Abstract
Sponges form symbiotic relationships with diverse and abundant microbial communities. Cyanobacteria are among the most important members of the microbial communities that are associated with sponges. Here, we performed a genus-wide comparative genomic analysis of the newly described marine benthic cyanobacterial genus Leptothoe (Synechococcales). We obtained draft genomes from Le. kymatousa TAU-MAC 1615 and Le. spongobia TAU-MAC 1115, isolated from marine sponges. We identified five additional Leptothoe genomes, host-associated or free-living, using a phylogenomic approach, and the comparison of all genomes showed that the sponge-associated strains display features of a symbiotic lifestyle. Le. kymatousa and Le. spongobia have undergone genome reduction; they harbored considerably fewer genes encoding for (i) cofactors, vitamins, prosthetic groups, pigments, proteins, and amino acid biosynthesis; (ii) DNA repair; (iii) antioxidant enzymes; and (iv) biosynthesis of capsular and extracellular polysaccharides. They have also lost several genes related to chemotaxis and motility. Eukaryotic-like proteins, such as ankyrin repeats, playing important roles in sponge-symbiont interactions, were identified in sponge-associated Leptothoe genomes. The sponge-associated Leptothoe stains harbored biosynthetic gene clusters encoding novel natural products despite genome reduction. Comparisons of the biosynthetic capacities of Leptothoe with chemically rich cyanobacteria revealed that Leptothoe is another promising marine cyanobacterium for the biosynthesis of novel natural products.
1. Introduction
Sponges host abundant and remarkable diverse microbial communities [1] that exhibit biological complexity similar to the human microbiome [2,3]. Cyanobacteria are an ancient lineage of photosynthetic prokaryotes demonstrating ecological key roles (e.g., oxygen production, nitrogen fixation, carbon flux) in a broad range of habitats, including sponges [4,5], with which they are often found in symbiosis (cyanobionts). Several approaches including whole-genome sequencing of symbiotic microbes and metagenomic binning have provided insights into the functional potential of the symbionts [6,7]. For instance, it has been shown that across sponge-associated bacteria taxa there are pathways involved in carbon fixation, B-vitamin synthesis, taurine metabolism, sulfite oxidation, and most steps of nitrogen metabolism [7,8]. Candidatus Synechococcus spongiarum, a widespread (yet uncultivated) sponge symbiont, has specific adaptations to life inside sponges [9]. This obligate cyanobiont showed extreme genome reduction [9], similarly to other bacterial sponge symbionts such as Candidatus Endohaliclona renieramycinifaciens [10]. Genome reduction is the major genomic feature of bacterial symbionts [11] and is thought to be a process that reduces the cost of genome replication [12].
Cyanobacteria are a prolific source of natural products with complex chemical structures and interesting bioactivities [13]. Advances in genomics have greatly expanded our knowledge and understanding of cyanobacterial natural product biosynthesis [13] with known natural products linked to biosynthetic gene clusters and new natural products discovered through genome mining (e.g., [14,15,16,17]). However, the majority of natural products from cyanobacteria are described from a relative limited number of genera [13]. This phenomenon is attributed to problems with cyanobacterial taxonomy that obfuscates the true distribution of natural products in marine cyanobacteria.
Marine sponges are also prolific sources of natural products of great interest for drug development, contributing to nearly 30% (more than 4850 compounds) of all marine natural products discovered [18]. Sponge-associated bacteria are widely thought to be responsible for sponge natural product diversity [2,3,19], with cyanobacteria being among the major producers [20].
Sponge-associated members of the newly described marine cyanobacterium Leptothoe [21,22] were found to be highly cytotoxic against human breast, skin, and colon cancer epithelial cells [23]. Extracts of Le. kymatousa TAU-MAC 1615 were found to have antibacterial activity [23]. In this study, we sequenced two draft Leptothoe genomes, Le. kymatousa TAU-MAC 1615 and Le. spongobia TAU-MAC 1115, previously isolated form the marine sponges Chondrilla nucula and Acanthella acuta, respectively [21,24]. We performed comparative genomic analyses of sponge-associated, other host-associated and free-living members of Leptothoe genus, and other marine cyanobacterial genera, aiming to (a) identify symbiosis factors and (b) gain insight into their natural product biosynthetic potential.
2. Results and Discussion
Two new sponge-associated cyanobacterial draft genomes belonging to the newly described Leptothoe genus were recovered from the isolates TAU-MAC 1615 and 1115 (Figure S1, Table 1). To date, most microbial isolates derived from sponges have been mainly affiliated with Proteobacteria [7,25,26]. A limited number of cyanobacterial strains have been isolated from sponges [24]. Indeed, only one genome assembly from a sponge-associated cyanobacterial strain is available in public databases for a Myxosarcina-like cyanobacterium isolated from Terpios hoshinota [27]. Recently, a considerable number of novel genera and species of cyanobacteria were found to be associated with Aegean Sea sponges [21,22].

Table 1.
Genome properties and quality metrics of Leptothoe strains used for comparative genomic analyses. Subsystem statistics were obtained using the RAST server [28] and SEED tool [29].
The size of the assembled sponge-associated Leptothoe genomes ranged between 4.06 Mb for Le. kymatousa TAU-MAC 1615 and 5.24 Mb for Le. spongobia TAU-MAC 1115, with G+C contents of 50.5% and 47.3%, respectively (Figure S1, Table 1). Table 1 summarizes the basic genome features of sponge-associated Leptothoe genomes, Leptothoe genomes associated with other macro-organisms (isolated from turfs), and free-living Leptothoe genomes used for comparative genomic analyses in this study after verification of genome-wide relatedness using phylogenomic approaches (Figure 1). The draft assemblies of sponge-associated strains showed the smallest genome sizes (Table 1). Although the genome coverage of sponge-associated strains was high (approximately ×450 and ×90 for TAU-MAC 1615 and TAU-MAC 1115, respectively), the genome completeness estimated by the marker sets used by CheckM (encode essential functions) was substantially complete (≈70%) (Table 1). The GC% contents of the strains were quite similar, varying from 47.3% (TAU-MAC 1115, CCMR0081) to 50.5% (TAU-MAC 1615). Concerning the number of scaffolds, the SIO3F4 metagenome showed the highest number (1508) and the lowest N50 (7412). The number of coding sequences (CDSs) of sponge-associated strains was considerably lower (3638–4790) than the rest of the Leptothoe strains, while the amount of CDSs categorized in RAST subsystems (13–17%) was quite similar (Table 1). The remaining 84–87% of CDSs were not classified in any subsystem.
