
Structure and Absolute Configuration of Phenanthro-perylene Quinone Pigments from the Deep-Sea Crinoid Hypalocrinus naresianus

 , ,
, ,
Abstract
:1. Introduction
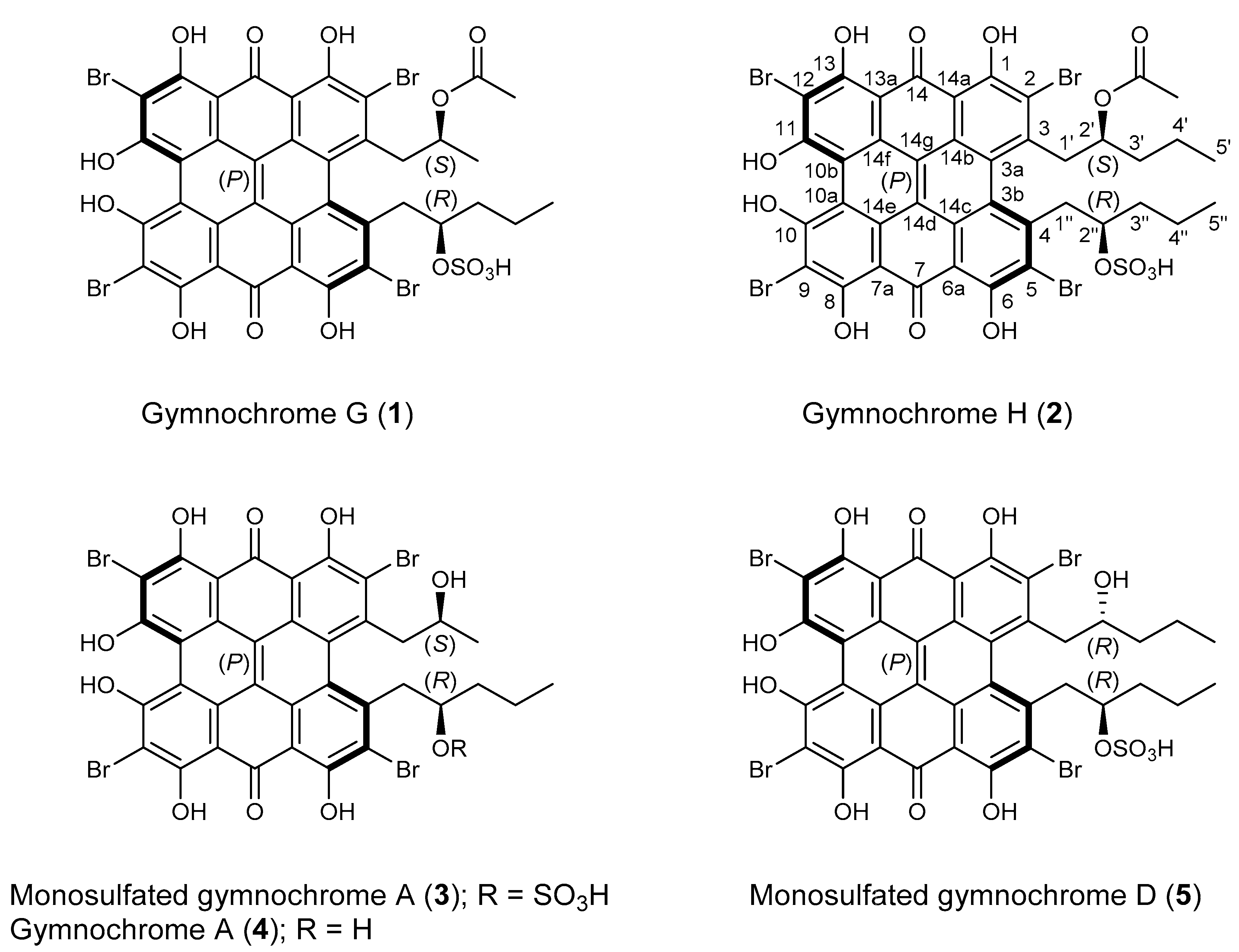
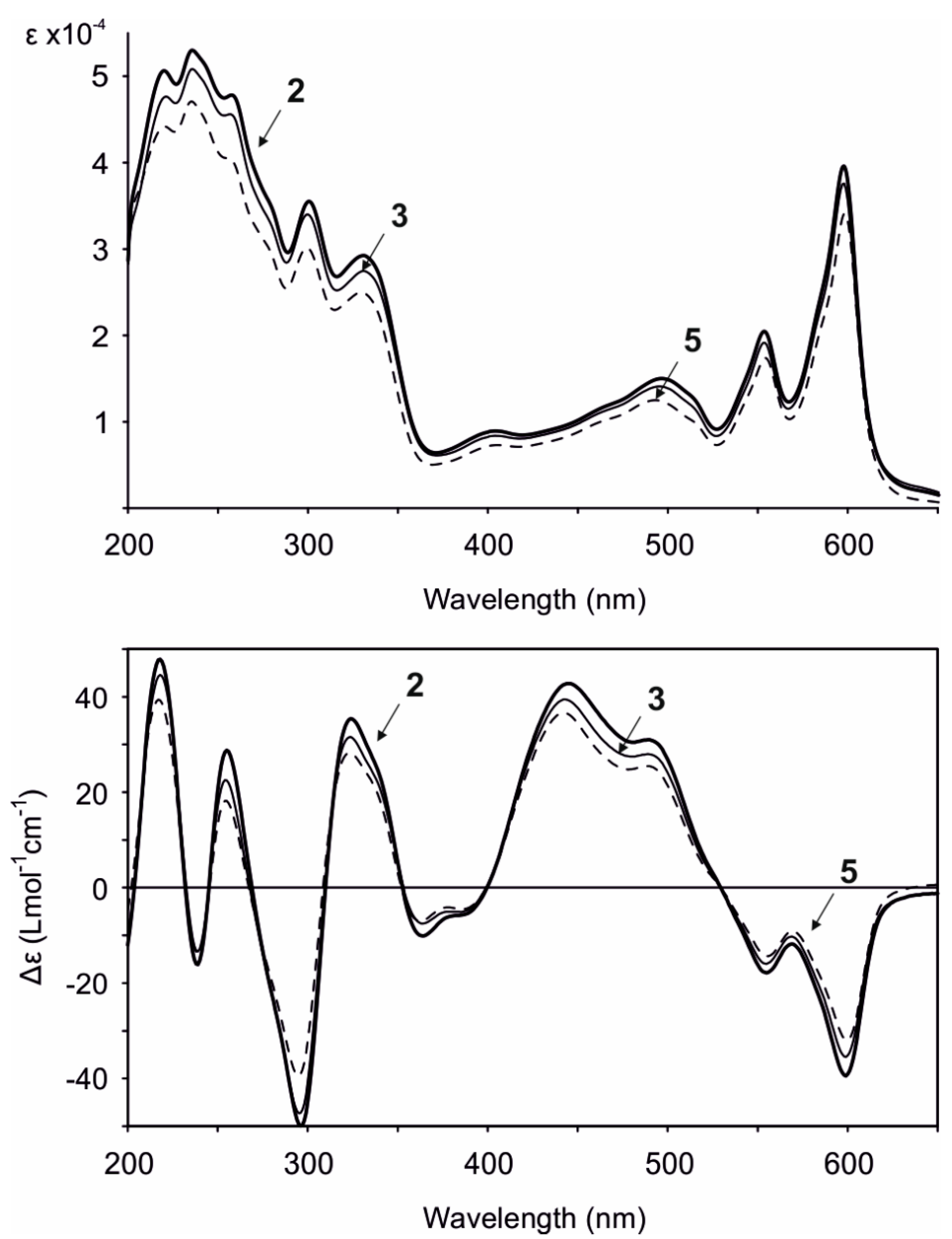
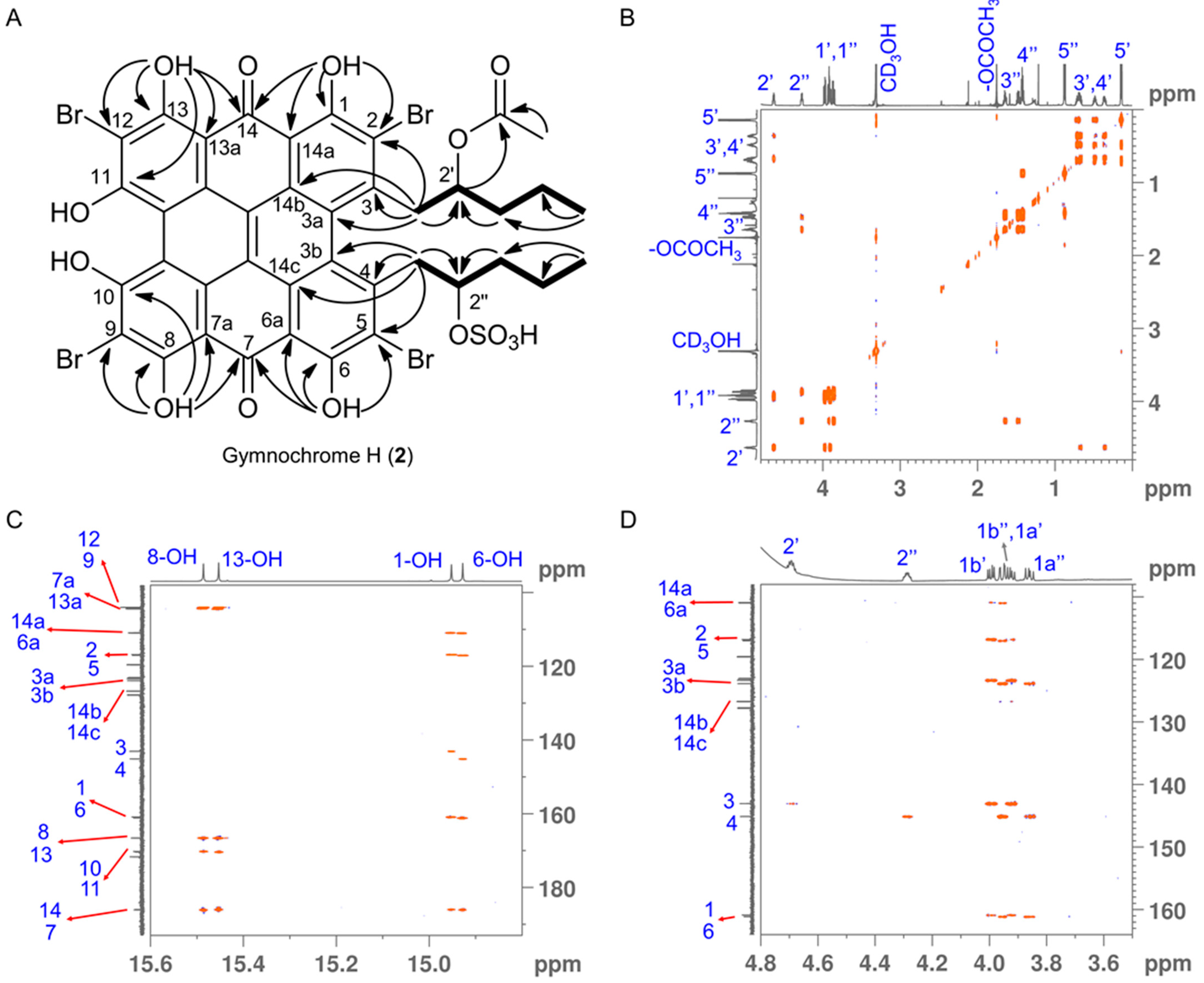
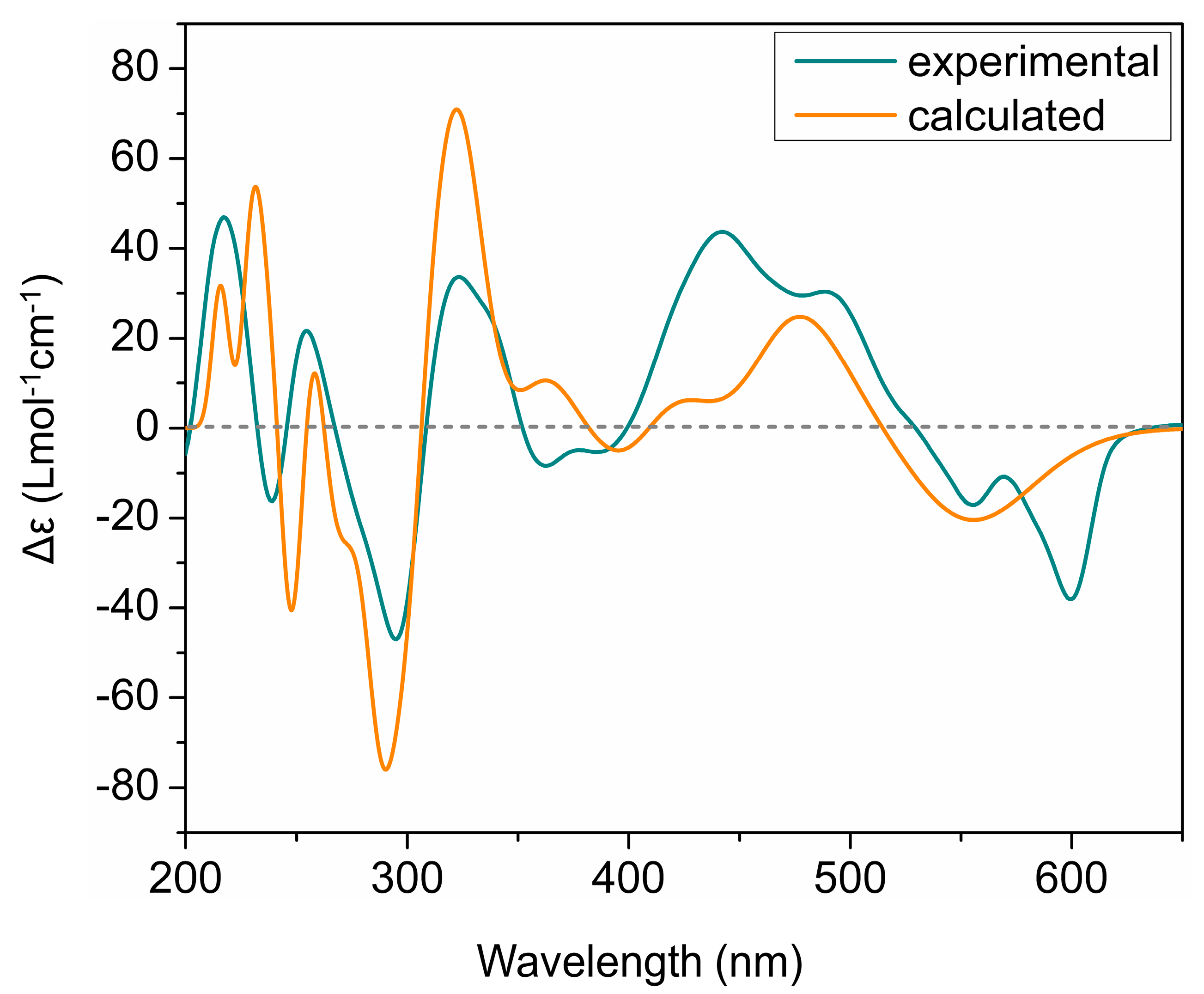
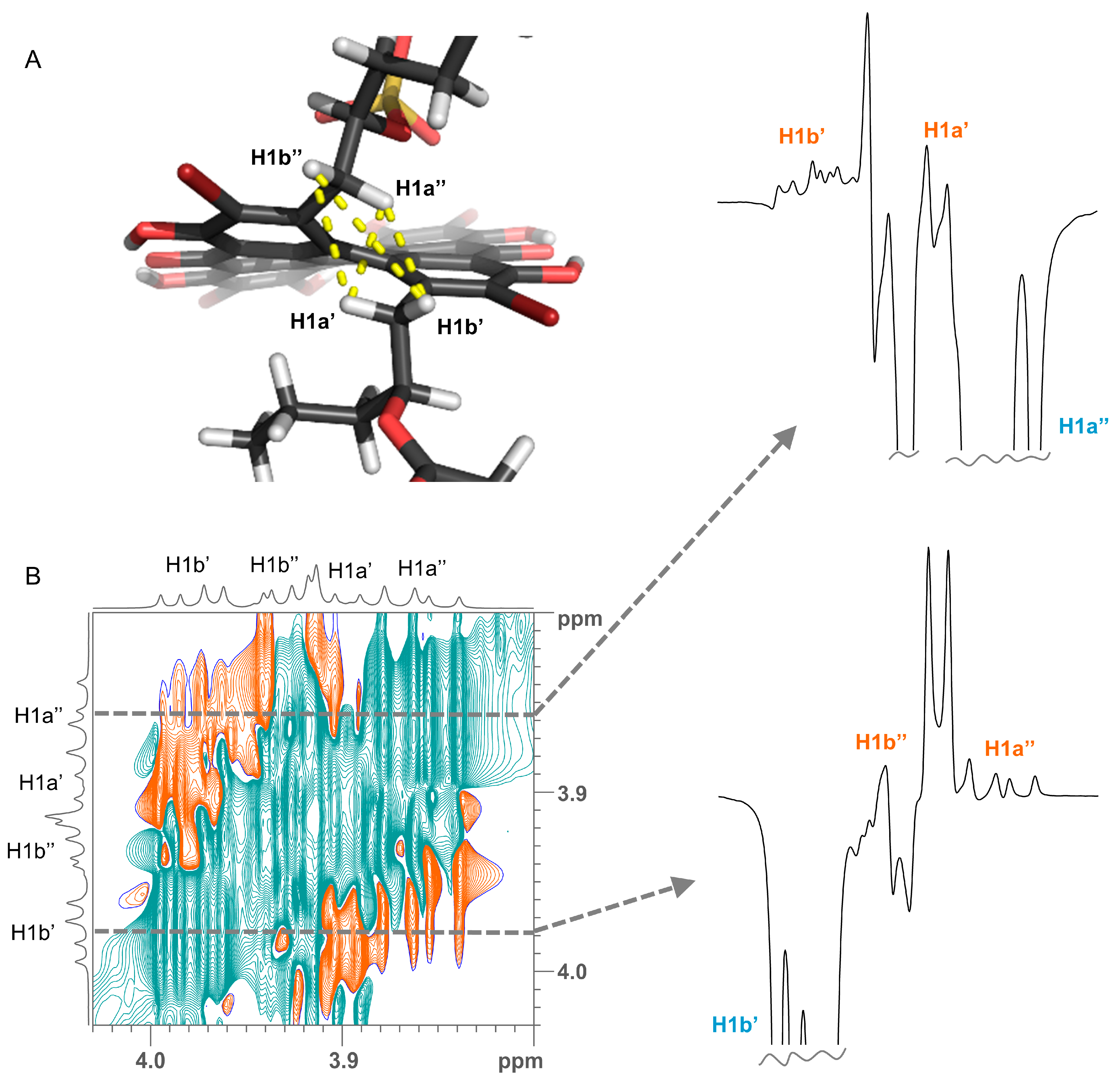
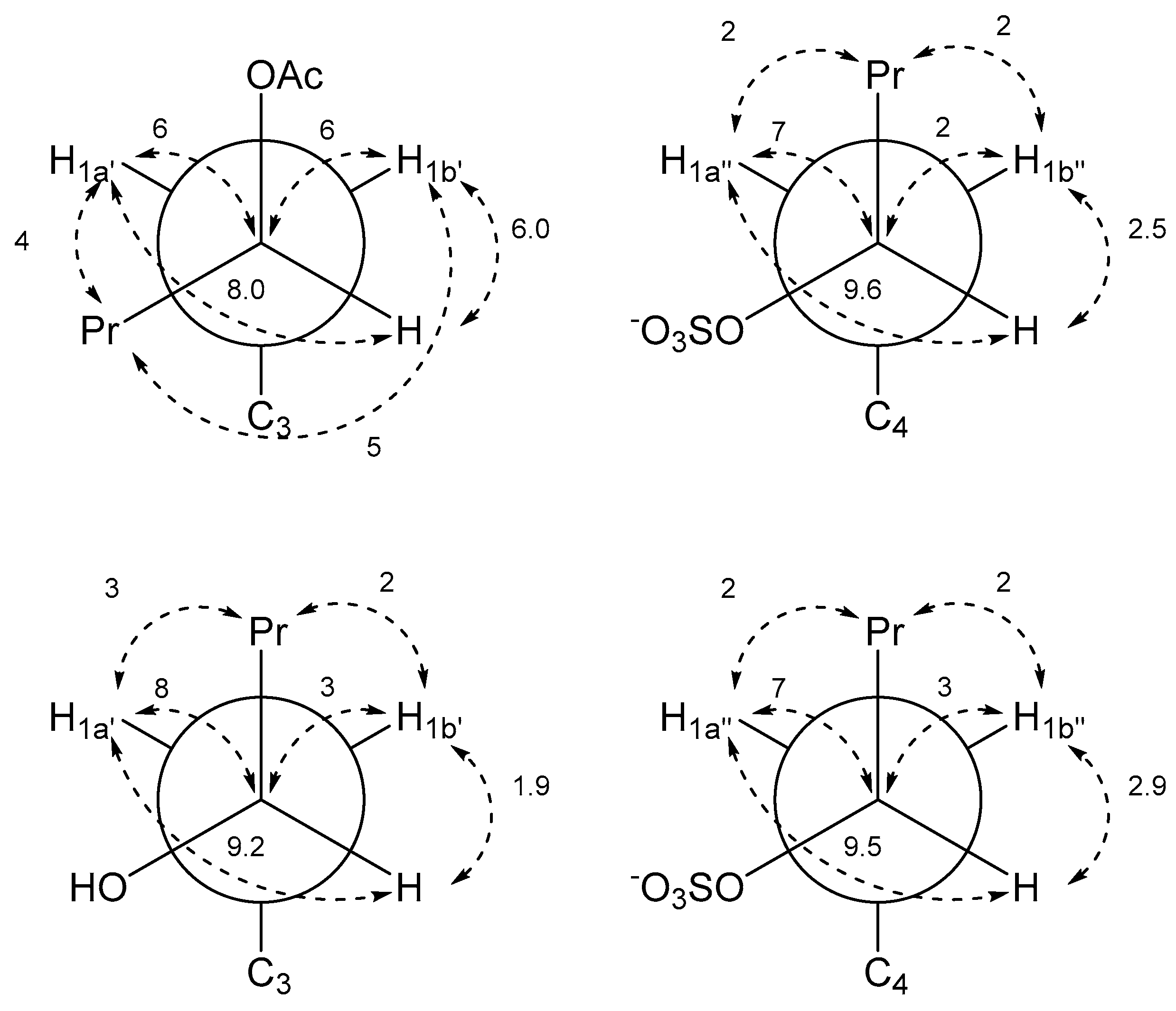
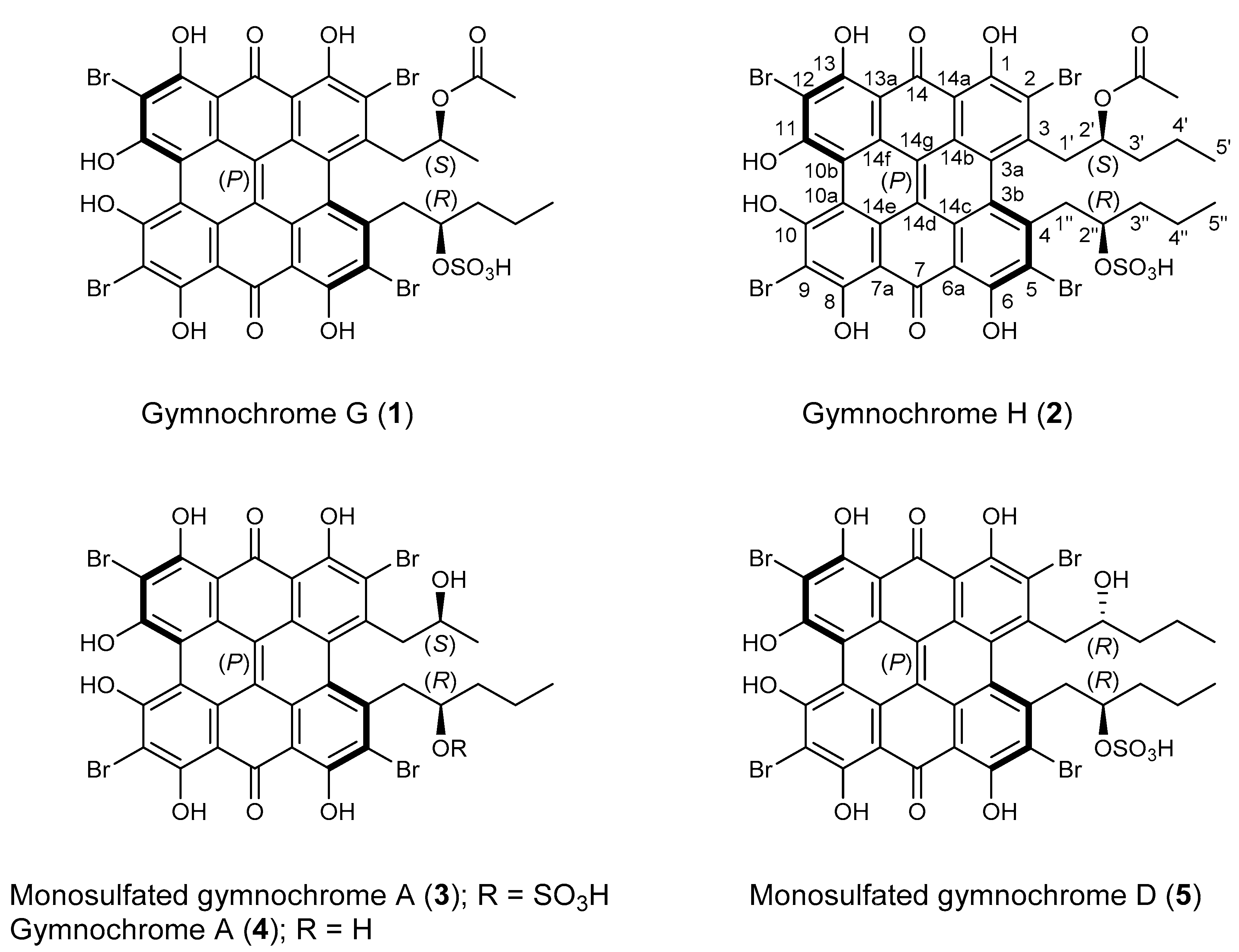
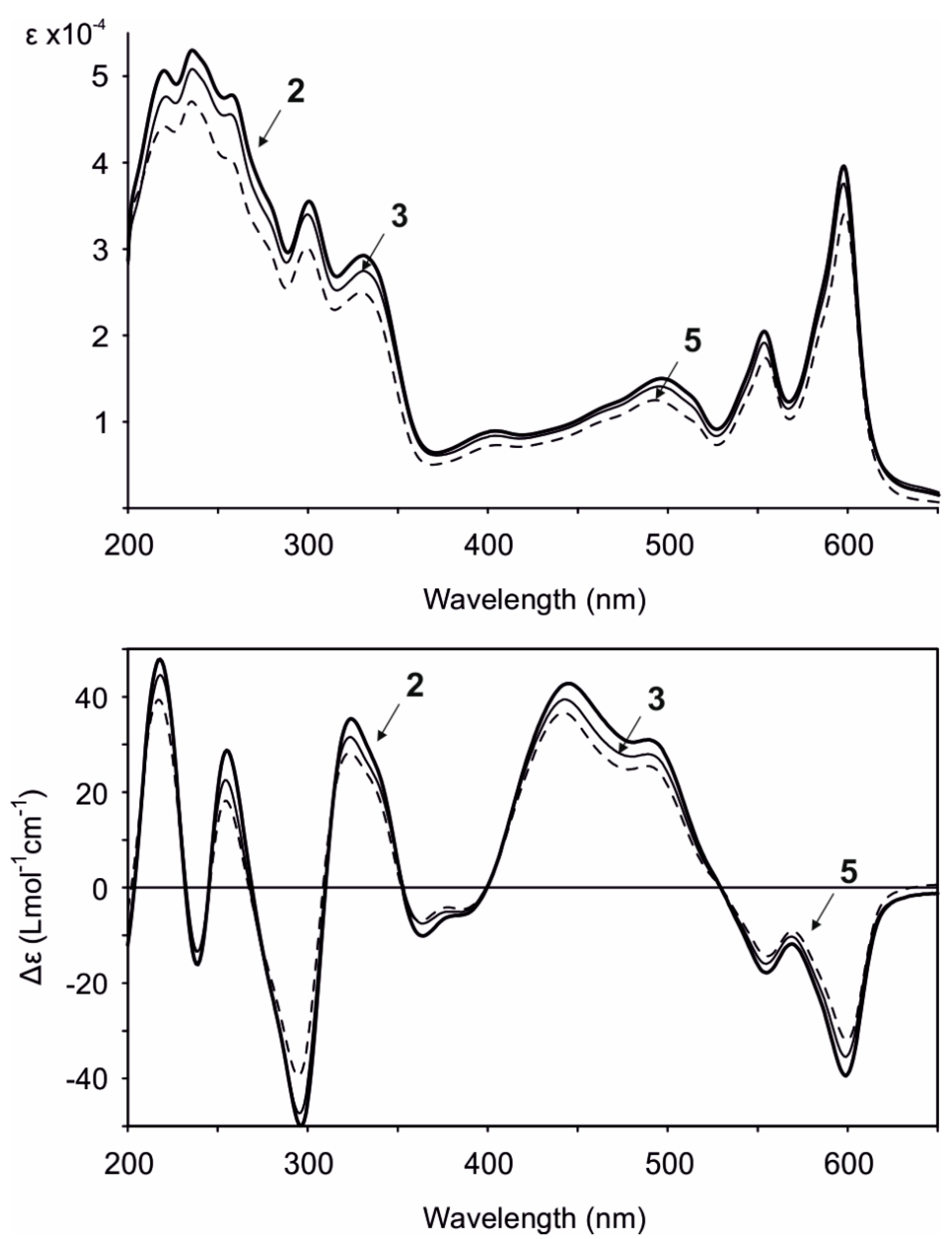
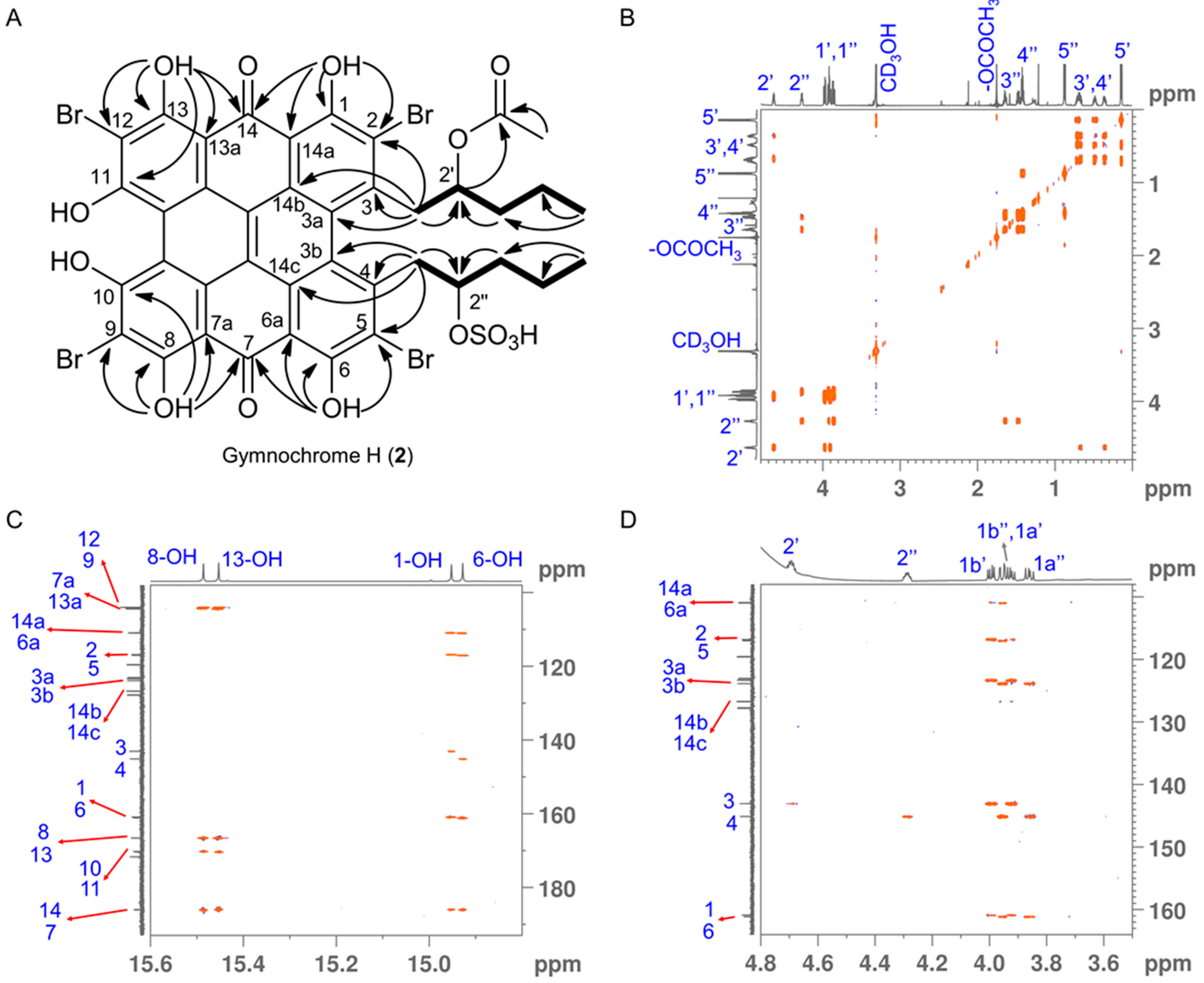
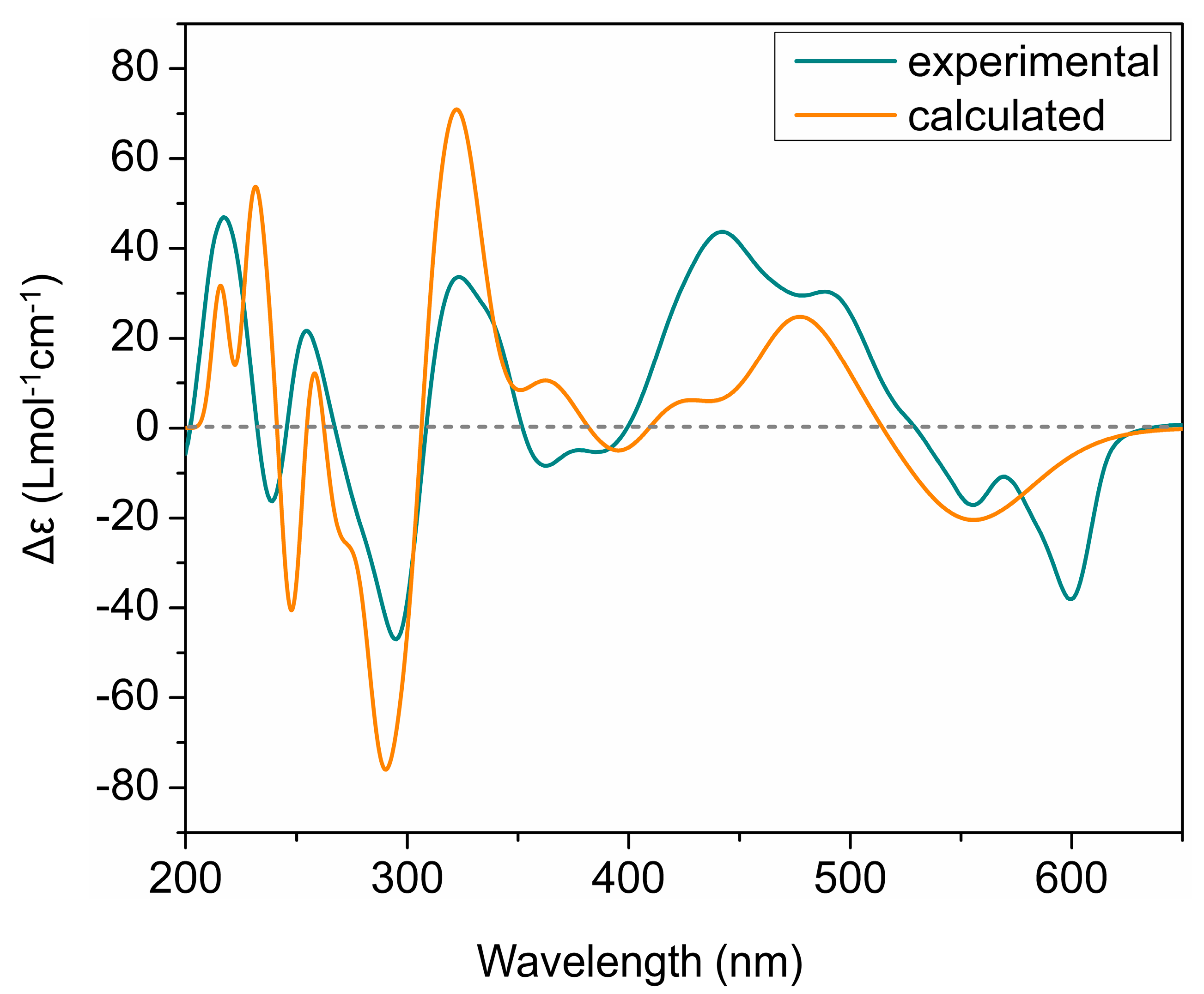
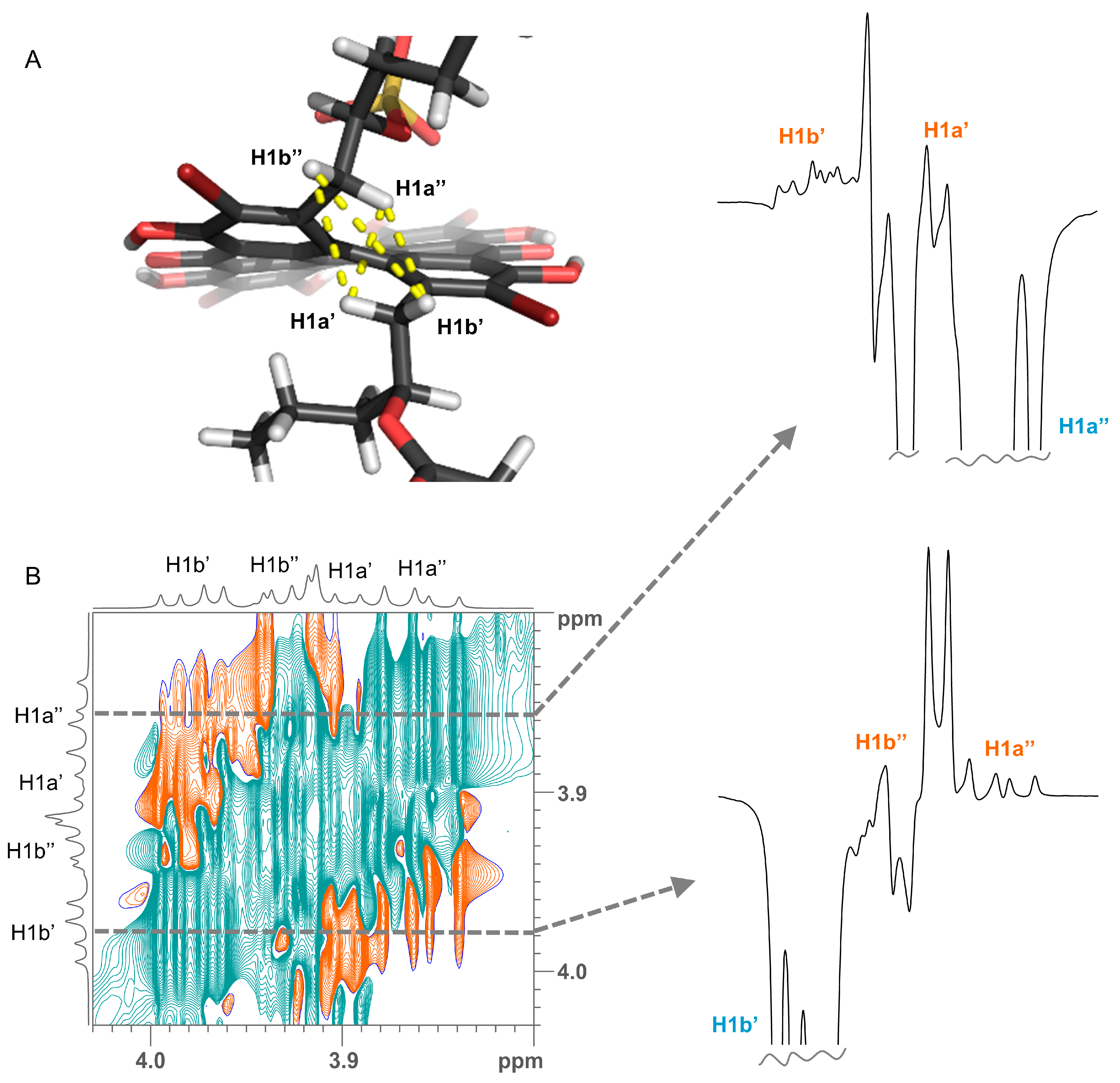
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. DP4+ Analysis
3.5. ECD Calculations
3.6. Biological Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolkenstein, K.; Fuentes-Monteverde, J.C.; Nath, N.; Oji, T.; Griesinger, C. Hypalocrinins, taurine