
Model-Free Approach for the Configurational Analysis of Marine Natural Products †


Abstract
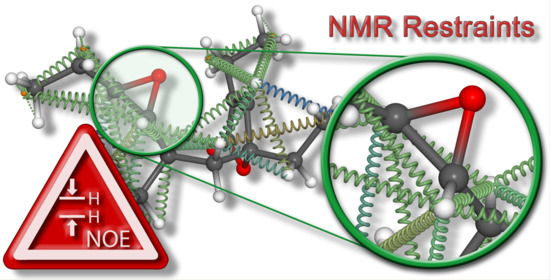
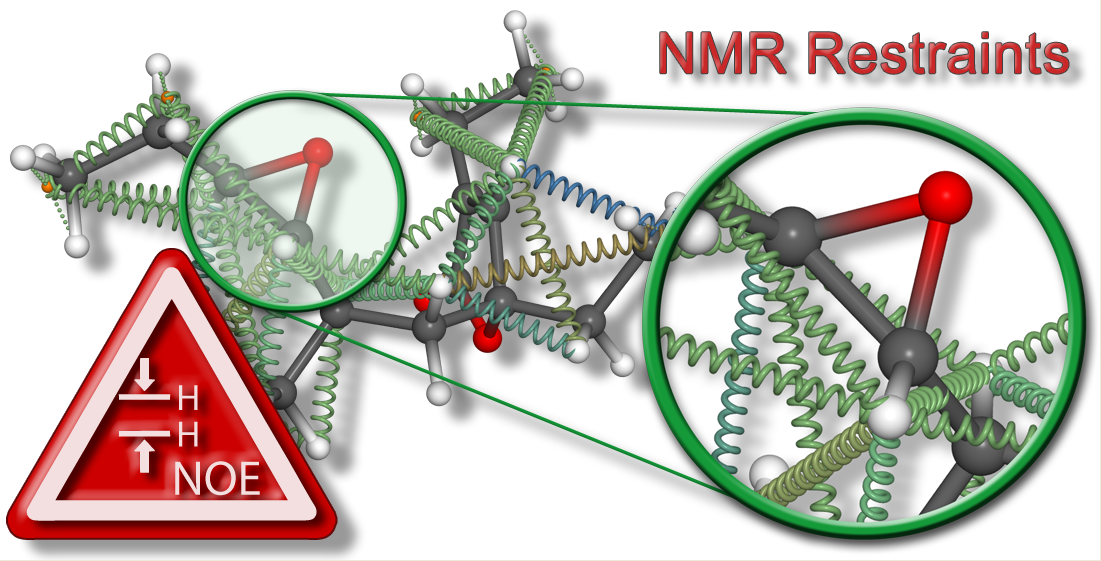{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
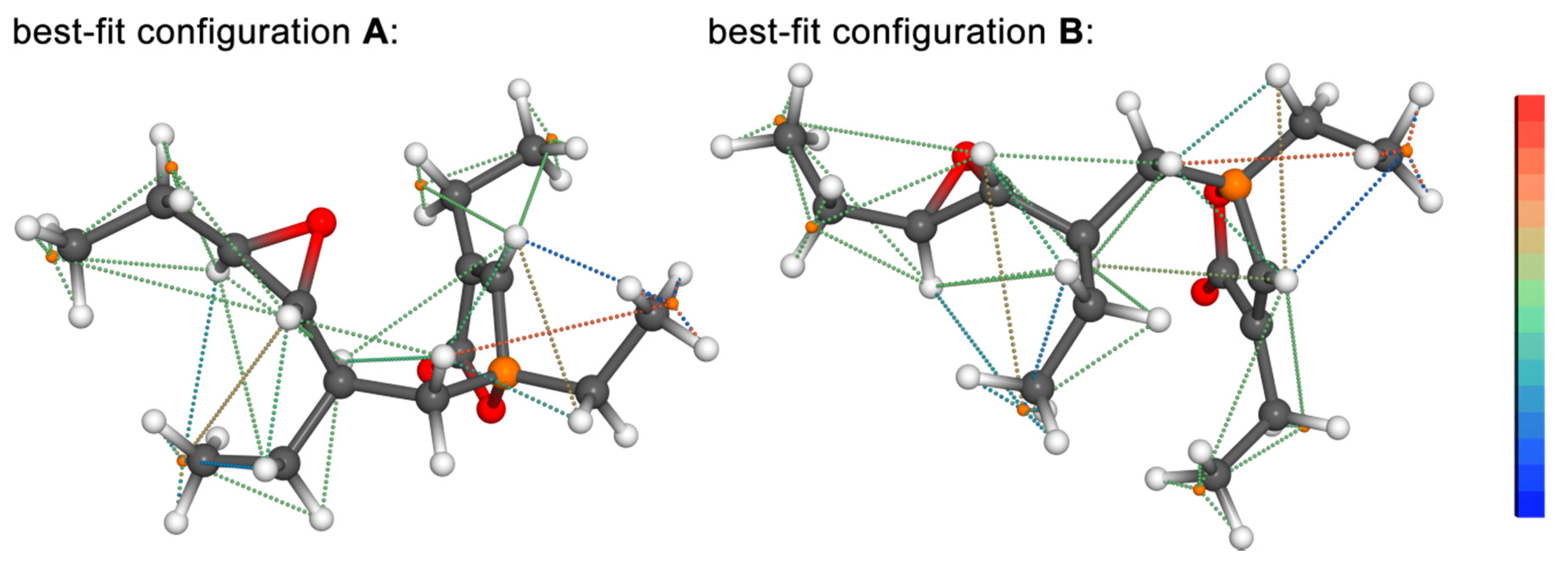{kind=link}
{kind=link}
{kind=link}
{kind=link}
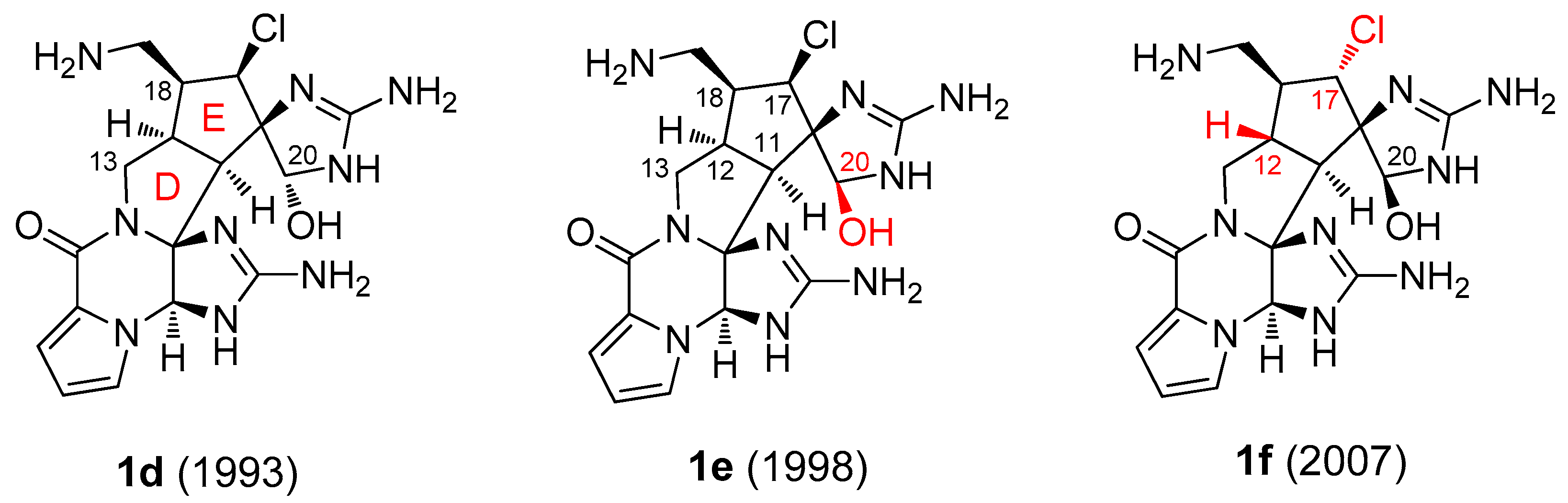{kind=link}
1. Introduction
2. Results and Discussion
2.1. Palau’amine Derivatives (1)
2.2. Plakilactone H (2)
2.3. Manzamine A (3)
3. Methods
3.1. NMR Data
3.2. DG/DDD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Köck, M.; Junker, J. Marine Natural Products—New Ways in the Constitutional Assignment. In New Aspects in Bioorganic Chemistry; Diederichsen, U., Lindhorst, T.K., Wessjohann, L., Westermann, B., Eds.; Wiley-VCH: Weinheim, Germany, 1999; pp. 365–378. [Google Scholar]
- Lindel, T.; Junker, J.; Köck, M. Cocon: From NMR correlation data to molecular constitutions. J. Mol. Model. 1997, 3, 364–368. [Google Scholar] [CrossRef]
- Meiler, J.; Sanli, E.; Junker, J.; Meusinger, R.; Lindel, T.; Will, M.; Maier, W.; Köck, M. Validation of structural proposals by substructure analysis and C-13 NMR chemical shift prediction. J. Chem. Inf. Comput. Sci. 2002, 42, 241–248. [Google Scholar] [CrossRef]
- Meiler, J.; Köck, M. Novel methods of automated structure elucidation based on C-13 NMR spectroscopy. Magn. Reson. Chem. 2004, 42, 1042–1045. [Google Scholar] [CrossRef]
- Smurnyy, Y.D.; Elyashberg, M.E.; Blinov, K.A.; Lefebvre, B.A.; Martin, G.E.; Williams, A.J. Computer-aided determination of relative stereochemistry and 3D models of complex organic molecules from 2D NMR spectra. Tetrahedron 2005, 61, 9980–9989. [Google Scholar] [CrossRef]
- Smurnyy, Y.D.; Blinov, K.A.; Churanova, T.S.; Elyashberg, M.E.; Williams, A.J. Toward more reliable C-13 and H-1 chemical shift prediction: A systematic comparison of neural-network and least-squares regression based approaches. J. Chem. Inf. Model. 2008, 48, 128–134. [Google Scholar] [CrossRef]
- Elyashberg, M.; Blinov, K.; Molodtsov, S.; Smurnyy, Y.; Williams, A.J.; Churanova, T. Computer-assisted methods for molecular structure elucidation: Realizing a spectroscopist’s dream. J. Cheminformatics 2009, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Elyashberg, M. Identification and structure elucidation by NMR spectroscopy. Trends Anal. Chem. 2015, 69, 88–97. [Google Scholar] [CrossRef]
