
An Integrative Bioinformatic Analysis for Keratinase Detection in Marine-Derived Streptomyces

 ,
,
Abstract
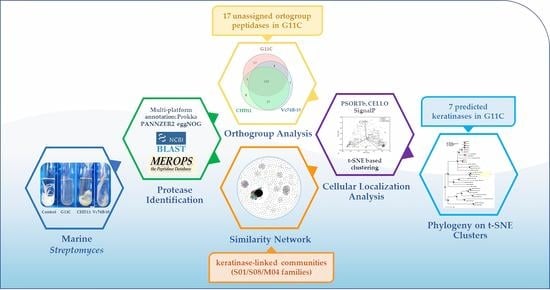
1. Introduction
2. Results
2.1. Genome Features of Streptomyces Strains with Differential Keratinolytic Activity
2.2. Comparative Genomics of Differential Keratinolytic Streptomycete Strains
2.2.1. Protease Search
2.2.2. Network Analysis
2.3. Filtering by Cellular Localization Scores
2.4. Phylogeny on t-SNE Groups
2.5. Summary of the Main Results of the Bioinformatic Pipeline
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Genomic DNA Extraction
4.3. Genome Sequencing, Assembly, and Annotation
4.4. Phylogenomic Tree Inference
4.5. Putative Protease Identification
4.6. Classification of Protease Families and Identification of Protease Orthogroups
4.7. Creation of Custom Databases: Functional Keratinases and Putative Non-Keratinases
4.8. Similarity Network
4.9. Cellular Localization and Dimension Reduction
4.10. Clustering and Phylogeny of t-SNE Groups
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korniłłowicz-Kowalska, T.; Bohacz, J. Biodegradation of keratin waste: Theory and practical aspects. Waste Manag. 2011, 31, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, A. Bacterial keratinases: Useful enzymes for bioprocessing agroindustrial wastes and beyond. Food Bioprocess Technol. 2008, 1, 105–116. [Google Scholar] [CrossRef]
- Lange, L.; Huang, Y.; Busk, P.K. Microbial decomposition of keratin in nature—A new hypothesis of industrial relevance. Appl. Microbiol. Biotechnol. 2016, 100, 2083–2096. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, M.H.; Zheng, W.; Moyer, A.E.; Sjövall, P.; Lindgren, J. Preservation potential of keratin in deep time. PLoS ONE 2018, 13, e0206569. [Google Scholar] [CrossRef] [PubMed]
- Moyer, A.E.; Zheng, W.; Schweitzer, M.H. Keratin durability has implications for the fossil record: Results from a 10 year feather degradation experiment. PLoS ONE 2016, 11, e0157699. [Google Scholar] [CrossRef] [PubMed]
- De Souza Carvalho, I.; Novas, F.E.; Agnolín, F.L.; Isasi, M.P.; Freitas, F.I.; Andrade, J.A. A Mesozoic bird from Gondwana preserving feathers. Nat. Commun. 2015, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Busk, P.K.; Herbst, F.A.; Lange, L. Genome and secretome analyses provide insights into keratin decomposition by novel proteases from the non-pathogenic fungus Onygena corvina. Appl. Microbiol. Biotechnol. 2015, 99, 9635–9649. [Google Scholar] [CrossRef] [PubMed]
- Mazotto, A.M.; Couri, S.; Damaso, M.C.T.; Vermelho, A.B. Degradation of feather waste by Aspergillus niger keratinases: Comparison of submerged and solid-state fermentation. Int. Biodeterior. Biodegrad. 2013, 85, 189–195. [Google Scholar] [CrossRef]
- Balaji, S.; Senthil Kumar, M.; Karthikeyan, R.; Kumar, R.; Kirubanandan, S.; Sridhar, R.; Sehgal, P.K. Purification and characterization of an extracellular keratinase from a hornmeal-degrading Bacillus subtilis MTCC (9102). World J. Microbiol. Biotechnol. 2008, 24, 2741–2745. [Google Scholar] [CrossRef]
- Cheng, S.W.; Hu, H.M.; Shen, S.W.; Takagi, H.; Asano, M.; Tsai, Y.C. Production and characterization of keratinase of a feather-degrading Bacillus licheniformis PWD-1. Biosci. Biotechnol. Biochem. 1995, 59, 2239–2243. [Google Scholar] [CrossRef]
