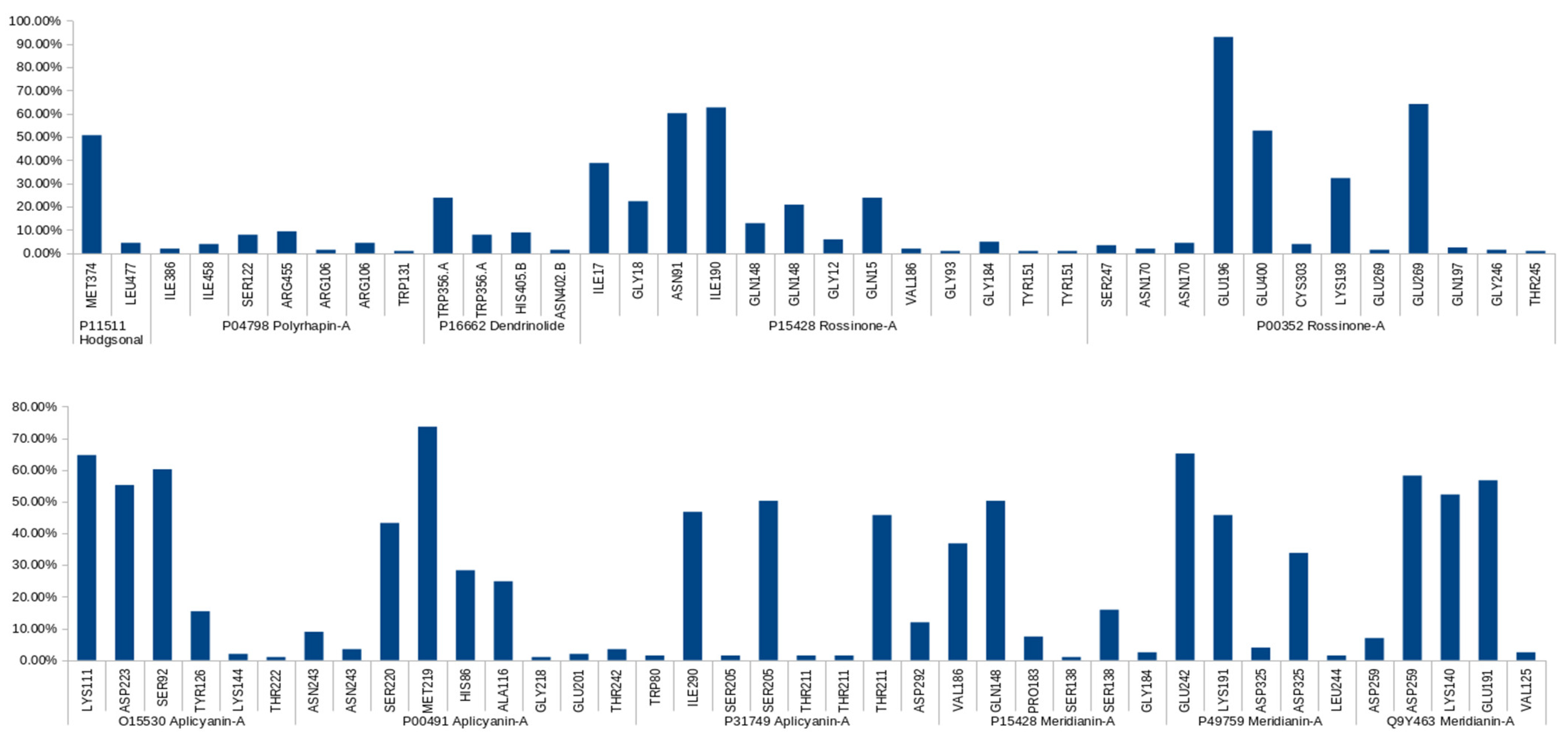
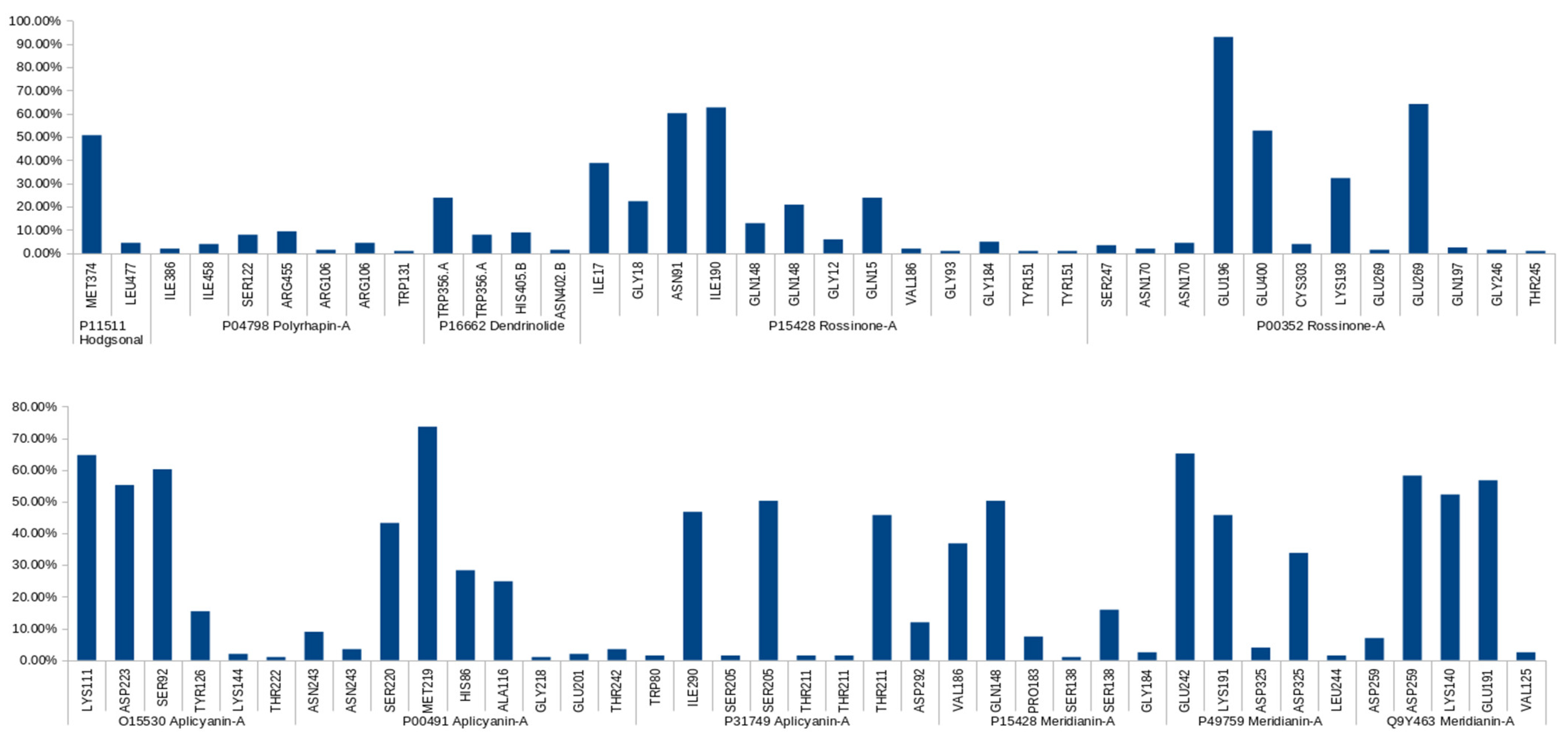
2.3.2. Binding Mode Analysis. Hydrogen Bonding
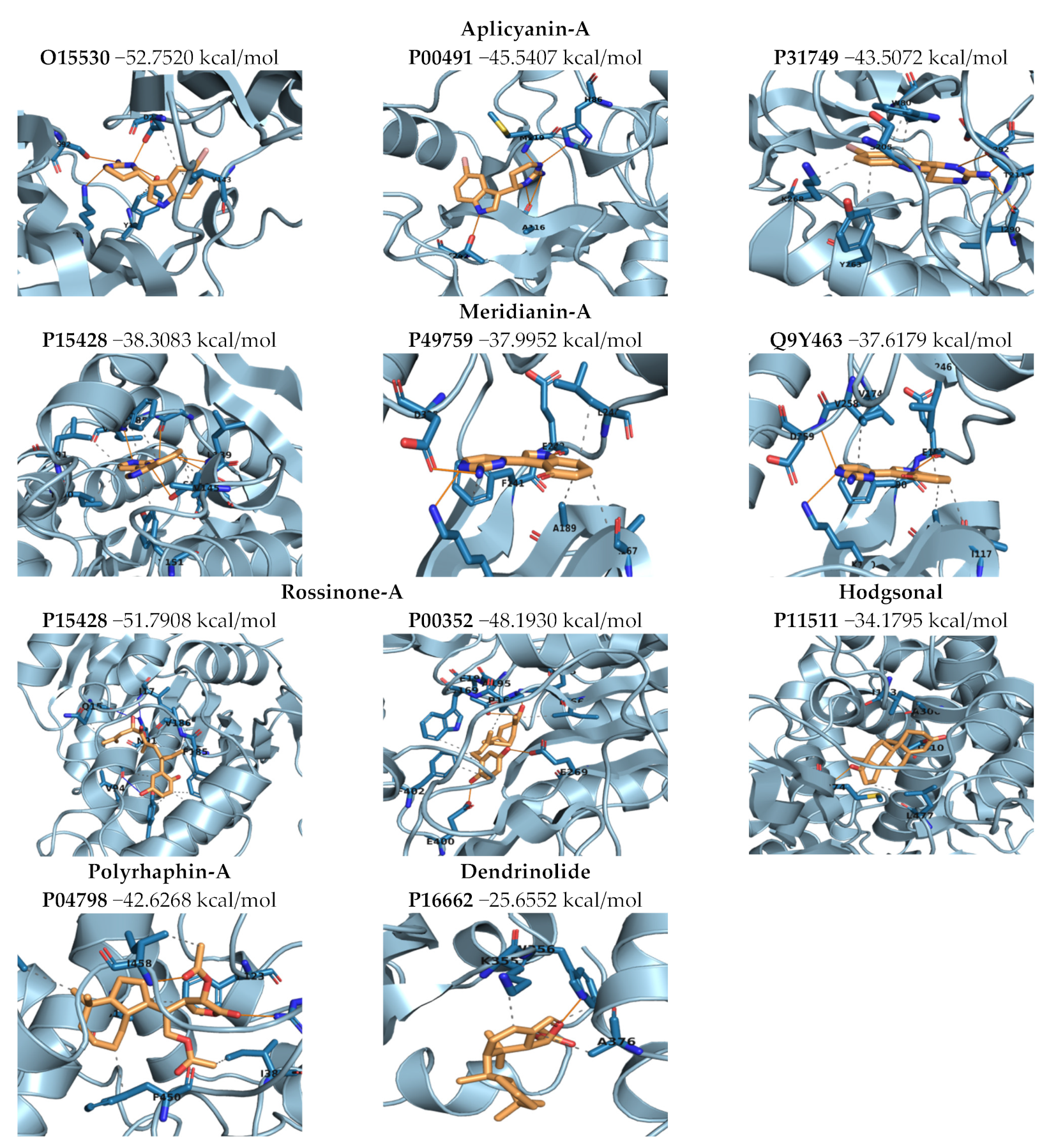
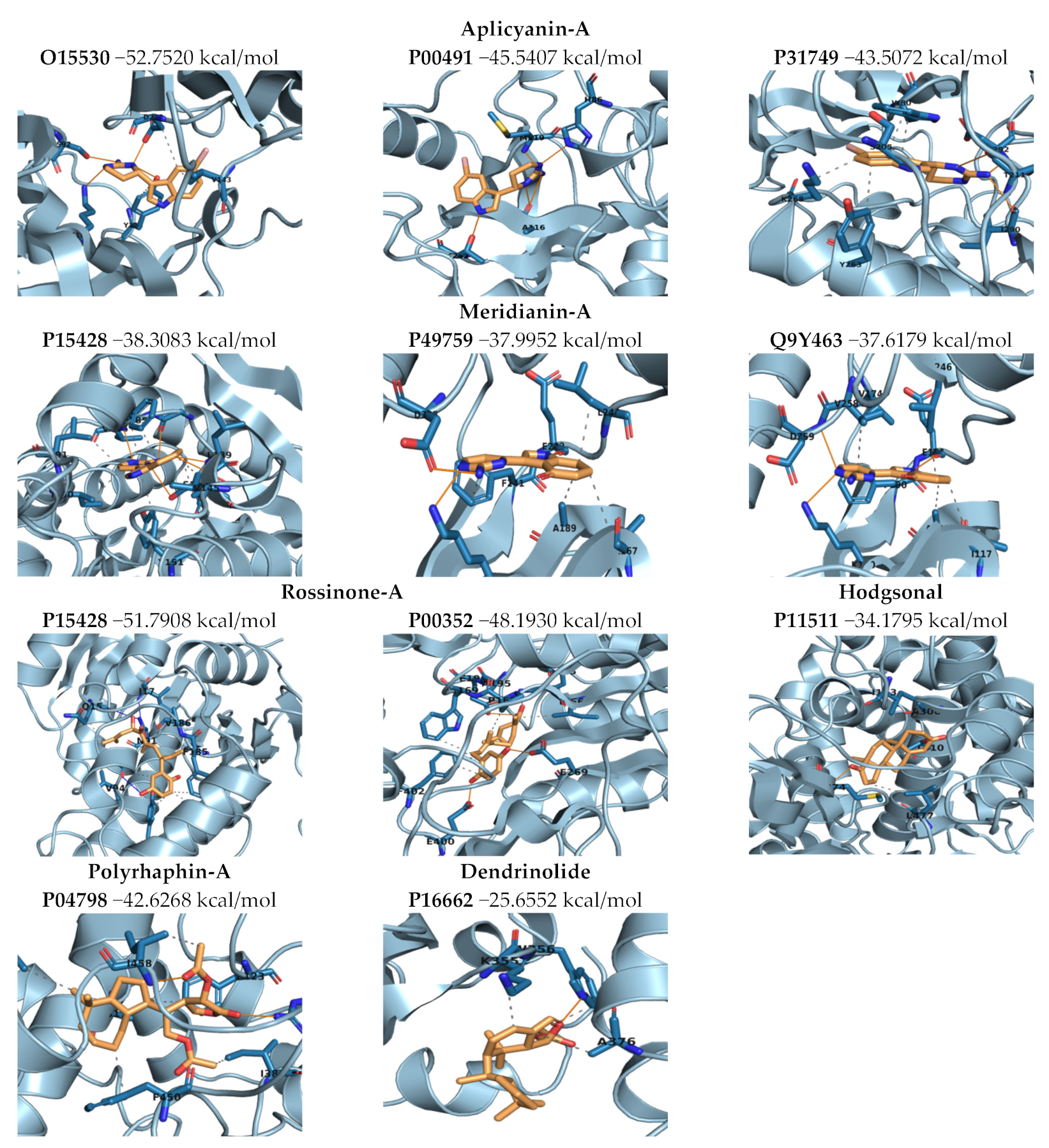
2.3.3. Binding Mode Analysis. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA). Overall Molecule–Target Association
2.3.4. Complexes Prioritization
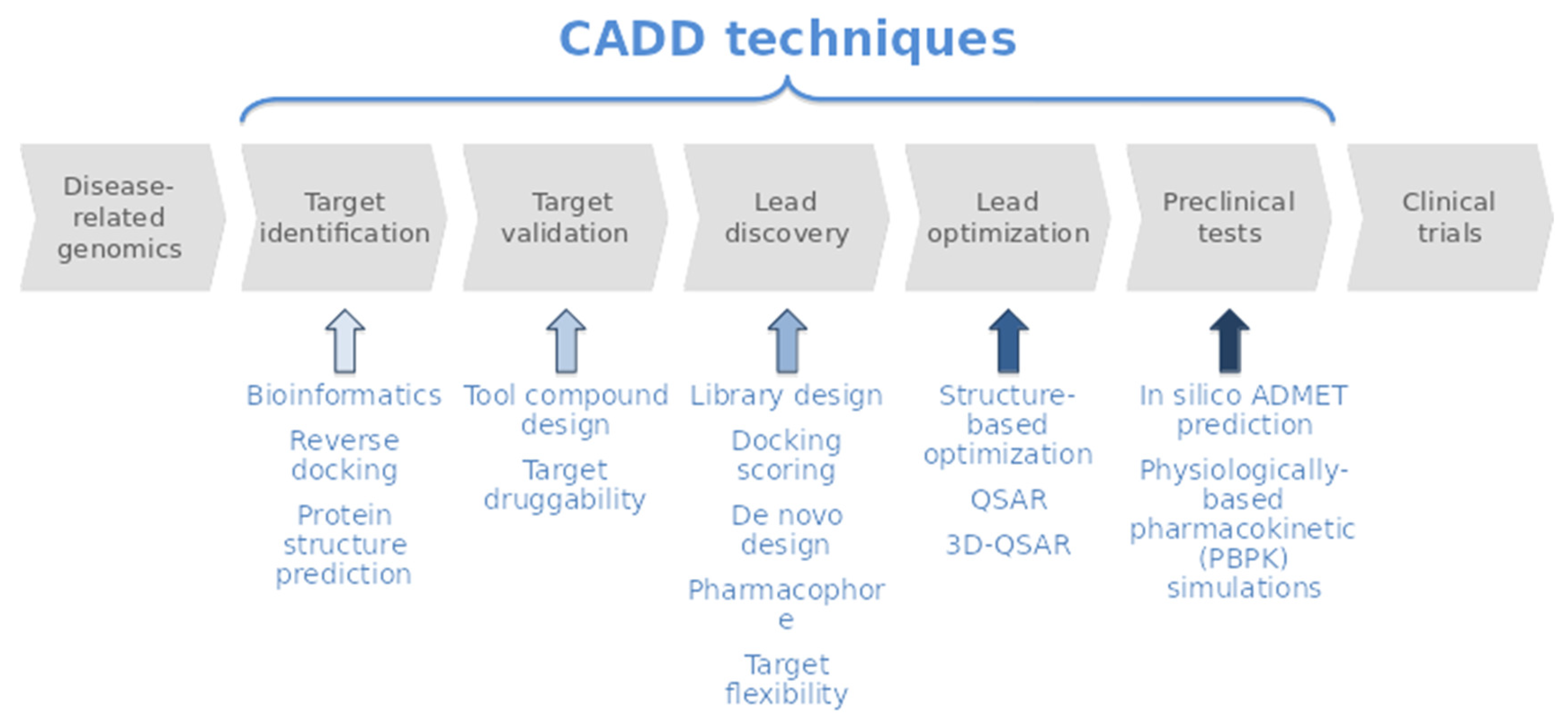
3. Materials and Methods
3.1. Initial Dataset
3.2. Virtual Profiling
3.3. Target Selection
3.4. Target Modelling
3.5. Toxicology Prediction
3.6. Drug Likeness Evaluation
3.7. Docking Calculations
3.8. Molecular Dynamics Simulation
3.9. Molecular Dynamics Analysis
3.10. MM/Generalized Born Surface Area
3.11. Graphical Representations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve RD productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Ou-Yang, S.-S.; Lu, J.-Y.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W.E. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Casares, J.R.; Quintero, J.; Jorba, G.; Junet, V.; Martínez, V.; Pozo-Rubio, T.; Oliva, B.; Daura, X.; Mas, J.M.; Montoto, C. Methods to Develop an in silico Clinical Trial: Computational Head-to-Head Comparison of Lisdexamfetamine and Methylphenidate. Front. Psychiatry 2021, 12, 1–21. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Sydow, D.; Burggraaff, L.; Szengel, A.; Van Vlijmen, H.W.T.; Ijzerman, A.P.; Van Westen, G.J.P.; Volkamer, A. Advances and Challenges in Computational Target Prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Brroijmans, N.; Kuntz, I.D. Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef]
- De Vivo, M.; Cavalli, A. Recent advances in dynamic docking for drug discovery. WIREs Comput. Mol. Sci. 2017, 7, e1320. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A.; De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A.; de Vivo, M.; Masetti, M.; et al. The Role of Molecular Dynamics and Related Methods in Drug Discovery The Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Shen, C.; Hu, X.; Gao, J.; Li, D.; Cao, D.; Hou, T. Combined strategies in structure-based virtual screening. Phys. Chem. Chem. Phys. 2020, 22, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Pereira, F. Expert Opinion on Drug Discovery Have marine natural product drug discovery efforts been productive and how can we improve their efficiency ? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Li, J.W.H.; Vederas, J.C. Drug Discovery and Natural Products: End of an Era or an Endless Frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.R.; Keyzers, R.A.; Copp, B.R.; Davis, R.A. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef]
- Avila, C.; Angulo-preckler, C. A Minireview on Biodiscovery in Antarctic Marine Benthic Invertebrates. Front. Mar. Sci. 2021, 8, 1–15. [Google Scholar] [CrossRef]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about clinically approved and Preclinically investigated marine natural products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Lu, W.Y.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Application of marine natural products in drug research. Bioorganic Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Worachartcheewan, A.; Shoombuatong, W.; Songtawee, N.; Simeon, S.; Prachayasittikul, V.; Nantasenamat, C. Computer-Aided Drug Design of Bioactive Natural Products. Curr. Top. Med. Chem. 2015, 15, 1780–1800. [Google Scholar] [CrossRef] [PubMed]
- Reker, D.; Perna, A.M.; Rodrigues, T.; Schneider, P.; Reutlinger, M.; Mönch, B.; Koeberle, A.; Lamers, C.; Gabler, M.; Steinmetz, H.; et al. Revealing the macromolecular targets of complex natural products. Nat. Chem. 2014, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.B.; The International Natural Products Science Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-Aided Drug Design Applied to Marine Drug Discovery: Meridianins as Alzheimer’s Disease Therapeutic Agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, H.; El Sayed, K. Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds. Mar. Drugs 2016, 14, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, P.; Schneider, G.; Kapoor, S.; Waldmann, H.; Ziegler, S.; Schneider, P.; Schneider, G.; Lima, A.N.; Philot, E.A.; Trossini, G.H.; et al. De-orphaning the marine natural product (±)-marinopyrrole A by computational target prediction and biochemical validation. Chem. Commun. 2017, 24, 3232. [Google Scholar] [CrossRef]
- Cuyàs, E.; Castillo, D.; Llorach-Parés, L.; Lozano-Sánchez, J.; Verdura, S.; Nonell-Canals, A.; Brunet, J.; Bosch-Barrera, J.; Joven, J.; Valdés, R.; et al. Computational de-orphanization of the olive oil biophenol oleacein: Discovery of new metabolic and epigenetic targets. Food Chem. Toxicol. 2019, 131, 110529. [Google Scholar] [CrossRef]
- Friedrich, L.; Cingolani, G.; Ko, Y.H.; Iaselli, M.; Miciaccia, M.; Perrone, M.G.; Neukirch, K.; Bobinger, V.; Merk, D.; Hofstetter, R.K.; et al. Learning from Nature: From a Marine Natural Product to Synthetic Cyclooxygenase-1 Inhibitors by Automated De Novo Design. Adv. Sci. 2021, 8, 1–12. [Google Scholar] [CrossRef]
- Qiang, B.; Lai, J.; Jin, H.; Zhang, L.; Liu, Z. Target prediction model for natural products using transfer learning. Int. J. Mol. Sci. 2021, 22, 4632. [Google Scholar] [CrossRef]
- Mayr, F.; Möller, G.; Garscha, U.; Fischer, J.; Castaño, P.R.; Inderbinen, S.G.; Temml, V.; Waltenberger, B.; Schwaiger, S.; Hartmann, R.W.; et al. Finding new molecular targets of familiar natural products using in silico target prediction. Int. J. Mol. Sci. 2020, 21, 7102. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Aires-de-Sousa, J. Computational methodologies in the exploration of marine natural product leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Santamaria-Navarro, E.; Felix, E.; Nonell-Canals, A. Cabrakan. Available online: https://www.mindthebyte.com/ (accessed on 10 December 2018).