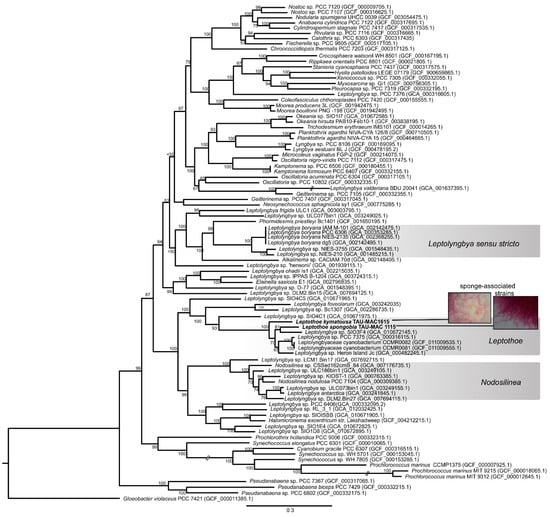
Figure 1.
Maximum likelihood phylogenomic tree based on 120 conserved proteins in cyanobacterial genomes. The sponge-associated Leptothoe strains sequenced in the present study are shown in bold. Accession numbers of the sequences are presented in parentheses.
The phylogenomic reconstruction based on 120 bacterial single-copy conserved marker genes indicated that our two sponge-associated strains were placed inside Leptothoe clade and grouped together with 5 of the 35 public cyanobacterial genomes identified as Leptolyngbya or Leptolyngbyaceae (Figure 1). Leptothoe is a thin marine filamentous cyanobacterium with benthic lifestyle being epilithic, epizoic, or epiphytic, recently separated from the polyphyletic filamentous genus Leptolynbya by a taxonomic re-evaluation [21].
The remaining 30 cyanobacteria, identified as Leptolyngbya or Leptolyngbyaceae, were grouped in different clades spread across a variety of cyanobacteria orders (Figure 1). This analysis further supported the evolutionary divergence of Leptothoe from Leptolyngbya sensu stricto and other thin filamentous genera separated from Leptolyngbya such as Nodosilinea and Elainella (Figure 1). The analysis also supported the evolutionary distinction of the two sponge-associated strains, TAU-MAC 1115 and TAU-MAC 1615, which belong to different species Le. spongobia and Le. kymatousa (Figure 1). The free-living strains PCC 7375 and Heron Island J were placed separately within a subclade along with two strains (CCMR0081 and CCMR0081) isolated from turf samples growing over corals, while the metagenome-assembled genome SIO3F4 isolated from turfs assembles in Panama was placed in the same subclade with Le. spongobia TAU-MAC 1115 (Figure 1). Further, our phylogenomic analysis showed that the members of genus Leptothoe (seven in total) were not grouped on the basis of the isolation source (host-associated or free-living), indicating the lack of host-specific clustering. Other marine bacterial genera, including free-living and host-associated members such as Pseudovibrio, have shown a lack of host-specific clustering [30]. However, the sponge-associated Le. kymatousa TAU-MAC 1615 was placed separately in the phylogenomic tree, likely suggesting its independent evolution from other host-associated strains of the genus. These results might indicate distinct patterns of evolution among members of genus Leptothoe.
2.1. Genomic Hallmarks of a Symbiotic Lifestyle
2.1.1. Genome Reduction of Sponge-Associate Strains
Leptothoe kymatousa TAU-MAC 1615 and Leptothoe spongobia TAU-MAC 1115 are not obligate sponge symbionts and can sustain growth in pure cultures. However, these two strains showed considerable smaller genome size (Figure 2a). This pattern of genome reduction was also observed for the obligate sponge cyanobiont Candidatus Synechococcus spongiarum [9,31]. Genome reduction is widely observed mainly in obligate bacterial symbionts [11], although signs of genome reduction have been identified in facultative symbionts too [32]. Both cyanobionts are slow growing in culture conditions, and thus they could be considered as facultative symbionts that are generally not essential for the host’s survival, although they may contribute to host fitness. Recently evolved symbionts, which have undergone genome reduction, are generally characterized by a proliferation of pseudogenes and mobile elements [11]. The two Leptothoe sponge-associated genomes reported here showed lower numbers of those traits compared to the other members of the genus (Table 1); on the basis of these data, we hypothesized that sponges did not recently acquire the Leptothoe symbionts.
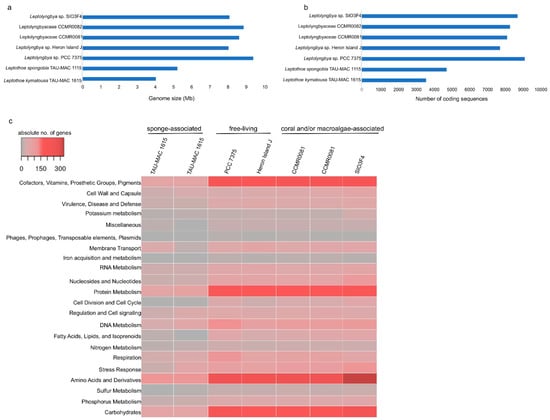
Figure 2.
Comparison of genome size (a), number of coding sequences (b), and number of genes in each RAST subsystem [28] (c) of the analyzed Leptothoe genomes.
The number of coding sequences of the sponge-associated strains were also half compared to the remaining strains (Figure 2b). An overview of different subsystems obtained using the RAST and SEED annotation showed that the sponge-associated Leptothoe strains harbored considerably fewer genes related to several essential functions (Figure 2c, Table S1). Remarkably, Le. kymatousa TAU-MAC 1615 and Le. spongobia TAU-MAC 1115 have undergone extreme reduction of the number of genes encoding for cofactors, vitamins, prosthetic groups, pigments, proteins, and amino acid biosynthesis (Figure 2c, Table S1). Sponge-associated strains may be dependent on co-occurring microbes for lost metabolic capacities [33]. A complete loss of genes involved in DNA recombination and fewer genes involved in DNA repair were observed in their genomes, while they retained approximately the same number of genes for DNA replication as the rest of the Leptothoe genomes. The sponge-associated strains have lost several genes involved in potassium, sulfur, and phosphorus metabolism (Table S1). They were also found to have fewer genes related to stress response, mainly genes coding for antioxidant enzymes, as well as fewer genes responsible for the biosynthesis of capsular polysaccharide (CPS) and extracellular polysaccharides (Table S1). Our Leptothoe genomes were characterized by a complete or near complete lack of chemotaxis and motility traits, which are among the most depleted functions in sponge-associated bacteria genomes [34].