- Cedrola, S.M.L.; de Melo, A.C.N.; Mazotto, A.M.; Lins, U.; Zingali, R.B.; Rosado, A.S.; Peixoto, R.S.; Vermelho, A.B. Keratinases and sulfide from Bacillus subtilis SLC to recycle feather waste. World J. Microbiol. Biotechnol. 2012, 28, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Jaouadi, B.; Abdelmalek, B.; Fodil, D.; Ferradji, F.Z.; Rekik, H.; Zaraî, N.; Bejar, S. Purification and characterization of a thermostable keratinolytic serine alkaline proteinase from Streptomyces sp. strain AB1 with high stability in organic solvents. Bioresour. Technol. 2010, 101, 8361–8369. [Google Scholar] [CrossRef]
- Mabrouk, M.E.M. Feather degradation by a new keratinolytic Streptomyces sp. MS-2. World J. Microbiol. Biotechnol. 2008, 24, 2331–2338. [Google Scholar] [CrossRef]
- Bressollier, P.; Letourneau, F.; Urdaci, M.; Verneuil, B. Purification and characterization of a keratinolytic serine proteinase from Streptomyces albidoflavus. Appl. Environ. Microbiol. 1999, 65, 2570–2576. [Google Scholar] [CrossRef]
- Chitte, R.R.; Nalawade, V.K.; Dey, S. Keratinolytic activity from the broth of a feather-degrading thermophilic Streptomyces thermoviolaceus strain SD8. Lett. Appl. Microbiol. 1999, 28, 131–136. [Google Scholar] [CrossRef]
- Böckle, B.; Galunsky, B.; Muller, R. Characterization of a Keratinolytic Serine Proteinase from Streptomyces Pactum DSM 40530. Appl. Environ. Microbiol. 1995, 61, 3705–3710. [Google Scholar] [CrossRef]
- Nam, G.W.; Lee, D.W.; Lee, H.S.; Lee, N.J.; Kim, B.C.; Choe, E.A.; Hwang, J.K.; Suhartono, M.T.; Pyun, Y.R. Native-feather degradation by Fervidobacterium islandicum AW-1, a newly isolated keratinase-producing thermophilic anaerobe. Arch. Microbiol. 2002, 178, 538–547. [Google Scholar] [CrossRef]
- El-Naghy, M.A.; El-Ktatny, M.S.; Fadl-Allah, E.M.; Nazeer, W.W. Degradation of chicken feathers by Chrysosporium georgiae. Mycopathologia 1998, 143, 77–84. [Google Scholar] [CrossRef]
- Macedo, A.J.; Gava, R.; Driemeier, D.; Termignoni, C. Novel Keratinase from Bacillus subtilis S14 Exhibiting Remarkable Dehairing Capabilities. Appl. Environ. Microbiol. 2005, 71, 594–596. [Google Scholar] [CrossRef]
- Thys, R.C.S.; Brandelli, A. Purification and properties of a keratinolytic metalloprotease from Microbacterium sp. J. Appl. Microbiol. 2006, 101, 1259–1268. [Google Scholar] [CrossRef]
- Wu, W.L.; Chen, M.Y.; Tu, I.F.; Lin, Y.C.; Eswarkumar, N.; Chen, M.Y.; Ho, M.C.; Wu, S.H. The discovery of novel heat-stable keratinases from Meiothermus taiwanensis WR-220 and other extremophiles. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barman, N.C.; Zohora, F.T.; Das, K.C.; Mowla, M.G.; Banu, N.A.; Salimullah, M.; Hashem, A. Production, partial optimization and characterization of keratinase enzyme by Arthrobacter sp. NFH5 isolated from soil samples. AMB Express 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Habbeche, A.; Saoudi, B.; Jaouadi, B.; Haberra, S.; Kerouaz, B.; Boudelaa, M.; Badis, A.; Ladjama, A. Purification and biochemical characterization of a detergent-stable keratinase from a newly thermophilic actinomycete Actinomadura keratinilytica strain Cpt29 isolated from poultry compost. J. Biosci. Bioeng. 2014, 117, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Jaouadi, N.Z.; Rekik, H.; Badis, A.; Trabelsi, S.; Belhoul, M.; Yahiaoui, A.B.; Aicha, H.B.; Toumi, A.; Bejar, S.; Jaouadi, B. Biochemical and Molecular Characterization of a Serine Keratinase from Brevibacillus brevis US575 with Promising Keratin-Biodegradation and Hide-Dehairing Activities. PLoS ONE 2013, 8, e76722. [Google Scholar] [CrossRef]