conjugated anthraquinone and biaryl pigments from the deep sea crinoid. Hypalocrinus Naresianus J. Nat. Prod. 2019, 82, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Wolkenstein, K. Persistent and widespread occurrence of bioactive quinone pigments during post-Paleozoic crinoid diversification. Proc. Natl. Acad. Sci. USA 2015, 112, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Karschin, N.; Wolkenstein, K.; Griesinger, C. Magnetically induced alignment of natural products for stereochemical structure determination via NMR. Angew. Chem. Int. Ed. 2020, 59, 15860–15864. [Google Scholar] [CrossRef]
- Altmann, R.; Etzlstorfer, C.; Falk, H. Chiroptical properties and absolute configurations of the hypericin chromophore propeller enantiomers. Monatsh. Chem. 1997, 128, 785–793. [Google Scholar] [CrossRef]
- De Riccardis, F.; Iorizzi, M.; Minale, L.; Riccio, R.; Richer de Forges, B.; Debitus, C. The gymnochromes: Novel marine brominated phenanthroperylenequinone pigments from the stalked crinoid Gymnocrinus Richeri. J. Org. Chem. 1991, 56, 6781–6787. [Google Scholar] [CrossRef]
- Horeau, A. Principe et applications d’une nouvelle methode de determination des configurations dite “par dedoublement partiel”. Tetrahedron Lett. 1961, 2, 506–512. [Google Scholar] [CrossRef]
- Nasini, G.; Merlini, L.; Andreetti, G.D.; Bocelli, G.; Sgarabotto, P. Stereochemistry of cercosporin. Tetrahedron 1982, 38, 2787–2796. [Google Scholar] [CrossRef]
- Kemami Wangun, H.V.; Wood, A.; Fiorilla, C.; Reed, J.K.; McCarthy, P.J.; Wright, A.E. Gymnochromes E and F, cytotoxic phenanthroperylenequinones from a deep-water crinoid, Holopus Rangii. J. Nat. Prod. 2010, 73, 712–715. [Google Scholar] [CrossRef] [Green Version]