The number of COGs (clusters of orthologous groups of proteins) per genome ranged from 6594 in free-living PCC 7375 strain to 2925 in the sponge-associated TAU-MAC 1615 strain. Leptothoe strains shared > 80% average amino acid identity (AAI) (Figure S2), while only 1602 COG entries that account for approximately half (for sponge-associated strains) or even lower (≈25% for the rest of the strains) of the total number of COG entries were common in all genomes (Figure 3), likely suggesting specific adaptations for different lifestyles and for different symbiont types. Genome streamlining process forces adaptations of cyanobacterial genomes to specific niches that are also reflected in their different functional capacities [12]. Previously, sponge-associated and free-living Synechococcus genomes have also been found to share half of their total number of COGs, suggesting variability and specific adaptations of each member of the genus [9]. Comparisons based on COG categories among the sponge-associated, coral, and/or macroalgae-associated and free-living Leptothoe revealed a relative lower abundance of genes belonging to the different functional categories in the sponge-associated strains. This analysis identified an overrepresentation of functional categories ‘J’ (translation, ribosomal structure, and biogenesis), ‘L’ (replication, recombination, and repair), ‘O’ (posttranslational modification, protein turnover, chaperones), and ‘P’ (inorganic ion transport and metabolism) and was observed in the genomes of the coral and/or macroalgae-associated strains (Figure S3). A uniform distribution of genes belonging to COG functional categories between the two sponge-associated strains was detected.
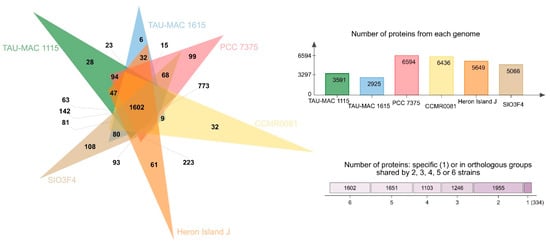
Figure 3.
Analysis of homologous protein clusters in the genomes of Le. kymatousa TAU-MAC 1615, Le. spongobia TAU-MAC 1115, Leptolyngbyaceae sp. CCMR0081, Leptolyngbyaceae sp. CCMR0082, Leptolyngbya sp. SIO3F4 Leptolyngbya sp. PCC 7375, and Leptolyngbya sp. Hero Island J.
The sponge-associated strains possess biosynthetic gene clusters (BGCs) encoding for natural products despite undergoing genome reduction (Figure 4a,b). It has been proposed that maintenance of such clusters sustains the symbiotic interaction [35]. Natural product BGCs were previously detected in another cyanobacterial sponge symbiont, Hormoscilla spongilae, suggesting that these biosynthetic capacities to produce metabolically expensive natural products may contribute to host fitness [36].
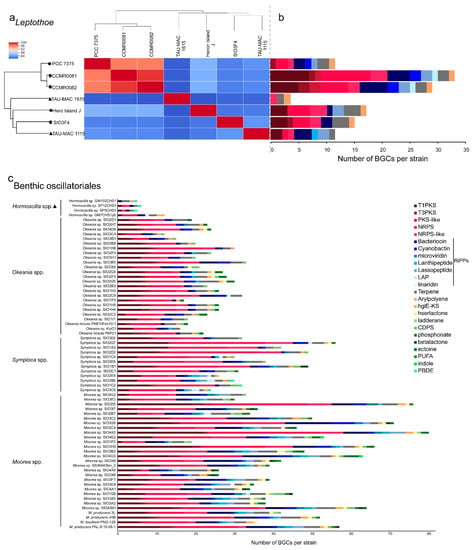
Figure 4.
Average nucleotide identity heatmap (a) and composition of BGCs identified in Leptothoe genomes (b) and composition of BGCs identified in marine benthic filamentous Oscillatoriales (c). The absolute number of BGCs per strain assigned to each BGC class is shown. Triangle, sponge associated strains; polygon, coral and/or macroalgae-associated strains; circle, free-living strains.
2.1.2. Eukaryotic-Like Protein (ELP)-Encoding Genes
Genes encoding eukaryotic-like proteins (ELPs) such as ankyrin-like domains (ANKs), tetratricopeptide repeats (TPRs), Leucine-rich repeat (LRR) protein, and pyrroloquinoline quinone (PQQ) were detected in the two sponge-associated Leptothoe strains (Table 2); ELPs are often detected in facultative or obligate symbionts and play a key role in the modulation of cellular protein–protein interactions [37,38]. In particular, abundance of ANKs seems to be a major genomic feature of sponge symbionts [6,26] as they have been thought to be involved in preventing phagocytosis by the sponge host. Indeed, the role of ANKs in modulating the amoebal phagocytosis in sponge symbionts was experimentally validated [39]. Genome analyses of sponge-associated Alphaproteobacteria [7,26,30], Deltaproteobacteria [40], and Poribacteria [41] have revealed the presence of ELPs, as well as the particular abundance of ANKs. The obligate sponge symbionts Hormoscilla spongeliae and Candidatus Synechococcus spongiarum had a great number of ELP repeats, while different free-living cyanobacteria taxa such as Nodosilinea, Leptolyngbya, Synechococcus, Prochlorococcus, and Cyanobium gracile almost lacked ANK domains, the typical genomic signature of sponge symbionts (Table 2). We also detected the different ELP types in varying proportions in other host-associated and free-living members of the genus Leptothoe. Similarly, ELPs have been previously detected in host-associated and free-living members of Alphaproteobacteria, likely suggesting the ability of strains to infect a different range of marine hosts and attach to various marine niches [26,30].
Table 2.
Eukaryotic-like protein repeats across symbiotic and free-living cyanobacterial genomes.