- Syed, D.G.; Lee, J.C.; Li, W.J.; Kim, C.J.; Agasar, D. Production, characterization and application of keratinase from Streptomyces gulbargensis. Bioresour. Technol. 2009, 100, 1868–1871. [Google Scholar] [CrossRef]
- Mitsuiki, S.; Ichikawa, M.; Oka, T.; Sakai, M.; Moriyama, Y.; Sameshima, Y.; Goto, M.; Furukawa, K. Molecular characterization of a keratinolytic enzyme from an alkaliphilic Nocardiopsis sp. TOA-1. Enzyme Microb. Technol. 2004, 34, 482–489. [Google Scholar] [CrossRef]
- Gupta, R.; Ramnani, P. Microbial keratinases and their prospective applications: An overview. Appl. Microbiol. Biotechnol. 2006, 70, 21–33. [Google Scholar] [CrossRef]
- Daroit, D.J.; Brandelli, A. A current assessment on the production of bacterial keratinases. Crit. Rev. Biotechnol. 2014, 34, 372–384. [Google Scholar] [CrossRef]
- Inada, S.; Watanabe, K. Draft genome sequence of Meiothermus ruber H328, which degrades chicken feathers, and identification of proteases and peptidases responsible for degradation. Genome Announc. 2013, 1, 3–4. [Google Scholar] [CrossRef]
- Yong, B.; Yang, B.; Zhao, C.; Feng, H. Draft Genome Sequence of Bacillus subtilis Strain S1-4, Which Degrades Feathers Efficiently. Genome Announc. 2013, 1, 13–14. [Google Scholar] [CrossRef]
- Park, G.-S.; Hong, S.-J.; Lee, C.-H.; Khan, A.R.; Ullah, I.; Jung, B.K.; Choi, J.; Kwak, Y.; Back, C.-G.; Jung, H.-Y.; et al. Draft Genome Sequence of Chryseobacterium sp. Strain P1-3, a Keratinolytic Bacterium Isolated from Poultry Waste. Genome Announc. 2014, 2, 10–11. [Google Scholar] [CrossRef]
- Pereira, J.Q.; Ambrosini, A.; Sant’Anna, F.H.; Tadra-Sfeir, M.; Faoro, H.; Pedrosa, F.O.; Souza, E.M.; Brandelli, A.; Passaglia, L.M.P. Whole-genome shotgun sequence of the keratinolytic bacterium Lysobacter sp. A03, isolated from the Antarctic environment. Genome Announc. 2015, 3, 1–2. [Google Scholar] [CrossRef]
- Kim, E.-M.; Hwang, K.H.; Park, J.-S. Complete Genome Sequence of Chryseobacterium camelliae Dolsongi-HT1, a Green Tea Isolate with Keratinolytic Activity. Genome Announc. 2018, 6, 1–2. [Google Scholar] [CrossRef]
- Li, B.; Liu, F.; Ren, Y.; Ding, Y.; Li, Y.; Tang, X.-F.; Tang, B. Complete Genome Sequence of Thermoactinomyces vulgaris Strain CDF, a Thermophilic Bacterium Capable of Degrading Chicken Feathers. Genome Announc. 2019, 8, 1–2. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, J.; Du, G.; Chen, J. Keratin Waste Recycling Based on Microbial Degradation: Mechanisms and Prospects. ACS Sustain. Chem. Eng. 2019, 7, 9727–9736. [Google Scholar] [CrossRef]
- Li, Z.W.; Liang, S.; Ke, Y.; Deng, J.J.; Zhang, M.S.; Lu, D.L.; Li, J.Z.; Luo, X.C. The feather degradation mechanisms of a new Streptomyces sp. isolate SCUT-3. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Cumsille, A.; Undabarrena, A.; González, V.; Claverías, F.; Rojas, C.; Cámara, B. Biodiversity of actinobacteria from the South Pacific and the assessment of Streptomyces chemical diversity with metabolic profiling. Mar. Drugs 2017, 15, 286. [Google Scholar] [CrossRef]