- Buevich, A.V.; Elyashberg, M.E. Synergistic Combination of CASE Algorithms and DFT Chemical Shift Predictions: A Powerful Approach for Structure Elucidation, Verification, and Revision. J. Nat. Prod. 2016, 79, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Troche-Pesqueira, E.; Anklin, C.; Gil, R.R.; Navarro-Vázquez, A. Computer-Assisted 3D Structure Elucidation of Natural Products using Residual Dipolar Couplings. Angew. Chem. Int. Ed. 2017, 56, 3660–3664. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Vázquez, A.; Gil, R.R.; Blinov, K. Computer-Assisted 3D Structure Elucidation (CASE-3D) of Natural Products Combining Isotropic and Anisotropic NMR Parameters. J. Nat. Prod. 2018, 81, 203–210. [Google Scholar] [CrossRef]
- Milanowski, D.J.; Oku, N.; Cartner, L.K.; Bokesch, H.R.; Williamson, R.T.; Sauri, J.; Liu, Y.Z.; Blinov, K.A.; Ding, Y.Q.; Li, X.C.; et al. Unequivocal determination of caulamidines A and B: Application and validation of new tools in the structure elucidation tool box. Chem. Sci. 2018, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.J.; Garcia, M.E.; Padron, J.M.; Navarro-Vázquez, A.; Gil, R.R.; Nicotra, V.E. Phytochemical Study of Senecio volckmannii Assisted by CASE-3D with Residual Dipolar Couplings and Isotropic H-1/C-13 NMR Chemical Shifts. J. Nat. Prod. 2018, 81, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Imperatore, C.; Mangoni, A.; Della Sala, G.; Taglialatela-Scafati, O. Challenges in the configuration assignment of natural products. A case-selective perspective. Nat. Prod. Rep. 2019, 36, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B. The nuclear Overhauser effect from a quantitative perspective. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 78, 1–46. [Google Scholar] [CrossRef]
- Neuhaus, D.; Williamson, M.P. The Nuclear Overhauser Effect in Structural and Conformational Analysis; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Köck, M.; Reggelin, M.; Immel, S. The Advanced Floating Chirality Distance Geometry Approach—How Anisotropic NMR Parameters Can Support the Determination of the Relative Configuration of Natural Products. Mar. Drugs 2020, 18, 330. [Google Scholar] [CrossRef]