2.2. Biosynthetic Potential of Leptothoe
The genomic repertoire for secondary metabolism of seven Leptothoe genomes was predicted using antiSMASH (Figure 4a,b). Leptothoe genomes were found to harbor a considerable number of BGCs (121), the majority of which have no known end product. BGCs with unknown end products are present in almost all cyanobacterial genomes [42], and on the other hand, there are still natural products for which a biosynthetic origin is unknown [43]. The vast majority of BGCs in Leptothoe genomes were predicted to encode non-ribosomal peptide synthetases (NRPS) (24 BGCs), followed by type I polyketide synthase (T1PKS) (20 BGCs) and bacteriocin (17 BGCs) (Figure 4, Table S2). Previously, genome-mining efforts have revealed that a major fraction of cyanobacterial natural products is produced using NRPS or PKS enzymes systems [43,44]. Bacteriocin BGCs, which are widespread in cyanobacterial genomes [45], were detected in almost all Leptothoe genomes (except for Le. kymatousa TAU-MAC 1615). Bacteriocins have been mainly reported to exhibit antimicrobial activity [46], but are also promising as antivirals, plant protection agents, and anticancer agents [47]. Further, it is suggested that bacteriocins may be involved in shaping bacterial communities through inter- and intra-specific interactions [47]. In addition, lassopeptide and terpene synthase BGCs were detected with high relative abundance in almost all the Leptothoe genomes, while cyanobactin and arylpolyene BGCs were rarely found in some of the genomes. Terpene BGCs, reported in a wide variety of bacteria including cyanobacteria [48], were also present in all Leptothoe genomes. The considerably similar Leptothoe strains CCMR0081 and CCMR0081 (96% average nucleotide identity) associated with corals and macroalgae (isolated from turfs) showed the highest number of natural product BGCs (Figure 4a,b, Table S2). In contrast, the two sponge-associated Leptothoe species (sharing ≈84% average nucleotide identity) showed the lowest number of natural product BGCs. Interestingly Le. spongobia strain harbored a BGC encoding for a lanthipeptide (Figure 4b). Lanthipeptides are ribosomally synthesized and post-translationally modified peptides (RiPPs) that display a wide variety of biological activities [49], while their detection and isolation are restricted to bacteria [43]. Lanthipeptide BGCs are particularly found in the genomes of many genera of Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Cyanobacteria [50].
We assigned producers of known natural products to the Leptothoe lineage by combining 16S rRNA phylogenetic analysis with the “Comprehensive database of secondary metabolites from cyanobacteria ‘CyanoMetDB’” [51]. We searched the database for entries attributed to Leptolyngbya, Pseudanabaena (Pseudanabaena persicina = Leptolyngbya ectocarpi), Phormidium (Phormidium ectocarpi = Leptolyngbya ectocarpi), or thin filamentous strains of pinkish color, and where a sequence was available, it was included in our phylogenetic analysis (Table S3, Figure 5). This analysis demonstrated that two strains previously assigned to Leptolyngbya, Leptolyngbya ectocarpi SAG 60.90, and Leptolyngbya sp. RS03, reported to produce compounds such as hierridin B, grassypeptolides D and E, a lyngbyastatin analogue, and dolastatin 12, belong to the Leptothoe genus (Table S3, Figure 5). However, the natural product BGCs involved in the biosynthesis of the abovementioned compounds have not been studied as the genomes of these Leptothoe strains have not been sequenced yet. Our phylogenetic analysis revealed that Leptothoe was closely affiliated with three other marine benthic cyanobacteria, Salileptolyngbya and two strains with unknown taxonomic status (Cyanobacterium csf1 and Filamentous cyanobacterium FLK9); csf1 was found to produce two new cyclic depsipeptides, companeramides A and B (Figure 5; CyanoMetDB). Further, in our analysis, other genera of marine origin with benthic lifestyle and often with a reddish to pinkish thallus color, known for the production of natural products such as linear and cyclic peptides, linear and cyclic non-peptides, and linear and cyclic depsipeptides, were extracted from the CyanoMetDB database. These chemically rich genera—Moorea, Caldora, Symploca, Okeania, and Hormoscilla—were placed in separate clades inside Oscillatoriales and were found to be distantly related to Leptothoe (Figure 5).
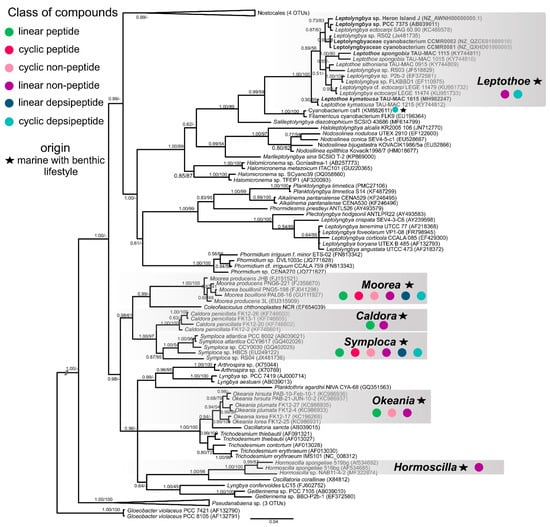
Figure 5.
Phylogenetic relationships of Leptothoe strains based on the 16S rRNA gene sequence, in relationship to representative strains of other marine filamentous cyanobacteria with benthic lifestyle, with Gloeobacter violaceus as outgroup. The tree was constructed with the Bayesian inference (ΒΙ) method and the maximum-likelihood (ML) method; BI topology is demonstrated. Support values are indicated as posterior probability for Bayesian inference and bootstrap support for maximum likelihood analysis. The bar represents 0.04 nucleotide substitutions per site.
Most of the natural products from marine cyanobacteria have been isolated from Moorea [52], which occur in high densities in marine environments of tropical and sub-tropical regions, making the harvest of biomass easily accessible [52]. Similarly, Symploca, Caldora, and Okeania form large populations attached to hard substrates in marine habitats [53,54] and yield a great number of natural products. Novel compounds with strong anticancer properties such as apratoxins, grassypeptolides, wewakazole B, odoamide, and caldoramide are isolated from these marine genera [55,56,57,58,59]. Interestingly, cyclic depsipeptides are the main peptides with cytotoxic effects isolated from marine cyanobacteria, including 76 compounds [60]. Genome-mining analysis conducted in the present study for >70 marine cyanobacteria also highlighted the high metabolic potential of the well-studied Oscillatoriales; numerous BGCs were identified in their genomes (Figure 4c). On the other hand, other marine filamentous cyanobacteria with smaller trichomes and a slower growth rate, such as Leptolyngbya-like or Pseudanabaena-like, have been overlooked [52], as well as the members of Leptothoe genus according to our analysis. Herein, we revealed another promising benthic marine cyanobacterium for novel natural products biosynthesis, Leptothoe, that warrants further exploration.