- Undabarrena, A.; Beltrametti, F.; Claverías, F.P.; González, M.; Moore, E.R.; Seeger, M.; Cámara, B. Exploring the diversity and antimicrobial potential of marine actinobacteria from the comau fjord in Northern Patagonia, Chile. Front. Microbiol. 2016, 7, 1–16. [Google Scholar] [CrossRef]
- Claverías, F.P.; Undabarrena, A.; Gonzalez, M.; Seeger, M.; Camara, B. Culturable diversity and antimicrobial activity of Actinobacteria from marine sediments in Valparaíso bay, Chile. Front. Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- González, V.; Vargas-Straube, M.J.; Beys-da-Silva, W.O.; Santi, L.; Valencia, P.; Beltrametti, F.; Cámara, B. Enzyme Bioprospection of Marine-Derived Actinobacteria from the Chilean Coast and New Insight in the Mechanism of Keratin Degradation in Streptomyces sp. G11C. Mar. Drugs 2020, 18, 537. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Huang, Y.; Zhang, Y.Q.; Liu, Z.H. Streptomyces emeiensis sp. nov., a novel streptomycete from soil in China. Int. J. Syst. Evol. Microbiol. 2007, 57, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Hain, T.; Ward-Rainey, N.; Kroppenstedt, R.M.; Stackebrandt, E.; Rainey, F.A. Discrimination of Streptomyces albidoflavus strains based on the size and number of 16S-23S ribosomal DNA intergenic spacers. Int. J. Syst. Bacteriol. 1997, 47, 202–206. [Google Scholar] [CrossRef][Green Version]
- Jain, C.; Rodriguez-R., L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Nguyen, T.T.H.; Myrold, D.D.; Mueller, R.S. Distributions of extracellular peptidases across prokaryotic genomes reflect phylogeny and habitat. Front. Microbiol. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Page, M.J.; Di Cera, E. Evolution of peptidase diversity. J. Biol. Chem. 2008, 283, 30010–30014. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D. Bacterial calpains and the evolution of the calpain (C2) family of peptidases. Biol. Direct 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Hahsler, M.; Piekenbrock, M.; Doran, D. Dbscan: Fast density-based clustering with R. J. Stat. Softw. 2019, 91, 1–30. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Mooers, A. Effects of tree shape on the accuracy of maximum likelihood-based ancestor reconstructions. Syst. Biol. 2004, 53, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Litsios, G.; Salamin, N. Effects of phylogenetic signal on ancestral state reconstruction. Syst. Biol. 2012, 61, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Pike, R.N. Bacterial peptidases. Contrib. Microbiol. 2005, 12, 132–180. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Carter, B.R.; Feely, R.A.; Lauvset, S.K.; Olsen, A. Surface ocean pH and buffer capacity: Past, present and future. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Rao, M.B.; Tanksale, A.M.; Ghatge, M.S.; Deshpande, V.V. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 1998, 62, 597–635. [Google Scholar] [CrossRef]
- Rawlings, N.D. Peptidase specificity from the substrate cleavage collection in the MEROPS database and a tool to measure cleavage site conservation. Biochimie 2016, 122, 5–30. [Google Scholar] [CrossRef]
- Brandelli, A.; Daroit, D.J.; Riffel, A. Biochemical features of microbial keratinases and their production and applications. Appl. Microbiol. Biotechnol. 2010, 85, 1735–1750. [Google Scholar] [CrossRef]
- Williams, C.M.; Richter, C.S.; Mackenzie, J.M.; Shih, J.C.H. Isolation, Identification, and Characterization of a Feather-Degrading Bacterium. Appl. Environ. Microbiol. 1990, 56, 1509–1515. [Google Scholar] [CrossRef]