- Crippen, G.M.; Havel, T.F. Distance Geometry and Molecular Conformation; John Wiley & Sons: Hoboken, NJ, USA, 1988. [Google Scholar]
- de Vlieg, J.; Scheek, R.M.; van Gunsteren, W.F.; Berendsen, H.J.C.; Kaptein, R.; Thomason, J. Combined procedure of distance geometry and restrained molecular dynamics techniques for protein structure determination from nuclear magnetic resonance data: Application to the DNA binding domain of lac repressor from Escherichia coli. Proteins Struct. Funct. Genet. 1988, 3, 209–218. [Google Scholar] [CrossRef]
- Havel, T.F.; Kuntz, I.D.; Crippen, G.M. The theory and practice of distance geometry. Bull. Math. Biol. 1983, 45, 665–720. [Google Scholar] [CrossRef]
- Kaptein, R.; Boelens, R.; Scheek, R.M.; van Gunsteren, W.F. Protein structures from NMR. Biochemistry 1988, 27, 5389–5395. [Google Scholar] [CrossRef]
- Scheek, R.M.; van Gunsteren, W.F.; Kaptein, R. Molecular dynamics simulation techniques for determination of molecular structures from nuclear magnetic resonance data. Methods Enzymol. 1989, 177, 204–218. [Google Scholar]
- Köck, M.; Junker, J. Determination of the relative configuration of organic compounds using NMR and DG: A systematic approach for a model system. J. Mol. Model. 1997, 3, 403–407. [Google Scholar] [CrossRef]
- Köck, M.; Schmidt, G.; Seiple, I.B.; Baran, P.S. Configurational Analysis of Tetracyclic Dimeric Pyrrole-Imidazole Alkaloids Using a Floating Chirality Approach. J. Nat. Prod. 2012, 75, 127–130. [Google Scholar] [CrossRef]
- Reggelin, M.; Hoffmann, H.; Köck, M.; Mierke, D.F. Determination of Conformation and Relative Configuration of a Small, Rapidly Tumbling Molecule in Solution by Combined Application of NOESY and Restrained MD Calculations. J. Am. Chem. Soc. 1992, 114, 3272–3277. [Google Scholar] [CrossRef]
- Mierke, D.F.; Reggelin, M. Simultaneous determination of conformation and configuration using distance geometry. J. Org. Chem. 1992, 57, 6365–6367. [Google Scholar] [CrossRef]
- Reggelin, M.; Köck, M.; Conde-Frieboes, K.; Mierke, D. Determination of the Relative Configuration by Distance Geometry Calculations with Proton-Proton Distances from NOESY Spectra. Angew. Chem. Int. Ed. 1994, 33, 753–755. [Google Scholar] [CrossRef]