3. Materials and Methods
3.1. Sponge-Associated Strains and Growth Conditions
Leptothoe kymatousa TAU-MAC 1615 and Le. spongobia TAU-MAC 1115 were previously isolated from the marine sponges Chondrilla nucula and Acanthella acuta, accordingly, from a rocky sublittoral zone of the North Aegean Sea [21,24]. The strains were purified, and an axenic culture was obtained only for Le. kymatousa TAU-MAC 1615 due to difficulties in producing pure cultures stemming from tightly associated heterotrophic bacteria. Axenic and mono-clonal cultures were grown in MN medium [61] for 20–25 days at 20–25 °C, at a photo irradiance of 8–15 μmol photons m−2 s−1. These strains are maintained in the Microalgae and Cyanobacteria Collection (TAU-MAC) of the Aristotle University of Thessaloniki [62].
3.2. Total Genomic DNA Extraction
The genomic DNA of Leptothoe kymatousa TAU-MAC 1615 was extracted using a DNA extraction kit (E.Z.N.A.® SP Plant DNA Mini Kit Protocol—Fresh/Frozen Samples, Omega Bio-Tek, Norcross, GA, USA). We harvested 50 mL cultures by centrifugation at 7000× g for 10 min, and the pellets were transferred to microcentrifuge tubes. A total of 200 μL of glass beads (two different sizes: 425 to 600 μm and 710 to 1180 μm; Sigma-Aldrich, St. Louis, MO, USA) and SP buffer were added, and the cells were disrupted mechanically with a FastPrep-24 homogenizer (MP Biomedicals, Irvine, CA, USA) at 6.5 m/s for 30 s (2 cycles). The sample of lysed cells was extracted as described in the manufacturers’ protocol.
A total of 50 mL cultures of Le. spongobia TAU-MAC 1115 were harvested by centrifugation at 7000× g for 10 min, washed twice with washing buffer (50 mM Tris-HCl, 100 mM EDTA, 100 mM NaCl), and transferred to microcentrifuge tubes. After centrifugation (at 7000× g for 4 min), the supernatant was discarded, glass beads (two different sizes: 425 to 600 μm and 710 to 1180 μm; Sigma-Aldrich, St. Louis, MO, USA) were added, and the cells were frozen at −80 °C. The sample was thawed at 64 °C and 800 μL of GOS buffer (100 mM TrisHCl (pH 8), 1.5% SDS, 10 mM EDTA, 1% deoxycholate, 1% Igepal-CA630, 5 mM thiourea, 10 mM dithiothreitol) [63] was added. Disruption of the cells was performed using FastPrep at 5 m s−1 for 30 s (2 cycles). The rest of the extraction procedure was performed as previously described in detail [63].
The purity, concentration, and quality of the DNA were determined using a Nanodrop ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE USA), gel electrophoresis, and an Agilent TapeStation (Agilent Technologies, Lexington, MA, USA).
3.3. Genome Sequencing and Assembly of Sponge-Associated Cyanobacteria
High-molecular-weight DNA was subjected to library construction (Illumina TruSeq PCR-free 150 bp) and sequenced by the Illumina HiSeq 2500 platform, with a paired-end 100-cycle run (Macrogen Europe, Amsterdam, the Netherlands). The quality of the raw data was initially assessed using FastQC v0.10.1 [64]. Prinseq [65] was used to perform quality filtering, and genome assembly was performed with SPAdes 3.5.0 [66], followed by scaffolding and gap-closing performed with Platanus 1.2.1 [67]. Scaffolds from culture contaminants were identified by Kraken 1.0 [68] and removed using ZEUSS 1.0.2 [69]. Genome assembly statistics were obtained using Assemblathon 2 [70]. Completeness and contamination of the genomes were accessed using CheckM v1.1.3 [71].
3.4. Phylogenomic and Phylogenetic Analysis
An alignment of 120 bacterial single-copy conserved marker genes was generated with the Genome Taxonomy Database GTDB-Tk [72] from 90 cyanobacterial genomes, including the two newly sequenced sponge-associated Leptothoe genomes as well as 35 genomes registered in GenBank as Leptolyngbya or Leptolyngbyaceae and representative taxa of Nostocales, Oscillatoriales, and Chroococcales. A maximum-likelihood phylogenomic tree was constructed by RAxML [73] that was based on the nucleotide substitution model LG +I +G assigned by a BIC calculation in ProtTest [74], with 1000 rapid bootstrap searches.
For 16S rRNA phylogenetic analysis, a dataset consisting of gene sequences belonging to Leptothoe genus (>94.5% sequence similarity via BLASTn searches) along with sequences of closely affiliated genera (such as Salileptolyngbya, Nodosilinea, Halomicronema), as well as sequences of other filamentous cyanobacteria was generated. Multiple sequence alignment was performed in MEGA v. 7.0 [75] using ClustalW [76]. The phylogenetic tree was constructed using maximum likelihood (ML) and Bayesian inference (BI). Two 16S rRNA gene sequences of the cyanobacterium Gloeobacter violaceus were used as outgroups (GenBank acc. no. AF132790, AF132791). The GTR+I+G model was determined by a BIC calculation in jModelTest 0.1.1 [77] as the most appropriate. The ML analysis was performed in MEGA v. 7 [75]. Bootstrap resampling was performed on 1000 replicates. Bayesian analysis was conducted using MrBayes 3.2.6 [78]. Four Metropolis-coupled MCMC chains (three heated chains and one cold) were run for 10,000,000 generations, the first 2,500,000 generations were discarded as burn-in, and the following datasets were sampled every 1000th generation. Phylogenomic and phylogenetic tree were visualized using the FigTree (V1.4.3) software (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 12 March 2021).