- Mitsuiki, S.; Sakai, M.; Moriyama, Y.; Goto, M.; Furukawa, K. Purification and Some Properties of a Keratinolytic Enzyme from an Alkaliphilic Nocardiopsis sp. TOA-1. Biosci. Biotechnol. Biochem. 2002, 66, 164–167. [Google Scholar] [CrossRef][Green Version]
- Riffel, A.; Lucas, F.; Heeb, P.; Brandelli, A. Characterization of a new keratinolytic bacterium that completely degrades native feather keratin. Arch. Microbiol. 2003, 179, 258–265. [Google Scholar] [CrossRef]
- Tatineni, R.; Doddapaneni, K.K.; Potumarthi, R.C.; Vellanki, R.N.; Kandathil, M.T.; Kolli, N.; Mangamoori, L.N. Purification and characterization of an alkaline keratinase from Streptomyces sp. Bioresour. Technol. 2008, 99, 1596–1602. [Google Scholar] [CrossRef]
- Qiu, J.; Wilkens, C.; Barrett, K.; Meyer, A.S. Microbial enzymes catalyzing keratin degradation: Classification, structure, function. Biotechnol. Adv. 2020, 44, 1–22. [Google Scholar] [CrossRef]
- Gerlt, J.A. Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. [Google Scholar] [CrossRef]
- Yamamura, S.; Morita, Y.; Hasan, Q.; Yokoyama, K.; Tamiya, E. Keratin degradation: A cooperative action of two enzymes from Stenotrophomonas sp. Biochem. Biophys. Res. Commun. 2002, 294, 1138–1143. [Google Scholar] [CrossRef]
- Huang, Y.; Łężyk, M.; Herbst, F.A.; Busk, P.K.; Lange, L. Novel keratinolytic enzymes, discovered from a talented and efficient bacterial keratin degrader. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Wang, S.L.; Hsu, W.T.; Liang, T.W.; Yen, Y.H.; Wang, C.L. Purification and characterization of three novel keratinolytic metalloproteases produced by Chryseobacterium indologenes TKU014 in a shrimp shell powder medium. Bioresour. Technol. 2008, 99, 5679–5686. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, T.; Song, N.; Li, Q.; Wang, Z.; Zhang, X.; Lu, X.; Fang, J.; Chen, J. Purification and characterization of four key enzymes from a feather-degrading Bacillus subtilis from the gut of tarantula Chilobrachys guangxiensis. Int. Biodeterior. Biodegrad. 2014, 96, 26–32. [Google Scholar] [CrossRef]
- Monod, M.; Capoccia, S.; Léchenne, B.; Zaugg, C.; Holdom, M.; Jousson, O. Secreted proteases from pathogenic fungi. Int. J. Med. Microbiol. 2002, 292, 405–419. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files—Version 1.33. 2011. Available online: https://github.com/najoshi/sickle (accessed on 8 June 2020).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Nouioui, I.; Carro, L.; García-López, M.; Meier-Kolthoff, J.P.; Woyke, T.; Kyrpides, N.C.; Pukall, R.; Klenk, H.P.; Goodfellow, M.; Göker, M. Genome-based taxonomic classification of the phylum Actinobacteria. Front. Microbiol. 2018, 9, 1–119. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Barrett, A.J.; McDonald, J.K. Nomenclature: Protease, proteinase and peptidase. Biochem. J. 1986, 237, 935. [Google Scholar] [CrossRef]
- HMMER-Based MEROPS Web-Server. Available online: https://www.ebi.ac.uk/merops/submit_searches.shtml. (accessed on 14 April 2020).
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Larsson, J. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R package version 6.1.0. 2020. Available online: https://cran.r-project.org/package=eulerr (accessed on 18 March 2021).