- Köck, M.; Junker, J. How many NOE derived restraints are necessary for a reliable determination of the relative configuration of an organic compound? Application to a model system. J. Org. Chem. 1997, 62, 8614–8615, Erratum in 1998, 63, 2409–2409. [Google Scholar] [CrossRef]
- Kaptein, R.; Zuiderweg, E.R.P.; Scheek, R.M.; Boelens, R.; van Gunsteren, W.F. A protein structure from nuclear magnetic resonance data. The lac repressor headpiece. J. Mol. Biol. 1985, 182, 179–182. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M.; Brünger, A.T.; Karplus, M. Solution Conformation of a Heptadecapeptide Comprising the DNA-Binding Helix F of the Cyclic AMP Receptor Protein of Escherichia coli—Combined Use of H-1 Nuclear Magnetic-Resonance and Restrained Molecular Dynamics. J. Mol. Biol. 1985, 186, 435–455. [Google Scholar] [CrossRef]
- Weber, P.L.; Morrison, R.; Hare, D. Determining stereo-specific H-1 nuclear magnetic-resonance assignments from distance geometry calculations. J. Mol. Biol. 1988, 204, 483–487. [Google Scholar] [CrossRef]
- Holak, T.A.; Gondol, D.; Otlewski, J.; Wilusz, T. Determination of the complete three-dimensional structure of the trypsin inhibitor from squash seeds in aqueous solution by nuclear magnetic resonance and a combination of distance geometry and dynamical simulated annealing. J. Mol. Biol. 1989, 210, 635–648. [Google Scholar] [CrossRef]
- Havel, T.F. An evaluation of computational strategies for use in the determination of protein-structure from distance contraints obtained by nuclear magnetic resonance Prog. Biophys. Mol. Biol. 1991, 56, 43–78. [Google Scholar] [CrossRef]
- van Schaik, R.C.; Berendsen, H.J.C.; Torda, A.E.; van Gunsteren, W.F. A Structure Refinement Method Based On Molecular-Dynamics In Four Spatial Dimensions. J. Mol. Biol. 1993, 234, 751–762. [Google Scholar] [CrossRef]
- A pseudo-atom is a virtual atom whose position is the geometrical average of two (isochronous methylene groups or ortho-/meta-CHs in aromatic systems), three (methyl group), or more hydrogen atoms. A pseudo-atom is applied if an assignment to one proton is not possible and therefore its position is used in order to consider the distance information.
- Immel, S.; Köck, M.; Reggelin, M. Configurational Analysis by Residual Dipolar Coupling Driven Floating Chirality Distance Geometry Calculations. Chem. Eur. J. 2018, 24, 13918–13930. [Google Scholar] [CrossRef]