3.5. Annotation and Comparative Analyses of Genomes
Open reading frames (ORFs) prediction and annotation were performed using the draft genomes of the two sponge-associated Leptothoe strains in the RAST (Rapid Annotation using Subsystem Technology) prokaryotic genome annotation server (version 2.0) [28] with standard procedures. For comparative analysis, five genomes of strains belonging to genus Leptothoe were identified in NCBI’s GenBank [79] and selected. Prior to the comparative genomic analyses, all genome datasets were re-annotated using RAST [28] and PROKKA [80]. Subsystems annotation of all seven genomes was performed with the RAST server [28] and SEED tool [29]. CDSs (predicted using RAST) of all seven genomes were subjected to annotation on the basis of clusters of orthologous groups (COGs) of proteins using the on-line server WebMGA (e-value = 0.001) [81]. Pseudogenes were calculated using NCBI’s annotation pipeline. The different classes of mobile elements were analyzed separately. PHASTER [82] was used for phage detection, TransposonPSI (http://transposonpsi.sourceforge.net/, accessed on 12 March 2021) for transposon identification, and ISEScan [83] for identification of insertion sequence elements. In order to detect eukaryotic-like proteins (ELPs) such as ankyrin repeats (ANKs), tetratricopeptide repeat (TPRs), leucine-rich repeats, WD40 proteins, and pyrroloquinoline quinone (PQQ), we searched the annotation files manually using the key words ‘repeats’, ‘Ankyrin’, ‘Tetratricopeptide’, ‘leucine’, and ‘PQQ’ (similar to Karimi et al. [7,34]). Heatmaps for average nucleotide and amino acid identities were estimated using the program GET_HOMOLOGUES [84]. All seven genomes were searched for in terms of the presence of natural product biosynthetic gene clusters (BGCs) using antiSMASH 5.1.1 [85], which was done in order to gain further insight to their metabolic potential.
3.6. Data Availability
The Leptothoe kymatousa TAU-MAC 1615 Whole Genome project was deposited at DDBJ/ENA/GenBank under the accession number JADOER000000000. The Leptothoe spongobia TAU-MAC 1115 Whole Genome Shotgun project was deposited at DDBJ/ENA/GenBank under the accession number JADOES000000000.
4. Conclusions
Here, we report the first two draft genomes of sponge-associated filamentous Synechococcales. Our comparative genomic analyses revealed symbiosis signatures of sponge-associated Leptothoe such as reduction of their gene content, functional dissimilarities to other host-associated and free-living members of Leptothoe, and presence of ELP repeats. Moreover, genome-mining analysis revealed the unique biosynthetic potential of Leptothoe with more than 100 natural product BGCs, emerging as an unexplored source of potent marine natural products. Additional studies are needed to identify and characterize these produced compounds. Future research to address the actual functioning of sponge-associated cyanobacteria should include metagenomics, metabolomics, and metatranscriptomics.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/md19060298/s1, Table S1. Number of genes in subsystems (obtained by RAST server and SEED tool) per Leptothoe strain. Table S2. Biosynthetic gene clusters per Leptothoe genome (antiSMASH results). Table S3. Compounds produced by Leptothoe cyanobacteria extracted from the CyanoMetDB (Jones et al., 2020). Figure S1. Circular view of the genomes of two sponge-associated Leptothoe species, generated with CGview (Stothard and Wishart, 2005). Circles from interior to exterior represent GC content and GC skew. Blue circles denote the coding sequences on forward and reverse strands. Figure S2. Average amino acid identity heatmap of Leptothoe genomes. Figure S3. Distribution of the categories of functional genes present in seven Leptothoe strains on the basis of the cluster of orthologous groups (COGs) of proteins. The alphabetic code for the COG categories is as follows: C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms.
Author Contributions
Conceptualization, D.K. and S.G.; methodology, D.K., R.V.P., and D.P.F.; validation, D.K., D.P.F., and S.G.; formal analysis, D.K. and R.V.P.; investigation, D.K.; resources, S.G., D.P.F., and K.S.; data curation, D.K.; writing—original draft preparation, D.K.; writing—review and editing, all authors.; visualization, D.K.; supervision, S.G. and D.P.F.; project administration, S.G.; funding acquisition, S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the General Secretariat for Research and Technology (GSRT) and the Hellenic Foundation for Research and Innovation (HFRI), grant number 938 (http://www.elidek.gr/en/homepage/, accessed on 12 March 2021) to D.K. S.G. would like to acknowledge co-funding of this work by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH—CREATE—INNOVATE (project code: T1EDK-02681). R.V.P. received funding from the Doctoral Program in Microbiology and Biotechnology from the University of Helsinki.
Data Availability Statement
The Leptothoe kymatousa TAU-MAC 1615 Whole Genome project was deposited at DDBJ/ENA/GenBank under the accession number JADOER000000000. The Leptothoe spongobia TAU-MAC 1115 Whole Genome Shotgun project was deposited at DDBJ/ENA/GenBank under the accession number JADOES000000000.