- Santamaria-Navarro, E.; Nonell-Canals, A. Hurakan. Available online: https://www.mindthebyte.com/ (accessed on 10 December 2018).
- Pinero, J.; Queralt-Rosinach, N.; Bravo, A.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 12 March 2021).
- Krahn, A.I.; Wells, C.; Drewry, D.H.; Beitel, L.K.; Durcan, T.M.; Axtman, A.D. Defining the Neural Kinome: Strategies and Opportunities for Small Molecule Drug Discovery to Target Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 1871–1886. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Neurodegenerative drug discovery: Building on the past, looking to the future. Future Med. Chem. 2017, 9, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Catanesi, M.; Caioni, G.; Castelli, V.; Benedetti, E.; d’Angelo, M.; Cimini, A. Benefits under the Sea: The Role of Marine Compounds in Neurodegenerative Disorders. Mar. Drugs 2021, 19, 24. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1 (accessed on 12 March 2021).
- Figtree, G.A.; Broadfoot, K.; Casadei, B.; Califf, R.; Crea, F.; Drummond, G.R.; Freedman, J.E.; Guzik, T.J.; Harrison, D.; Hausenloy, D.J.; et al. A call to action for new global approaches to cardiovascular disease drug solutions. Eur. Heart J. 2021, 42, 1464–1475. [Google Scholar] [CrossRef]
- Liang, B.; Cai, X.-Y.; Gu, N. Marine Natural Products and Coronary Artery Disease. Front. Cardiovasc. Med. 2021, 8, 739932. [Google Scholar] [CrossRef]
- Zhou, J.-B.; Luo, R.; Zheng, Y.-L.; Pang, J.-Y. Recent Advances in the Discovery and Development of Marine Natural Products with Cardiovascular Pharmacological Effects. Mini-Rev. Med. Chem. 2018, 18, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, E.; Manganaro, A.; Gini, G. VEGA-QSAR: AI inside a platform for predictive toxicology. In Proceedings of the Popularize Artificial Intelligence 2013, Turin, Italy, 5 December 2013. [Google Scholar]
- Avila, C. Chemical War in Marine Animal Forests: Natural Products and Chemical Interactions. In Perspectives on the Marine Animal Forests of the World; Springer Nature: Cham, Switzerland, 2020; pp. 239–307. ISBN 9783030570545. [Google Scholar]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.; Ruiz-carmona, S.; Barril, X. An investigation of structural stability in protein-ligand complexes reveals the balance between order and disorder. Commun. Chem. 2019, 2, 110. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Yang, H.; Lv, W.; Zhang, X.; Zhai, H. Prediction of the inhibitory concentrations of chloroquine derivatives using Deep Neural Networks models. J. Biomol. Struct. Dyn. 2021, 39, 672–680. [Google Scholar] [CrossRef]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef] [Green Version]
- Viciano, I.; Castillo, R.; Martí, S. QM/MM modeling of the hydroxylation of the androstenedione substrate catalyzed by cytochrome P450 aromatase (CYP19A1). J. Comput. Chem. 2015, 36, 1736–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viciano, I.; Martí, S. Theoretical Study of the Mechanism of Exemestane Hydroxylation Catalyzed by Human Aromatase Enzyme. J. Phys. Chem. B 2016, 120, 3331–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlanson, D.A.; Arndt, J.W.; Cancilla, M.T.; Cao, K.; Elling, R.A.; English, N.; Friedman, J.; Hansen, S.K.; Hession, C.; Joseph, I.; et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg. Med. Chem. Lett. 2011, 21, 3078–3083. [Google Scholar] [CrossRef]
- Medina, J.R.; Becker, C.J.; Blackledge, C.W.; Duquenne, C.; Feng, Y.; Grant, S.W.; Heerding, D.; Li, W.H.; Miller, W.H.; Romeril, S.P.; et al. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J. Med. Chem. 2011, 54, 1871–1895. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Fu, Q.; Ni, D.; Zhang, J.; Li, X.; Lu, S. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J. Enzym. Inhib. Med. Chem. 2019, 34, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busschots, K.; Lopez-Garcia, L.A.; Lammi, C.; Stroba, A.; Zeuzem, S.; Piiper, A.; Alzari, P.M.; Neimanis, S.; Arencibia, J.M.; Engel, M.; et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem. Biol. 2012, 19, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Rettenmaier, T.J.; Sadowsky, J.D.; Thomsen, N.D.; Chen, S.C.; Doak, A.K.; Arkin, M.R.; Wells, J.A. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc. Natl. Acad. Sci. USA 2014, 111, 18590–18595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Yilmaz, O.G.; Olmez, E.O.; Ulgen, K.O. Targeting the Akt1 allosteric site to identify novel scaffolds through virtual screening. Comput. Biol. Chem. 2014, 48, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Canduri, F.; Dos Santos, D.M.; Pereira, J.H.; Dias, M.V.B.; Silva, R.G.; Mendes, M.A.; Basso, L.A.; Palma, M.S.; Santos, D.S. Structural basis for inhibition of human PNP by immucillin-H. Biochem. Biophys. Res. Commun. 2003, 309, 917–922. [Google Scholar] [CrossRef]
- Decherchi, S.; Berteotti, A.; Bottegoni, G.; Rocchia, W.; Cavalli, A. The ligand binding mechanism to purine nucleoside phosphorylase elucidated via molecular dynamics and machine learning. Nat. Commun. 2015, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Suarez, J.; Haapalainen, A.M.; Cahill, S.M.; Ho, M.C.; Yan, F.; Almo, S.C.; Schramm, V.L. Catalytic site conformations in human PNP by 19F-NMR and crystallography. Chem. Biol. 2013, 20, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, D.M.; Canduri, F.; Pereira, J.H.; Dias, M.V.B.; Silva, R.G.; Mendes, M.A.; Palma, M.S.; Basso, L.A.; De Azevedo, W.F.; Santos, D.S. Crystal structure of human purine nucleoside phosphorylase complexed with acyclovir. Biochem. Biophys. Res. Commun. 2003, 308, 553–559. [Google Scholar] [CrossRef]
- Saen-oon, S.; Schramm, V.L.; Schwartz, S.D. Transition path sampling study of the reaction catalyzed by purine nucleoside phosphorylase. Z. Phys. Chem. 2008, 222, 1359–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassera, M.B.; Hazleton, K.Z.; Merino, E.F.; Obaldia, N.; Ho, M.C.; Murkin, A.S.; DePinto, R.; Gutierrez, J.A.; Almo, S.C.; Evans, G.B.; et al. Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS ONE 2011, 6, e26916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.A.; Hurley, T.D. Development of a high-throughput in vitro assay to identify selective inhibitors for human ALDH1A1. Chem. Biol. Interact. 2014, 234, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.; Cho, H.; Tong, M.; Ding, Y. NAD+-Linked 15-Hydroxyprostaglandin Dehydrogenase: Structure and Biological Functions. Curr. Pharm. Des. 2006, 12, 955–962. [Google Scholar] [CrossRef]
- Niesen, F.H.; Schultz, L.; Jadhav, A.; Bhatia, C.; Guo, K.; Maloney, D.J.; Pilka, E.S.; Wang, M.; Oppermann, U.; Heightman, T.D.; et al. High-Affinity Inhibitors of Human NAD+-Dependent 15-Hydroxyprostaglandin Dehydrogenase: Mechanisms of Inhibition and Structure-Activity Relationships. PLoS ONE 2010, 5, e13719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Nagarajan, D.; Mukherjee, S.; Chandra, N. PLIC: Protein-ligand interaction clusters. Database 2014, 2014, bau029. [Google Scholar] [CrossRef] [Green Version]
- Antczak, M.I.; Zhang, Y.; Wang, C.; Doran, J.; Naidoo, J.; Voruganti, S.; Williams, N.S.; Markowitz, S.D.; Ready, J.M. Inhibitors of 15-Prostaglandin Dehydrogenase To Potentiate Tissue Repair. J. Med. Chem. 2017, 60, 3979–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.R.; Sharma, S.; Joshi, P.; Wani, A.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. Meridianin derivatives as potent Dyrk1A inhibitors and neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 2948–2952. [Google Scholar] [CrossRef]