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy 2008), Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]
- Atkinson, H.J.; Morris, J.H.; Ferrin, T.E.; Babbitt, P.C. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE 2009, 4, e4345. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, P10008. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Hu, Y. Efficient and High Quality Force-Directed Graph. Math. J. 2005, 10, 37–71. [Google Scholar]
- Fruchterman, T.M.J.; Reingold, E.M. Graph drawing by force-directed placement. Softw. Pract. Exp. 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Sanchez, G.; Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of Protein Subcellular Localization. PROTEINS Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Cenk Sahinalp, S.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Van Der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2625. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Bollback, J.P. SIMMAP: Stochastic character mapping of discrete traits on phylogenies. BMC Bioinform. 2006, 7, 1–7. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Data | Streptomyces sp. G11C | Streptomyces sp. CHD11 | Streptomyces sp. Vc74B-19 |
|---|---|---|---|
| Total number of paired reads | 7,253,694 | 9,168,658 | 8,356,602 |
| Total raw reads bases (Gbp) | 187.73 | 232.83 | 213.71 |
| Assembly size (bp) (≥500 bp contigs) | 6,873,298 | 7,469,836 | 7,625,040 |
| Number of contigs (≥500 bp) | 167 | 91 | 197 |
| Contigs (N50) (kb) | 204,147 | 98,291 | 76,195 |
| G + C content (%) | 73.15 | 71.67 | 72.33 |
| Predicted CDS | 5953 | 6661 | 6835 |
| CheckM completeness (%) | 99.53 | 100 | 99.24 |
| L50 statistics | 24 | 12 | 31 |
| Number of: | Streptomyces sp. G11C | Streptomyces sp. CHD11 | Streptomyces sp. Vc74B-19 |
|---|---|---|---|
| ORFs encoding putative proteases | 179 | 198 | 207 |
| Putative proteases classified into a peptidase family | 151 | 166 | 177 |
| p-orthogroups in each strain | 113 | 132 | 137 |
| “unassigned p-orthogroup” peptidases | 17 | 8 | 3 |
| Number of Peptidases Present in: | |||
|---|---|---|---|
| Category | t-SNE Group 0 | t-SNE Group 1 | t-SNE Group 2 |
| Functional keratinase | 40 | 5 | 10 |
| Keratinase-linked sequence—strain G11C | 1 | 9 | 1 |
| Keratinase-linked sequence—strain CHD11 | 4 | 8 | 1 |
| Keratinase-linked sequence—strain Vc74B-19 | 4 | 11 | 1 |
| three-strain category—strain G11C | 5 | 13 | 6 |
| three-strain category—strain CHD11 | 4 | 18 | 5 |
| three-strain category—strain Vc74B-19 | 8 | 24 | 7 |
| Putative non-keratinase | 2 | 3 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencia, R.; González, V.; Undabarrena, A.; Zamora-Leiva, L.; Ugalde, J.A.; Cámara, B. An Integrative Bioinformatic Analysis for Keratinase Detection in Marine-Derived Streptomyces. Mar. Drugs 2021, 19, 286. https://doi.org/10.3390/md19060286
Valencia R, González V, Undabarrena A, Zamora-Leiva L, Ugalde JA, Cámara B. An Integrative Bioinformatic Analysis for Keratinase Detection in Marine-Derived Streptomyces. Marine Drugs. 2021; 19(6):286. https://doi.org/10.3390/md19060286
Chicago/Turabian StyleValencia, Ricardo, Valentina González, Agustina Undabarrena, Leonardo Zamora-Leiva, Juan A. Ugalde, and Beatriz Cámara. 2021. "An Integrative Bioinformatic Analysis for Keratinase Detection in Marine-Derived Streptomyces" Marine Drugs 19, no. 6: 286. https://doi.org/10.3390/md19060286
APA StyleValencia, R., González, V., Undabarrena, A., Zamora-Leiva, L., Ugalde, J. A., & Cámara, B. (2021). An Integrative Bioinformatic Analysis for Keratinase Detection in Marine-Derived Streptomyces. Marine Drugs, 19(6), 286. https://doi.org/10.3390/md19060286