- Pachler, K.G.R. Nuclear magnetic resonance study of some α-amino acids—I: Coupling constants in alkaline and acidic medium. Spectrochim. Acta 1963, 19, 2085–2092. [Google Scholar] [CrossRef]
- Pachler, K.G.R. Nuclear magnetic resonance study of some α-amino acids—II. Rotational isomerism. Spectrochim. Acta 1964, 20, 581–587. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon−proton spin-coupling constants. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.J.; Gorka, A.P.; Bokesch, H.R.; Wamiru, A.; O’Keefe, B.R.; Schnermann, M.J.; Gustafson, K.R. Harnessing natural product diversity for fluorophore discovery: Naturally occurring fluorescent hydroxyanthraquinones from the marine crinoid Pterometra Venusta. J. Nat. Prod. 2018, 81, 2750–2755. [Google Scholar] [CrossRef]
- Laille, M.; Gerald, F.; Debitus, C. In vitro antiviral activity on dengue virus of marine natural products. Cell. Mol. Life Sci. 1998, 54, 167–170. [Google Scholar] [CrossRef]
- Laurent, D.; Baumann, F.; Benoit, A.G.; Mortelecqe, A.; Nitatpattana, N.; Desvignes, I.; Debitus, C.; Laille, M.; Gonzalez, J.-P.; Chungue, E. Structure-activity relationships of dengue antiviral polycyclic quinones. Southeast Asian J. Trop. Med. Public Health 2005, 36, 901–905. [Google Scholar] [PubMed]
- Hudson, J.B.; Delaey, E.; de Witte, P.A. Bromohypericins are potent photoactive antiviral agents. Photochem. Photobiol. 1999, 70, 820–822. [Google Scholar] [CrossRef]
- Takahashi, D.; Maoka, T.; Tsushima, M.; Fujitani, K.; Kozuka, M.; Matsuno, T.; Shingu, T. New quinone sulfates from the crinoids Tropiometra afra macrodiscus and Oxycomanthus japonicus. Chem. Pharm. Bull. 2002, 50, 1609–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolkenstein, K.; Schoefberger, W.; Müller, N.; Oji, T. Proisocrinins A–F, brominated anthraquinone pigments from the stalked crinoid Proisocrinus ruberrimus. J. Nat. Prod. 2009, 72, 2036–2039. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Khokhar, S.; Davis, R.A. Crinoids: Ancient organisms, modern chemistry. Nat. Prod. Rep. 2017, 34, 571–584. [Google Scholar] [CrossRef]
- Meissner, A.; Sørensen, O.W. Measurement of J(H,H) and long-range J(X,H) coupling constants in small molecules. Broadband XLOC and J-HMBC. Magn. Reson. Chem. 2001, 39, 49–52. [Google Scholar] [CrossRef]
- Edden, R.A.E.; Keeler, J. Development of a method for the measurement of long-range 13C–1H coupling constants from HMBC spectra. J. Magn. Reson. 2004, 166, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2019.