- Liu, Y.A.; Jin, Q.; Ding, Q.; Hao, X.; Mo, T.; Yan, S.; Zou, Y.; Huang, Z.; Zhang, X.; Gao, W.; et al. A Dual Inhibitor of DYRK1A and GSK3β for β-Cell Proliferation: Aminopyrazine Derivative GNF4877. ChemMedChem 2020, 15, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.A.; Jin, Q.; Zou, Y.; Ding, Q.; Yan, S.; Wang, Z.; Hao, X.; Nguyen, B.; Zhang, X.; Pan, J.; et al. Selective DYRK1A Inhibitor for the Treatment of Type 1 Diabetes: Discovery of 6-Azaindole Derivative GNF2133. J. Med. Chem. 2020, 63, 2958–2973. [Google Scholar] [CrossRef] [PubMed]
- Lee Walmsley, D.; Murray, J.B.; Dokurno, P.; Massey, A.J.; Benwell, K.; Fiumana, A.; Foloppe, N.; Ray, S.; Smith, J.; Surgenor, A.E.; et al. Fragment-Derived Selective Inhibitors of Dual-Specificity Kinases DYRK1A and DYRK1B. J. Med. Chem. 2021, 64, 8971–8991. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Karthikeyan, C.; Hari, N.S.; Moorthy, N.; Waiker, D.K.; Jain, A.K.; Trivedi, P. Human CDC2-Like Kinase 1 (CLK1): A Novel Target for Alzheimer’s Disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef]
- Walter, A.; Chaikuad, A.; Helmer, R.; Loaë, N.; Preu, L.; Ott, I.; Knapp, S.; Meijer, L.; Kunick, C. Molecular structures of cdc2-like kinases in complex with a new inhibitor chemotype. PLoS ONE 2018, 13, e0196761. [Google Scholar] [CrossRef] [Green Version]
- Miley, M.J.; Zielinska, A.K.; Keenan, J.E.; Bratton, S.M.; Radominska-Pandya, A.; Redinbo, M.R. Crystal Structure of the Cofactor-Binding Domain of the Human Phase II Drug-Metabolism Enzyme UDP-Glucuronosyltransferase 2B7. J. Mol. Biol. 2007, 369, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef] [Green Version]
- Bart, A.G.; Takahashi, R.H.; Wang, X.; Scott, E.E. Human cytochrome P450 1A1 adapts active site for atypical nonplanar substrate. Drug Metab. Dispos. 2020, 48, 86–92. [Google Scholar] [CrossRef]
- Joshi, P.; McCann, G.J.P.; Sonawane, V.R.; Vishwakarma, R.A.; Chaudhuri, B.; Bharate, S.B. Identification of Potent and Selective CYP1A1 Inhibitors via Combined Ligand and Structure-Based Virtual Screening and Their in Vitro Validation in Sacchrosomes and Live Human Cells. J. Chem. Inf. Model. 2017, 57, 1309–1320. [Google Scholar] [CrossRef]
- Tramonti, A.; Cuy, E.; Encinar, A.; Pietzke, M.; Paone, A.; Verdura, S.; Arbus, A.; Martin-castillo, B.; Giardina, G.; Joven, J.; et al. Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers 2021, 13, 4009. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Ivanochko, D.; Schapira, M. Methyltransferase inhibitors: Competing with, or exploiting the bound cofactor. Molecules 2019, 24, 4492. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.S.; Berteotti, A.; Ronsisvalle, S.; Rocchia, W.; Cavalli, A. Steered Molecular Dynamics Simulations for Studying Protein–Ligand Interaction in Cyclin-Dependent Kinase 5. J. Chem. Inf. Model. 2014, 54, 470–480. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, A.; Niesen, F.H.; Schultz, L.; Oppermann, U.; Maloney, D.J.; Simeonov, A. Potent and selective inhibitors of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase (HPGD). Probe Rep. NIH Mol. Libr. Progr. 2010, 147, 1–36. [Google Scholar]
- Morgan, C.A.; Hurley, T.D. Characterization of two distinct structural classes of selective aldehyde dehydrogenase 1A1 inhibitors. J. Med. Chem. 2015, 58, 1964–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Shortall, K.; Djeghader, A.; Magner, E.; Soulimane, T. Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 410. [Google Scholar] [CrossRef]
- Li, B.; Yang, K.; Liang, D.; Jiang, C.; Ma, Z. Discovery and development of selective aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors. Eur. J. Med. Chem. 2021, 209, 112940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Sun, Y.J.; Rose, J.; Chung, Y.J.; Hsiao, C.D.; Chang, W.R.; Kuo, I.; Perozich, J.; Lindahl, R.; Hempel, J.; et al. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 1997, 4, 317–326. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Rastelli, G.; Degliesposti, G.; Del Rio, A.; Sgobba, M. Binding estimation after refinement, a new automated procedure for the refinement and rescoring of docked ligands in virtual screening. Chem. Biol. Drug Des. 2009, 73, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Pinzi, L. Refinement and Rescoring of Virtual Screening Results. Front. Chem. 2019, 7, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emamian, E.S. AKT/GSK3 signaling pathway and schizophrenia. Front. Mol. Neurosci. 2012, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Chen, Y.W.; Wang, T.W.; Lai, W.S. Akting up in the GABA hypothesis of schizophrenia: Akt1 deficiency modulates GABAergic functions and hippocampus-dependent functions. Sci. Rep. 2016, 6, 33095. [Google Scholar] [CrossRef]
- Mora, A.; Davies, A.M.; Bertrand, L.; Sharif, I.; Budas, G.R.; Jovanović, S.; Mouton, V.; Kahn, C.R.; Lucocq, J.M.; Gray, G.A.; et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003, 22, 4666–4676. [Google Scholar] [CrossRef]
- Ito, K.; Akazawa, H.; Tamagawa, M.; Furukawa, K.; Ogawa, W.; Yasuda, N.; Kudo, Y.; Liao, C.H.; Yamamoto, R.; Sato, T.; et al. PDK1 coordinates survival pathways and β-adrenergic response in the heart. Proc. Natl. Acad. Sci. USA 2009, 106, 8689–8694. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, V.; Bogomolovas, J.; Ehler, E.; dos Remedios, C.G.; Yu, J.; Gao, C.; Lange, S. PKC and PKN in heart disease. J. Mol. Cell. Cardiol. 2019, 128, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Di, R.; Tao, F.; Chang, Z.; Lu, S.; Fan, W.; Shan, C.; Li, X.; Yang, Z. PDK1 Regulates Vascular Remodeling and Promotes Epithelial-Mesenchymal Transition in Cardiac Development. Mol. Cell. Biol. 2010, 30, 3711–3721. [Google Scholar] [CrossRef] [Green Version]
- Barile, E.; De, S.K.; Pellecchia, M. PDK1 inhibitors. Pharm. Pat. Anal. 2012, 1, 145–163. [Google Scholar] [CrossRef] [Green Version]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef] [Green Version]
- Hossen, M.J.; Kim, S.C.; Yang, S.; Kim, H.G.; Jeong, D.; Yi, Y.S.; Sung, N.Y.; Lee, J.O.; Kim, J.H.; Cho, J.Y. PDK1 disruptors and modulators: A patent review. Expert Opin. Ther. Pat. 2015, 25, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Modulators of Nucleoside Metabolism in the Therapy of Brain Diseases. Curr. Top. Med. Chem. 2011, 11, 1068–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantia, S. Purine nucleoside phosphorylase inhibitors—An immunotherapy with novel mechanism of action for the treatment of melanoma. J. Immunother. Cancer 2015, 3, P292. [Google Scholar] [CrossRef] [Green Version]
- La Sala, G.; Gunnarsson, A.; Edman, K.; Tyrchan, C.; Hogner, A.; Frolov, A. Unravelling Allosteric Cross-Talk between Co-Activator Peptide and Ligand Binding Site in Glucocorticoid Receptor. Biophys. J. 2021, 120, 298a. [Google Scholar] [CrossRef]
- Reyes, F.; Fernández, R.; Rodríguez, A.; Francesch, A.; Taboada, S.; Ávila, C.; Cuevas, C. Aplicyanins A-F, new cytotoxic bromoindole derivatives from the marine tunicate Aplidium cyaneum. Tetrahedron 2008, 64, 5119–5123. [Google Scholar] [CrossRef]
- Šíša, M.; Pla, D.; Altuna, M.; Francesch, A.; Cuevas, C.; Albericio, F.; Álvarez, M. Total synthesis and antiproliferative activity screening of (±)-aplicyanins A, B and E and related analogues. J. Med. Chem. 2009, 52, 6217–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from marine invertebrates as important leads for anticancer drugs discovery and development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimers. Dis. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dolan, P.J.; Johnson, G.V.W. The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Devel. 2010, 13, 595–603. [Google Scholar]