Acknowledgments
We thank Lyudmila Saari for her kind assistance in purifying the sponge-associated strains.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Björk, J.R.; Easson, C.; Astudillo-García, C.; Olson, J.B.; Erwin, P.M.; López-Legentil, S.; Luter, H.; et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016, 7, 11870. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-Associated Microorganisms: Evolution, Ecology, and Biotechnological Potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef]
- Hentschel, U.; Piel, J.; Degnan, S.M.; Taylor, M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012. [Google Scholar] [CrossRef]
- Adams, D.G.; Duggan, P.S.; Owen, J. Cyanobacterial Symbioses. In Ecology of Cyanobacteria II: Their Diversity in Space and Time; Whitton, B.A., Ed.; Spriger: Dordrecht, The Netherlands, 2012; pp. 593–636. [Google Scholar]
- Whitton, B.A.; Potts, M. Introduction to the Cyanobacteria. In Ecology of Cyanobacteria II: Their Diversity in Space and Time; Whitton, B.A., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 1–14. [Google Scholar]
- Fan, L.; Reynolds, D.; Liu, M.; Stark, M.; Kjelleberg, S.; Webster, N.S.; Thomas, T. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. USA 2012, 109, E1878–E1887. [Google Scholar] [CrossRef]
- Karimi, E.; Keller-Costa, T.; Slaby, B.M.; Cox, C.J.; da Rocha, U.N.; Hentschel, U.; Costa, R. Genomic blueprints of sponge-prokaryote symbiosis are shared by low abundant and cultivatable Alphaproteobacteria. Sci. Rep. 2019, 9, 1999. [Google Scholar] [CrossRef]
- Engelberts, J.P.; Robbins, S.J.; de Goeij, J.M.; Aranda, M.; Bell, S.C.; Webster, N.S. Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J. 2020, 14, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Burgsdorf, I.; Slaby, B.M.; Handley, K.M.; Haber, M.; Blom, J.; Marshall, C.W.; Gilbert, J.A.; Hentschel, U.; Steindler, L. Lifestyle evolution in cyanobacterial symbionts of sponges. mBio 2015, 6, e00391-15. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.; Balaich, J.N.; Donia, M.S. Localized production of defence chemicals by intracellular symbionts of Haliclona sponges. Nat. Microbiol. 2019, 4, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012, 10, 13–26. [Google Scholar] [CrossRef]
- Larsson, J.; Nylander, J.A.A.; Bergman, B. Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC Evol. Biol. 2011, 11. [Google Scholar] [CrossRef]
- Dittmann, E.; Gugger, M.; Sivonen, K.; Fewer, D.P. Natural Product Biosynthetic Diversity and Comparative Genomics of the Cyanobacteria. Trends Microbiol. 2015, 23, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Sampaio-Dias, I.E.; Castelo-Branco, R.; Scharfenstein, H.; Rezende De Castro, R.; Silva, A.; Schneider, M.P.C.; Araújo, M.J.; Martins, R.; Domingues, V.F.; et al. Structure of hierridin c, synthesis of hierridins b and c, and evidence for prevalent alkylresorcinol biosynthesis in picocyanobacteria. J. Nat. Prod. 2019, 82, 393–402. [Google Scholar] [CrossRef]
- Mattila, A.; Andsten, R.M.; Jumppanen, M.; Assante, M.; Jokela, J.; Wahlsten, M.; Mikula, K.M.; Sigindere, C.; Kwak, D.H.; Gugger, M.; et al. Biosynthesis of the Bis-Prenylated Alkaloids Muscoride A and B. ACS Chem. Biol. 2019, 14, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Pancrace, C.; Ishida, K.; Briand, E.; Pichi, D.G.; Weiz, A.R.; Guljamow, A.; Scalvenzi, T.; Sassoon, N.; Hertweck, C.; Dittmann, E.; et al. Unique Biosynthetic Pathway in Bloom-Forming Cyanobacterial Genus Microcystis Jointly Assembles Cytotoxic Aeruginoguanidines and Microguanidines. ACS Chem. Biol. 2019, 14, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Heinilä, L.M.P.; Fewer, D.P.; Jokela, J.K.; Wahlsten, M.; Jortikka, A.; Sivonen, K. Shared PKS Module in Biosynthesis of Synergistic Laxaphycins. Front. Microbiol. 2020, 11, 578878. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Lackner, G.; Peters, E.E.; Helfrich, E.J.N.; Piel, J. Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc. Natl. Acad. Sci. USA 2017, 114, E347–E356. [Google Scholar] [CrossRef]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar] [CrossRef]
- Konstantinou, D.; Voultsiadou, E.; Panteris, E.; Zervou, S.-K.; Hiskia, A.; Gkelis, S. Leptothoe, a new genus of marine cyanobacteria (Synechococcales) and three new species associated with sponges from the Aegean Sea. J. Phycol. 2019, 55. [Google Scholar] [CrossRef]
- Konstantinou, D.; Voultsiadou, E.; Panteris, E.; Gkelis, S. Revealing new sponge-associated cyanobacterial diversity: Novel genera and species. Mol. Phylogenet. Evol. 2021, 155, 106991. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, D.; Mavrogonatou, E.; Zervou, S.K.; Giannogonas, P.; Gkelis, S. Bioprospecting Sponge-Associated Marine Cyanobacteria to Produce Bioactive Compounds. Toxins 2020, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, D.; Gerovasileiou, V.; Voultsiadou, E.; Gkelis, S. Sponges-Cyanobacteria associations: Global diversity overview and new data from the Eastern Mediterranean. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Esteves, A.I.S.; Hardoim, C.C.P.; Xavier, J.R.; Gonçalves, J.M.S.; Costa, R. Molecular richness and biotechnological potential of bacteria cultured from Irciniidae sponges in the north-east Atlantic. FEMS Microbiol. Ecol. 2013, 85, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Antunes, A. Whole-genome comparisons among the genus shewanella reveal the enrichment of genes encoding ankyrin-repeats containing proteins in sponge-associated bacteria. Front. Microbiol. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Lu, C.K.; Su, H.M.; Chiang, T.Y.; Hwang, C.C.; Liu, T.; Chen, Y.M. Draft genome of myxosarcina sp. Strain Gi1, a baeocytous cyanobacterium associated with the marine sponge terpios hoshinota. Stand. Genom. Sci. 2015, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Alex, A.; Antunes, A. Genus-wide comparison of Pseudovibrio bacterial genomes reveal diverse adaptations to different marine invertebrate hosts. PLoS ONE 2018, 13, e0194368. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.M.; Wang, Y.; Tian, R.M.; Wong, Y.H.; Batang, Z.B.; Al-Suwailem, A.M.; Bajic, V.B.; Qian, P.Y. Symbiotic Adaptation Drives Genome Streamlining of the Cyanobacterial Sponge Symbiont “Candidatus Synechococcus spongiarum”. mBio 2014, 5, e00079-14. [Google Scholar] [CrossRef]
- Sloan, D.B.; Moran, N.A. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol. Biol. Evol. 2012, 29, 3781–3792. [Google Scholar] [CrossRef]