- Immel, S.; Köck, M.; Reggelin, M. Configurational analysis by residual dipolar couplings: A critical assessment of diastereomeric differentiabilities. Chirality 2019, 31, 384–400. [Google Scholar] [CrossRef]
- Reggelin, M.; Immel, S. Configurational Analysis by Residual Dipolar Couplings: Critical Assessment of “Structural Noise” from Thermal Vibrations. Angew. Chem. Int. Ed. 2021, 60, 3412–3416. [Google Scholar] [CrossRef]
- Kinnel, R.B.; Gehrken, H.P.; Scheuer, P.J. Palauamine—A Cytotoxic and Immunosuppressive Hexacyclic Bisguanidine Antibiotic from the Sponge Stylotella-Agminata. J. Am. Chem. Soc. 1993, 115, 3376–3377. [Google Scholar] [CrossRef]
- Di Micco, S.; Zampella, A.; D’Auria, M.V.; Festa, C.; De Marino, S.; Riccio, R.; Butts, C.P.; Bifulco, G. Plakilactones G and H from a marine sponge. Stereochemical determination of highly flexible systems by quantitative NMR-derived interproton distances combined with quantum mechanical calculations of 13C chemical shifts. Beilstein J. Org. Chem. 2013, 9, 2940–2949. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T. Manzamine A, a Novel Antitumor Alkaloid from a Sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Köck, M.; Grube, A.; Seiple, I.B.; Baran, P.S. The pursuit of Palau’amine. Angew. Chem. Int. Ed. 2007, 46, 6586–6594. [Google Scholar] [CrossRef]
- Kinnel, R.B.; Gehrken, H.P.; Swali, R.; Skoropowski, G.; Scheuer, P.J. Palau’amine and its congeners: A family of bioactive bisguanidines from the marine sponge Stylotella aurantium. J. Org. Chem. 1998, 63, 3281–3286. [Google Scholar] [CrossRef]
- Halford, B. Palau’amine reconsidered. Chem. Eng. News 2007, 85, 12–12. [Google Scholar] [CrossRef]
- Grube, A.; Köck, M. Structural Assignment of Tetrabromostyloguanidine: Does the Relative Configuration of the Palau’amines Need Revision? Angew. Chem. Int. Ed. 2007, 46, 2320–2324. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.S.; Carroll, A.R.; Addepalli, R.; Avery, V.M.; Hooper, J.N.A.; Quinn, R.J. Natural products, stylissadines A and B, specific antagonists of the P2X(7) receptor, an important inflammatory target. J. Org. Chem. 2007, 72, 2309–2317. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kitamura, K.; Nagai, K.; Nakao, Y.; Fusetani, N.; van Soest, R.W.M.; Matsunaga, S. Carteramine A, an inhibitor of neutrophil chemotaxis, from the marine sponge Stylissa carteri. Tetrahedron Lett. 2007, 48, 2127–2129. [Google Scholar] [CrossRef]