- Tell, V.; Hilgeroth, A. Recent developments of protein kinase inhibitors as potential AD therapeutics. Front. Cell. Neurosci. 2013, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Sinibaldi, L.; Harifi, G.; Bottillo, I.; Iannicelli, M.; El Hassani, S.; Brancati, F.; Dallapiccola, B. A novel homozygous splice site mutation in the HPGD gene causes mild primary hypertrophic osteoarthropathy. Clin. Exp. Rheumatol. 2010, 28, 153–157. [Google Scholar] [PubMed]
- Khan, A.K.; Muhammad, N.; Khan, S.A.; Ullah, W.; Nasir, A.; Afzal, S.; Ramzan, K.; Basit, S.; Khan, S. A novel mutation in the HPGD gene causing primary hypertrophic osteoarthropathy with digital clubbing in a Pakistani family. Ann. Hum. Genet. 2018, 82, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Mahesh, D.M.; Madabhavi, I. Digital clubbing. Lung India 2012, 29, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Rodriguez-Urgelles, E.; Nonell-Canals, A.; Alberch, J.; Avila, C.; Sanchez-Martinez, M.; Giralt, A. Meridianins and lignarenone B as potential GSK3β inhibitors and inductors of structural neuronal plasticity. Biomolecules 2020, 10, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, F.; Alves, G.; Debiton, E.; Nauton, L.; Théry, V.; Durieu, E.; Ferandin, Y.; Lozach, O.; Meijer, L.; Anizon, F.; et al. Synthesis, Protein Kinase Inhibitory Potencies, and in Vitro Antiproliferative Activities of Meridianin Derivatives. J. Med. Chem. 2011, 54, 4474–4489. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, R.A. Meridianins: Marine-Derived Potent Kinase Inhibitors. Mini-Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Singh, V.; Shin, H.-S.; Patel, R.V. Kinase Inhibitor Indole Derivatives as Anticancer Agents: A Patent Review. Recent Pat. Anticancer Drug Discov. 2016, 12, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [Green Version]
- Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Kororamides, convolutamines, and indole derivatives as possible tau and dual-specificity kinase inhibitors for Alzheimer’s disease: A computational study. Mar. Drugs 2018, 16, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauletti, P.M.; Cintra, L.S.; Braguine, C.G.; da Silva Filho, A.A.; Silva, M.L.A.E.; Cunha, W.R.; Januário, A.H. Halogenated indole alkaloids from marine invertebrates. Mar. Drugs 2010, 8, 1526–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarachana, T.; Xu, M.; Wu, R.C.; Hu, V.W. Sex hormones in autism: Androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS ONE 2011, 6, e17116. [Google Scholar] [CrossRef] [PubMed]
- Xiaojiang, Z.; Shuzheng, L.; Chengjun, G.; Conghong, S.; Yunpeng, C.; Lin, Z.; Guozhong, W.; Zhisheng, W. Gene- gene interaction between PPARG and CYP1A1 gene on coronary artery disease in the Chinese Han Population. Oncotarget 2017, 8, 34398–34404. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.D.; Xie, W.; Yu, Z.X. Impact of interaction between CYP1A1 genetic polymorphisms and smoking on coronary artery disease in the Han of China. Clin. Exp. Hypertens. 2017, 39, 339–343. [Google Scholar] [CrossRef]
- Radominska-Pandya, A.; Pokrovskaya, I.D.; Xu, J.; Little, J.M.; Jude, A.R.; Kurten, R.C.; Czernik, P.J. Nuclear UDP-glucuronosyltransferases: Identification of UGT2B7 and UGT1A6 in human liver nuclear membranes. Arch. Biochem. Biophys. 2002, 399, 37–48. [Google Scholar] [CrossRef]
- Tukey, R.H.; Strassburg, C.P. Human Udp-G Lucuronosyltransferases: Metabolism, Expression, and Disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef]
- Arbitrio, M.; Scionti, F.; Altomare, E.; Di Martino, M.T.; Agapito, G.; Galeano, T.; Staropoli, N.; Iuliano, E.; Grillone, F.; Fabiani, F.; et al. Polymorphic Variants in NR1I3 and UGT2B7 Predict Taxane Neurotoxicity and Have Prognostic Relevance in Patients With Breast Cancer: A Case-Control Study. Clin. Pharmacol. Ther. 2019, 106, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Ahern, T.P.; Christensen, M.; Cronin-Fenton, D.P.; Lunetta, K.L.; Siøland, H.; Gjerde, J.; Garne, J.P.; Rosenberg, C.L.; Silliman, R.A.; Srøensen, H.T.; et al. Functional polymorphisms in UDP-glucuronosyl transferases and recurrence in tamoxifen-treated breast cancer survivors. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1937–1943. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, Y.; Huang, W.; Wang, X.; Ni, X.; Lu, H.; Hu, J.; Deng, S.; Zhu, X.; Xie, H.; et al. Effects of Comedication and Genetic Factors on the Population Pharmacokinetics of Lamotrigine: A Prospective Analysis in Chinese Patients with Epilepsy. Front. Pharmacol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bastami, S.; Gupta, A.; Zackrisson, A.L.; Ahlner, J.; Osman, A.; Uppugunduri, S. Influence of UGT2B7, OPRM1 and ABCB1 gene polymorphisms on postoperative morphine consumption. Basic Clin. Pharmacol. Toxicol. 2014, 115, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, W.; Nishizawa, D.; Fukuda, K.; Kasai, S.; Hasegawa, J.; Wajima, K.; Nakagawa, T.; Ikeda, K. Association between UGT2B7 gene polymorphisms and fentanyl sensitivity in patients undergoing painful orthognathic surgery. Mol. Pain 2016, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, N.H.; Ismail, S. Inhibition of UGT2B7 enzyme activity in human and rat liver microsomes by herbal constituents. Molecules 2018, 23, 2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casida, J.E.; Ford, B.; Jinsmaa, Y.; Sullivan, P.; Cooney, A.; Goldstein, D.S. Benomyl, aldehyde dehydrogenase, DOPAL, and the catecholaldehyde hypothesis for the pathogenesis of parkinsons disease. Chem. Res. Toxicol. 2014, 27, 1359–1361. [Google Scholar] [CrossRef]
- Salas-Leal, A.C.; Sandoval-Carrillo, A.; Romero-Gutiérrez, E.; Castellanos-Juárez, F.X.; Méndez-Hernández, E.M.; La Llave-León, O.; Quiñones-Canales, G.; Arias-Carrión, O.; Salas-Pacheco, J.M. rs3764435 Associated with Parkinson’s Disease in Mexican Mestizos: Case-Control Study Reveals Protective Effects Against Disease Development and Cognitive Impairment. Front. Neurol. 2019, 10, 1066. [Google Scholar] [CrossRef] [PubMed]
- Kotraiah, V.; Pallares, D.; Toema, D.; Kong, D.; Beausoleil, E. Identification of aldehyde dehydrogenase 1A1 modulators using virtual screening. J. Enzyme Inhib. Med. Chem. 2013, 28, 489–494. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Sterzyńska, K.; Sosińska, P.; Andrzejewska, M.; Zawierucha, P.; Nowicki, M.; Zabel, M. Inhibition of ALDH1A1 activity decreases expression of drug transporters and reduces chemotherapy resistance in ovarian cancer cell lines. Int. J. Biochem. Cell Biol. 2016, 78, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Condello, S.; Wang, Y.; Swetzig, W.M.; Barber, E.; Hurley, T.; Matei, D. A novel aldh1a1 inhibitor targets cells with stem cell characteristics in ovarian cancer. Cancers 2019, 11, 502. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Song, H.; Jiang, L.; Qiao, Y.; Yang, D.; Wang, D.; Li, J. Silybin Prevents Prostate Cancer by Inhibited the ALDH1A1 Expression in the Retinol Metabolism Pathway. Front. Cell Dev. Biol. 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Llorach-Pares, L.; Fernández-Arroyo, S.; Luciano-Mateo, F.; Cabré, N.; Stursa, J.; Werner, L.; Martin-Castillo, B.; Viollet, B.; et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell 2018, 17, e12772. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, 2006–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, E.; Santamaría-Navarro, E.; Sanchez-Martinez, M.; Nonell-Canals, A. Itzamna. Available online: https://www.mindthebyte.com/ (accessed on 10 December 2018).