- Morris, J.; Lenski, R.E.; Zinser, E.R. The Black Queen Hypothesis: Evolution of Dependencies through Adaptive Gene Loss. mBio 2012, 3, e00036-12. [Google Scholar] [CrossRef] [PubMed]
- Karimi, E.; Slaby, B.M.; Soares, A.R.; Blom, J.; Hentschel, U.; Costa, R. Metagenomic binning reveals versatile nutrient cycling and distinct adaptive features in alphaproteobacterial symbionts of marine sponges. FEMS Microbiol. Ecol. 2018, 94, fiy074. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Peretó, J.; Gil, R.; Latorre, A. Learning how to live together: Genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 2008, 9, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Schorn, M.A.; Jordan, P.A.; Podell, S.; Blanton, J.M.; Agarwal, V.; Biggs, J.S.; Allen, E.E.; Moore, B.S. Comparative Genomics of Cyanobacterial Symbionts Reveals Distinct, Specialized Metabolism in Tropical Dysideidae Sponges. mBio 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Díez-Vives, C.; Moitinho-Silva, L.; Nielsen, S.; Reynolds, D.; Thomas, T. Expression of eukaryotic-like protein in the microbiome of sponges. Mol. Ecol. 2017, 26, 1432–1451. [Google Scholar] [CrossRef]
- Reynolds, D.; Thomas, T. Evolution and function of eukaryotic-like proteins from sponge symbionts. Mol. Ecol. 2016, 25, 5242–5253. [Google Scholar] [CrossRef]
- Nguyen, M.T.H.D.; Liu, M.; Thomas, T. Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 2014, 23, 1635–1645. [Google Scholar] [CrossRef]
- Liu, M.; Fan, L.; Zhong, L.; Kjelleberg, S.; Thomas, T. Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 2012, 6, 1515–1525. [Google Scholar] [CrossRef]
- Siegl, A.; Kamke, J.; Hochmuth, T.; Piel, J.; Richter, M.; Liang, C.; Dandekar, T.; Hentschel, U. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 2011, 5, 61–70. [Google Scholar] [CrossRef]
- Wang, H.; Fewer, D.P.; Holm, L.; Rouhiainen, L.; Sivonen, K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 9259–9264. [Google Scholar] [CrossRef]
- Welker, M.; Von Döhren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.C.; Monroe, E.A.; Eisman, E.B.; Gerwick, L.; Sherman, D.H.; Gerwick, W.H. The unique mechanistic transformations involved in the biosynthesis of modular natural products from marine cyanobacteria. Nat. Prod. Rep. 2010, 27, 1048–1065. [Google Scholar] [CrossRef]
- Wang, H.; Fewer, D.P.; Sivonen, K. Genome mining demonstrates the widespread occurrence of gene clusters encoding bacteriocins in cyanobacteria. PLoS ONE 2011, 6, e22384. [Google Scholar] [CrossRef] [PubMed]
- Piper, C.; Cotter, P.; Ross, R.; Hill, C. Discovery of Medically Significant Lantibiotics. Curr. Drug Discov. Technol. 2009, 6, 1–18. [Google Scholar] [CrossRef]
- Drider, D.; Bendali, F.; Naghmouchi, K.; Chikindas, M.L. Bacteriocins: Not Only Antibacterial Agents. Probiotics Antimicrob. Proteins 2016, 8, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Y.; Vélasquez, J.E.; Van Der Donk, W.A. Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. USA 2012, 109, 18361–18366. [Google Scholar] [CrossRef]
- Repka, L.M.; Chekan, J.R.; Nair, S.K.; Van Der Donk, W.A. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem. Rev. 2017, 117, 5457–5520. [Google Scholar] [CrossRef]
- Jones, M.; Pinto, E.; Torres, M.; Dörr, F.; Mazur-Marzec, H.; Szubert, K.; Tartaglione, L.; Dell’Aversano, C.; Miles, C.; Beach, D.; et al. Comprehensive database of secondary metabolites from cyanobacteria. Water Res. 2021, 196, 117017. [Google Scholar] [CrossRef]
- Martins, M.D.R.; Costa, M. Marine cyanobacteria compounds with anticancer properties: Implication of apoptosis. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.-K., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 621–647. ISBN 9783319071459. [Google Scholar]
- Engene, N.; Tronholm, A.; Salvador-Reyes, L.A.; Luesch, H.; Paul, V.J. Caldora penicillata gen. nov., comb. nov. (Cyanobacteria), a pantropical marine species with biomedical relevance. J. Phycol. 2015, 51, 670–681. [Google Scholar] [CrossRef]
- Engene, N.; Paul, V.J.; Byrum, T.; Gerwick, W.H.; Thor, A.; Ellisman, M.H. Five chemically rich species of tropical marine cyanobacteria of the genus Okeania gen. nov. (Oscillatoriales, Cyanoprokaryota). J. Phycol. 2013, 49, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.A.; McPhail, K.L. Apratoxin H and apratoxin A sulfoxide from the red sea cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef]
- Kwan, J.C.; Eksioglu, E.A.; Liu, C.; Paul, V.J.; Luesch, H. Grassystatins A-C from marine cyanobacteria, potent cathepsin E inhibitors that reduce antigen presentation. J. Med. Chem. 2009, 52, 5732–5747. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Imperial, L.; Garst, C.; Ratnayake, R.; Dang, L.H.; Paul, V.J.; Luesch, H. Caldoramide, a Modified Pentapeptide from the Marine Cyanobacterium Caldora penicillata. J. Nat. Prod. 2016, 79, 1867–1871. [Google Scholar] [CrossRef]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Lopez, J.A.V.; Al-Lihaibi, S.S.; Alarif, W.M.; Abdel-Lateff, A.; Nogata, Y.; Washio, K.; Morikawa, M.; Okino, T. Wewakazole B, a Cytotoxic Cyanobactin from the Cyanobacterium Moorea producens Collected in the Red Sea. J. Nat. Prod. 2016, 79, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Zhang, J.; He, S.; Yan, X. New peptides isolated from marine cyanobacteria, an overview over the past decade. Mar. Drugs 2017, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Rippka, R. Isolation and Purification of Cyanobacteria. Methods Enzymol. 1988, 167, 1–26. [Google Scholar]
- Gkelis, S.; Panou, M. Capturing biodiversity: Linking a cyanobacteria culture collection to the “scratchpads” virtual research environment enhances biodiversity knowledge. Biodivers. Data J. 2016, 4, e7965-1. [Google Scholar] [CrossRef] [PubMed]
- Jokela, J.; Heinilä, L.M.P.; Shishido, T.K.; Wahlsten, M.; Fewer, D.P.; Fiore, M.F.; Wang, H.; Haapaniemi, E.; Permi, P.; Sivonen, K. Production of high amounts of hepatotoxin nodularin and new protease inhibitors pseudospumigins by the brazilian benthic nostoc sp. CENA543. Front. Microbiol. 2017, 8, 1963. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; Mccue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments; BMC: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Alvarenga, D.O.; Fiore, M.F.; Varani, A.M. A metagenomic approach to cyanobacterial genomics. Front. Microbiol. 2017, 8, 809. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2013, 2, 10. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).