- Schrödinger Release 2017-4: Macromodel; Schrödinger, LLC: New York, NY, USA, 2019.
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W.C.; Still, W.C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mpw and mpw1pw models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good computational practice in the assignment of absolute configurations by TDDFT calculation of ECD spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Carlo, A.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Pescitelli, G. SpecDis Version 1.71, Berlin, Germany. 2017. Available online: http://specdis-software.jimdo.com (accessed on 20 November 2017).
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 3 | 5 | ||||
|---|---|---|---|---|---|---|
| Position | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) |
| 1/6 | 160.7, C/160.9, C | 160.8, C/160.9, C | 160.75, C/160.73, C | |||
| 2/5 | 116.7, C/116.9, C | 116.0, C/116.9, C | 116.2, C/116.6, C | |||
| 3/4 | 142.9, C/144.9, C | 144.1, C/145.4, C | 146.1, C/144.6, C | |||
| 3a/3b | 123.2, C/123.7, C | 123.4, C/124.0, C | 124.1, C/124.4, C | |||
| 6a/14a | 110.84, C/110.75, C | 110.7, C/110.6, C | 111.0, C/110.6, C | |||
| 7/14 | 185.9, C/185.8, C | 185.9, C/185.8, C | 185.9, C/185.5, C | |||
| 7a/13a | 104.2, C/104.3, C | 104.19, C/104.24, C | 104.1, C/104.0 C | |||
| 8/13 | 166.46, C/166.47, C | 166.4, C/166.4, C | 166.0, C/165.9, C | |||
| 9/12 | 103.81, C/103.80, C | 103.82, C/103.80, C | 103.6, C/103.3, C | |||
| 10/11 | 170.3, C/170.4, C | 170.2, C/170.3, C | 169.3, C/169.5, C | |||
| 10a/10b | 119.54, C b/119.48, C b | 119.44, C b/119.43, C b | 118.8, C b/118.8, C b | |||
| 14b/14c | 126.59, C/126.60, C | 126.45, C/126.47, C | 126.3, C/126.7, C | |||
| 14d/14g | 127.7, C b/127.6, C b | 127.7, C b/127.6, C b | 127.5, C b/127.4, C b | |||
| 14e/14f | 123.0, C b/122.9, C b | 122.94, C b/122.91, C b | 123.1, C b/122.9, C b | |||
| 1′ | 42.5 CH2 | 3.91 dd (13.6; 7.9), 3.98 dd (13.7; 6.1) | 48.8, CH2 | 3.62 dd (12.9; 9.8), 4.06 dd (12.8; 3.9) | 48.3, CH2 | 3.68 dd (13.9; 9.0), 3.93 dd (13.9; 1.3) |
| 2′ | 74.1, CH | 4.63 m | 68.2, CH | 3.69 m | 73.1, CH | 3.75 m |
| 2′-OCOCH3 | 171.5, C | |||||
| 2′-OCOCH3 | 20.6, CH3 | 1.75 s | ||||
| 3′ | 35.4, CH2 | 0.36 m, 0.68 m | 21.1, CH3 | –0.15 d (6.0) | 41.2, CH2 | 1.47 m, 1.56 m |
| 4′ | 18.1, CH2 | 0.48 m, 0.71 m | 19.9, CH2 | 1.36 m, 1.43 m | ||
| 5′ | 13.4, CH3 | 0.14 t (7.2) | 14.2, CH3 | 0.90 t (7.2) | ||
| 1″ | 44.2, CH2 | 3.86 dd (14.1; 9.6), 3.93 dd (14.2; 2.4) | 44.1, CH2 | 3.87 m, 3.92 m | 44.1, CH2 | 3.84 m, 3.89 m |
| 2″ | 80.0, CH | 4.27 m | 79.9, CH | 4.30 m | 80.0, CH | 4.27 m |
| 3″ | 38.8, CH2 | 1.47 m, 1.64 m | 38.8, CH2 | 1.49 m, 1.65 m | 38.6, CH2 | 1.39 m, 1.59 m |
| 4″ | 18.9, CH2 | 1.42 m | 18.9, CH2 | 1.43 m | 18.8, CH2 | 1.38 m |
| 5″ | 14.4, CH3 | 0.87 t (7.3) | 14.4, CH3 | 0.88 t (7.3) | 14.4, CH3 | 0.83 t (7.1) |