- Felix, E.; Nonell-Canals, A. Kin. Available online: https://www.mindthebyte.com/ (accessed on 10 December 2018).
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Jenson, C. Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: Seeking temperatures of maximum density. J. Comput. Chem. 1998, 19, 1179–1186. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Waskom, M. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucl. Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 31 December 2021).
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Concepción Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Z.; Cui, W. Marine-Derived Natural Compounds for the Treatment of Parkinson’s Disease. Mar. Drugs 2019, 17, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, M.A.; Dash, R.; Haque, M.N.; Mohibbullah, M.; Sohag, A.A.M.; Rahman, M.A.; Uddin, M.J.; Alam, M.; Moon, I.S. Neuroprotective Potentials of Marine Algae and Their Bioactive Metabolites: Pharmacological Insights and Therapeutic Advances. Mar. Drugs 2020, 18, 347. [Google Scholar] [CrossRef]
- Silva, M.; Seijas, P.; Otero, P. Exploitation of Marine Molecules to Manage Alzheimer’s Disease. Mar. Drugs 2021, 19, 373. [Google Scholar] [CrossRef]
- Bhandarkar, N.S.; Kumar, S.A.; Martin, J.; Brown, L.; Panchal, S.K. Attenuation of Metabolic Syndrome by EPA / DHA Ethyl Esters in Testosterone-Deficient Obese Rats. Mar. Drugs 2018, 16, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanan, P.; Davidson, N.C.; Schmidt, E.B.; Calder, P.C. Cardiovascular effects of marine omega-3 fatty acids. Lancet 2011, 376, 540–550. [Google Scholar] [CrossRef]
- Riccioni, G.; Orazio, N.D.; Franceschelli, S.; Speranza, L. Marine Carotenoids and Cardiovascular Risk Markers. Mar. Drugs 2011, 9, 1166–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
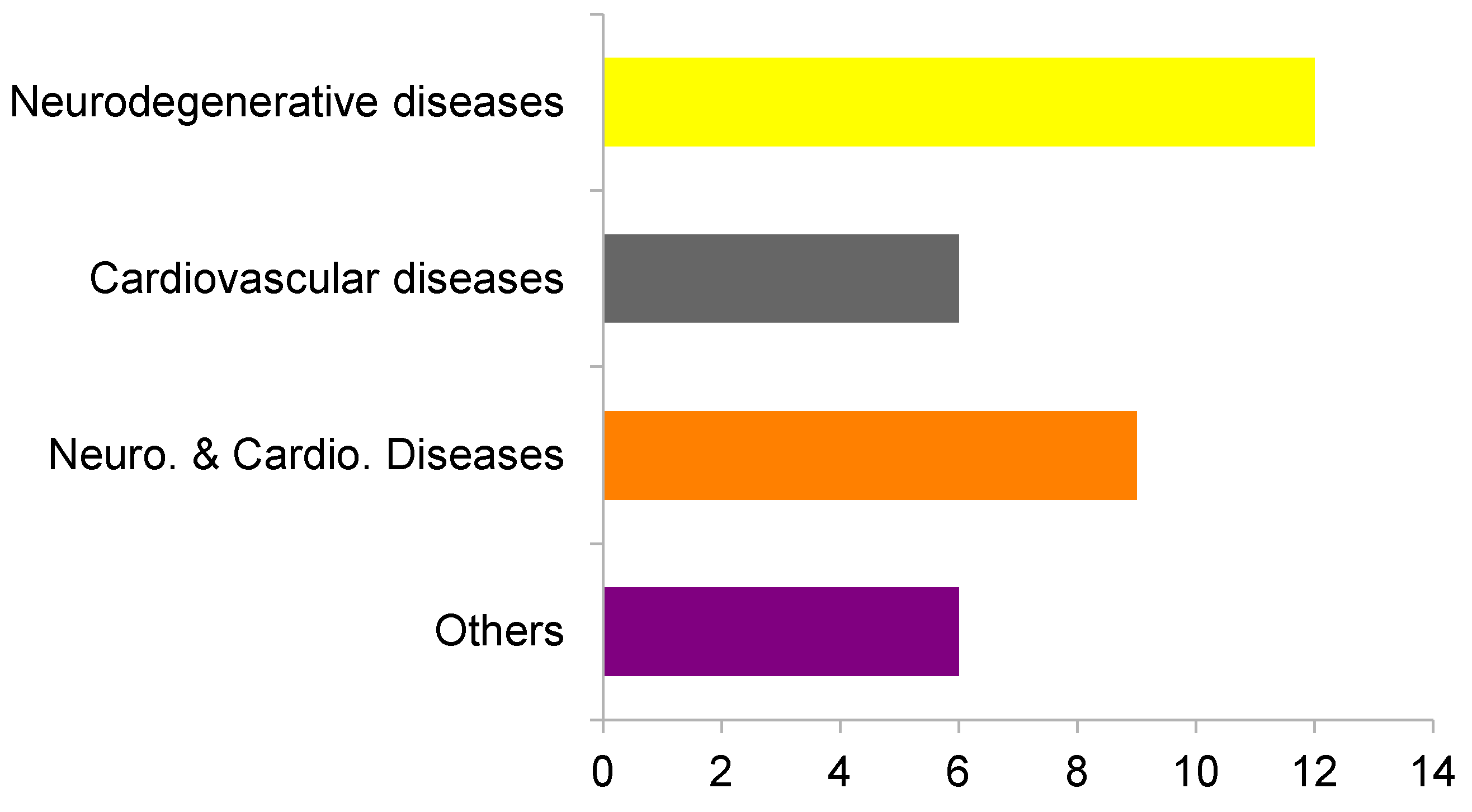

{kind=link}
{kind=link}
{kind=link}
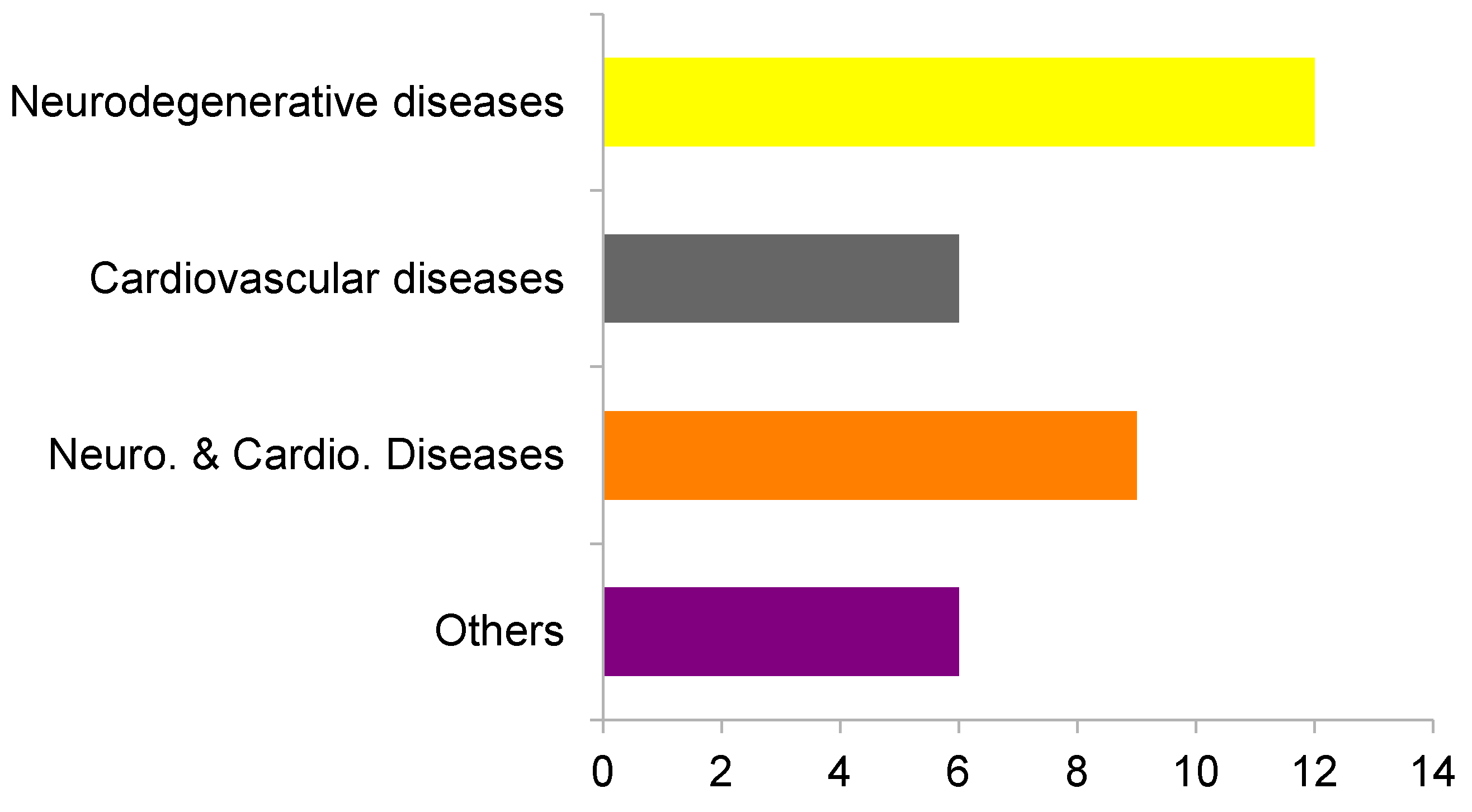{kind=link}
{kind=link}
{kind=link}
| Aplicyanin-A | Dendrinolide | Hodgsonal | Meridianin-A | Polyrhaphin-A | Pterenone | Rossinone-A |
|---|---|---|---|---|---|---|
| Q96KQ7 * | P16662 | P11511 | P49759 | P23416 | P01375 | P83916 * |
| Q16236 * | P83916 * | Q13627 | O75311 | P83916 * | Q96KQ7 * | |
| P09874 | Q96KQ7 * | Q96KQ7 * | P24046 | Q16236 * | Q07343 | |
| O15530 | Q16236 * | P46098 | P15428 * | Q16236 * | ||
| P31749 | P00374 | P14867 | P27815 | |||
| P00491 | P48730 | P04798 | Q08499 | |||
| Q13976 | P83916 | P15428 * | ||||