- Peplow, M. Chemists crack complex compound. Nature 2010, 463, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Seiple, I.B.; Su, S.; Young, I.S.; Lewis, C.A.; Yamaguchi, J.; Baran, P.S. Total Synthesis of Palau’amine. Angew. Chem. Int. Ed. 2010, 49, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Seiple, I.B.; Su, S.; Young, I.S.; Nakamura, A.; Yamaguchi, J.; Jørgensen, L.; Rodriguez, R.A.; O’Malley, D.P.; Gaich, T.; Köck, M.; et al. Enantioselective Total Syntheses of (−)-Palau’amine, (−)-Axinellamines, and (−)-Massadines. J. Am. Chem. Soc. 2011, 133, 14710–14726. [Google Scholar] [CrossRef]
- Lanman, B.A.; Overman, L.E.; Paulini, R.; White, N.S. On the structure of Palau’amine: Evidence for the revised relative configuration from chemical synthesis. J. Am. Chem. Soc. 2007, 129, 12896–12900. [Google Scholar] [CrossRef]
- Reinscheid, U.M.; Köck, M.; Cychon, C.; Schmidts, V.; Thiele, C.M.; Griesinger, C. The Absolute Configuration of Dibromopalau’amine. Eur. J. Org. Chem. 2010, 6900–6903. [Google Scholar] [CrossRef]
- Lindel, T.; Jacquot, D.E.N.; Zöllinger, M.; Kinnel, R.B.; McHugh, S.; Köck, M. Study on the absolute configuration of (−)-palau’amine. Tetrahedron Lett. 2010, 51, 6353–6355. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New York, NY, USA, 1986. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köck, M.; Reggelin, M.; Immel, S. Model-Free Approach for the Configurational Analysis of Marine Natural Products. Mar. Drugs 2021, 19, 283. https://doi.org/10.3390/md19060283
Köck M, Reggelin M, Immel S. Model-Free Approach for the Configurational Analysis of Marine Natural Products. Marine Drugs. 2021; 19(6):283. https://doi.org/10.3390/md19060283
Chicago/Turabian StyleKöck, Matthias, Michael Reggelin, and Stefan Immel. 2021. "Model-Free Approach for the Configurational Analysis of Marine Natural Products" Marine Drugs 19, no. 6: 283. https://doi.org/10.3390/md19060283
APA StyleKöck, M., Reggelin, M., & Immel, S. (2021). Model-Free Approach for the Configurational Analysis of Marine Natural Products. Marine Drugs, 19(6), 283. https://doi.org/10.3390/md19060283