| 1-OH | 14.92 s | 14.92 s | 14.88 s | |||
| 6-OH | 14.90 s | 14.91 s | 14.93 s | |||
| 8-OH | 15.48 s | 15.49 s | 15.47 s | |||
| 13-OH | 15.43 s | 15.46 s | 15.40 s | |||
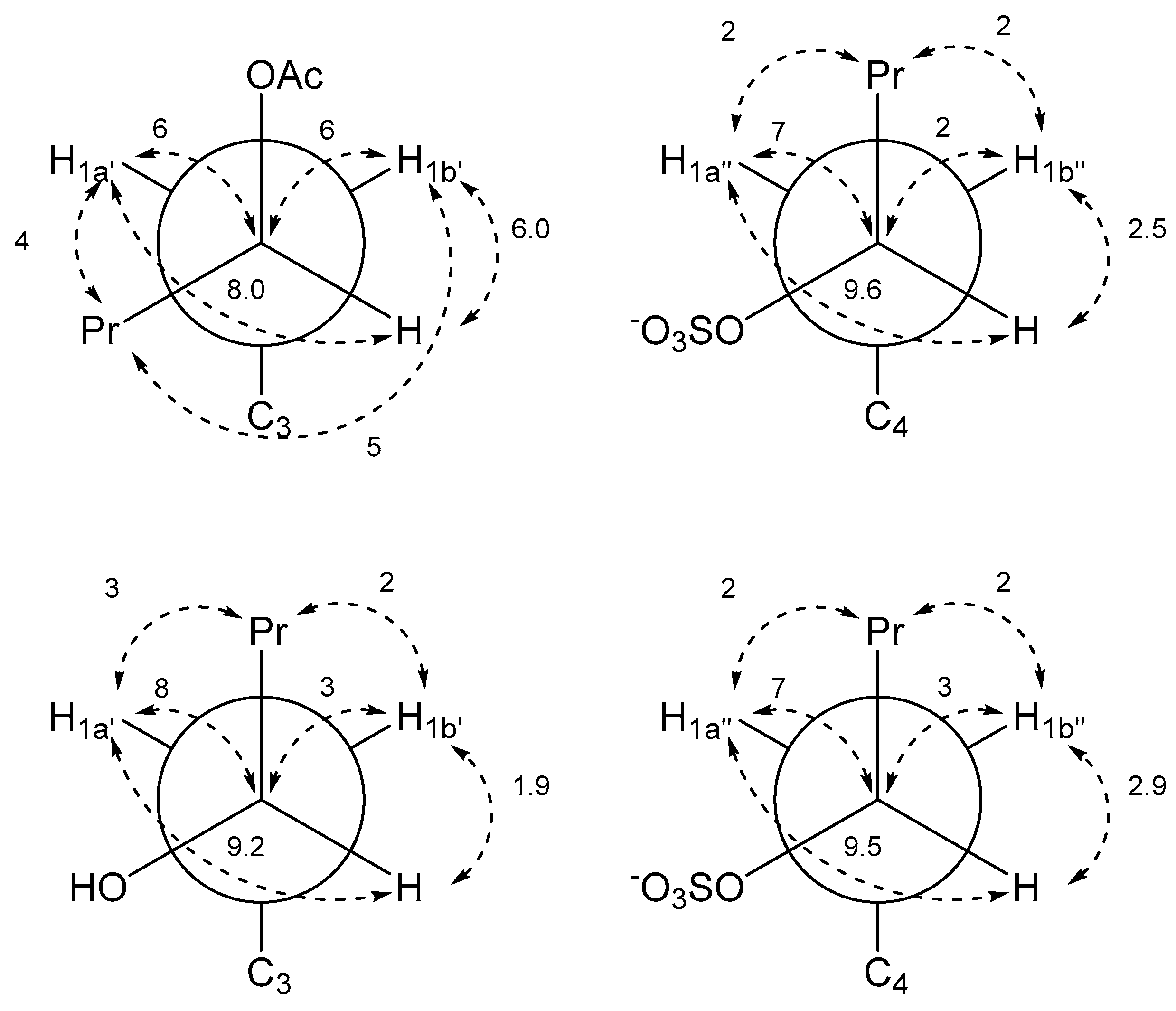
| Pentyl Acetate Side Chain | Pentyl Sulfate Side Chain | ||
|---|---|---|---|
| J-HMBC Correlation | J in Hz a | J-HMBC Correlation | J in Hz a |
| 3JC3a–H1a′ | 5 | 3JC3b–H1a″ | 4 |
| 3JC3a–H1b′ | 5 | 3JC3b–H1b″ | 5 |
| 3JC2–H1a′ | 8 | 3JC5–H1a″ | 8 |
| 3JC2–H1b′ | 5 | 3JC5–H1b″ | 5 |
| 3JH2′–H1a′ | 8.0 | 3JH2″–H1a″ | 9.6 |
| 3JH2′–H1b′ | 6.0 | 3JH2″–H1b″ | 2.5 |
| 3JC3–H2′ | 2 | 3JC4–H2″ | |
| 3JC3′–H1a′ | 4 | 3JC3″–H1a″ | 2 |
| 3JC3′–H1b′ | 5 | 3JC3″–H1b″ | 2 |
| 2JC2′–H1a′ | 6 | 2JC2″–H1a″ | 7 |
| 2JC2′–H1b′ | 6 | 2JC2″–H1b″ | 2 |
| Hydroxypentyl Side Chain | Pentyl Sulfate Side Chain | ||
|---|---|---|---|
| J-HMBC Correlation | J in Hz a | J-HMBC Correlation | J in Hz a |
| 3JC3a–H1a′ | 4 | 3JC3b–H1a″ | 4 |
| 3JC3a–H1b′ | 5 | 3JC3b–H1b″ | 5 |
| 3JC2–H1a′ | 8 | 3JC5–H1a″ | 8 |
| 3JC2–H1b′ | 4 | 3JC5–H1b″ | 4 |
| 3JH2′–H1a′ | 9.2 | 3JH2″–H1a″ | 9.5 |
| 3JH2′–H1b′ | 1.9 | 3JH2″–H1b″ | 2.9 |
| 3JC3–H2′ | 3JC4–H2″ | ||
| 3JC3′–H1a′ | 3 | 3JC3″–H1a″ | 2 |
| 3JC3′–H1b′ | 2 | 3JC3″–H1b″ | 2 |
| 2JC2′–H1a′ | 8 | 2JC2″–H1a″ | 7 |
| 2JC2′–H1b′ | 3 | 2JC2″–H1b″ | 3 |
| (2′R,2″R) | (2′R,2″S) | (2′S,2″R) | (2′S,2″S) | |
|---|---|---|---|---|
| Gymnochrome H (2) | 0.00 | 0.00 | 97.36 | 2.64 |
| Monosulfated gymnochrome A (3) | 0.00 | 0.00 | 100.00 | 0.00 |
| Monosulfated gymnochrome D (5) | 99.99 | 0.01 | 0.00 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vemulapalli, S.P.B.; Fuentes-Monteverde, J.C.; Karschin, N.; Oji, T.; Griesinger, C.; Wolkenstein, K. Structure and Absolute Configuration of Phenanthro-perylene Quinone Pigments from the Deep-Sea Crinoid Hypalocrinus naresianus. Mar. Drugs 2021, 19, 445. https://doi.org/10.3390/md19080445
Vemulapalli SPB, Fuentes-Monteverde JC, Karschin N, Oji T, Griesinger C, Wolkenstein K. Structure and Absolute Configuration of Phenanthro-perylene Quinone Pigments from the Deep-Sea Crinoid Hypalocrinus naresianus. Marine Drugs. 2021; 19(8):445. https://doi.org/10.3390/md19080445
Chicago/Turabian StyleVemulapalli, Sahithya Phani Babu, Juan Carlos Fuentes-Monteverde, Niels Karschin, Tatsuo Oji, Christian Griesinger, and Klaus Wolkenstein. 2021. "Structure and Absolute Configuration of Phenanthro-perylene Quinone Pigments from the Deep-Sea Crinoid Hypalocrinus naresianus" Marine Drugs 19, no. 8: 445. https://doi.org/10.3390/md19080445
APA StyleVemulapalli, S. P. B., Fuentes-Monteverde, J. C., Karschin, N., Oji, T., Griesinger, C., & Wolkenstein, K. (2021). Structure and Absolute Configuration of Phenanthro-perylene Quinone Pigments from the Deep-Sea Crinoid Hypalocrinus naresianus. Marine Drugs, 19(8), 445. https://doi.org/10.3390/md19080445