| P49841 | Q16236 * | P00352 | ||||
| P05129 | O00255 | |||||
| Q9Y463 | P07550 | |||||
| Q99714 |
| Molecule | Carcinogenicity | Mutagenicity | Developmental Toxicity | Skin Sensitization | Average Toxicity |
|---|---|---|---|---|---|
| Apliacyanin | LOW | NO | LOW | LOW | LOW |
| Dendrinolide | LOW | NO | LOW | LOW | LOW |
| Discorhabdin-B | LOW | LOW | LOW | LOW | LOW |
| Hodgsonal | MEDIUM | LOW | MEDIUM | HIGH | LOW |
| Meridianin-A | LOW | LOW | LOW | LOW | LOW |
| Polyrhaphin-A | MEDIUM | NO | MEDIUM | LOW | LOW |
| Pteroenone | MEDIUM | LOW | LOW | HIGH | LOW |
| Rossinone-A | LOW | LOW | LOW | MEDIUM | LOW |
| Pectinoside-B | LOW | NO | LOW | LOW | LOW |
| Complex | Predicted HBs Reported in the Literature | Literature Reference |
|---|---|---|
| Apliacynin-A-O15530 | Long-lived: ASP223, LYS111, SER92 | [57,58,59,60,61] |
| Aplicyanin-A-P00491 | Long-lived: MET219 Medium-lived: HIS86 Short-lived: ASN243, THR242, GLU201 | [64,65,66] |
| Apliacynin-A-P31749 | Long-lived: SER205 Medium-lived: ASP242 Short-lived: TRP80 | [62,63] |
| Meridianin-A-P15428 | Long-lived: GLN148 Medium-lived: SER138 Short-lived: GLU184 | [71,72,73,74] |
| Meridianin-A-P49759 | Long-lived: Glu242 Medium-lived: LYS191 Short-lived: LEU244 | [80,90,91] |
| Meridianin-A-Q9Y463 | Long-lived: LYS140, GLU191 | [75,76,77] |
| Rossinone-A-P15428 | Long-lived: ASN91 Medium-lived: GLN148, ILE17, GLN15 Short-lived: TYR151, GLY93, VAL186, GLY12 | [71,72,73,74] |
| Rossinone-A-P00352 | Long-lived: GLU196, GLU269, GLU400 Medium-lived: LYS193 | [70] |
| Hodgsonal-P11511 | Long-lived: MET374 Short-lived: LEU477 | [53,54,55,56] |
| Dendrinolide-P16662 | Medium-lived: TRP356 | [83] |
| Polyrhaphin-A-P04798 | Short-lived: ILE386, SER322 | [84,85,86,87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases. Mar. Drugs 2022, 20, 53. https://doi.org/10.3390/md20010053
Llorach-Pares L, Nonell-Canals A, Avila C, Sanchez-Martinez M. Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases. Marine Drugs. 2022; 20(1):53. https://doi.org/10.3390/md20010053
Chicago/Turabian StyleLlorach-Pares, Laura, Alfons Nonell-Canals, Conxita Avila, and Melchor Sanchez-Martinez. 2022. "Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases" Marine Drugs 20, no. 1: 53. https://doi.org/10.3390/md20010053
APA StyleLlorach-Pares, L., Nonell-Canals, A., Avila, C., & Sanchez-Martinez, M. (2022). Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases. Marine Drugs, 20(1), 53. https://doi.org/10.3390/md20010053