
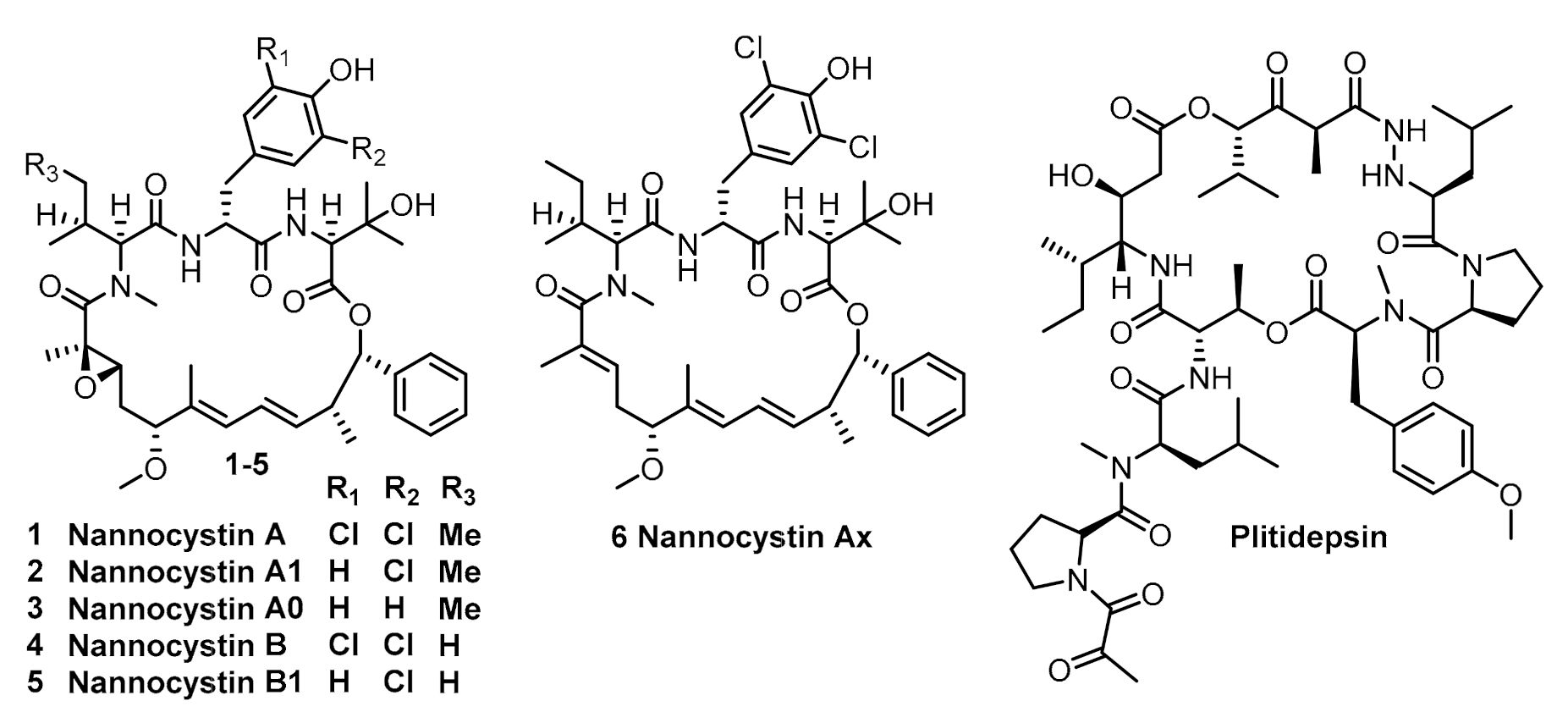
Optimization of Two Steps in Scale-Up Synthesis of Nannocystin A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
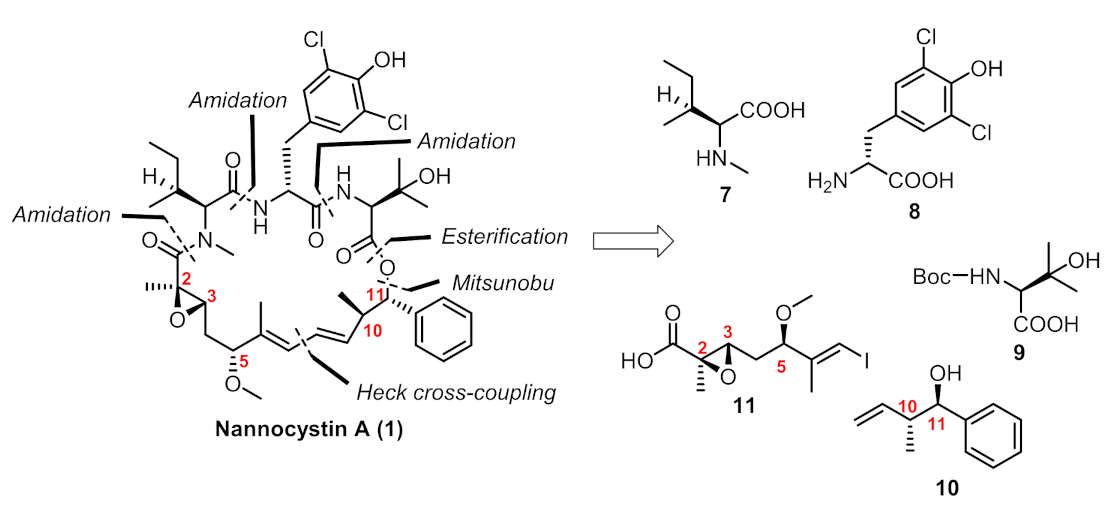
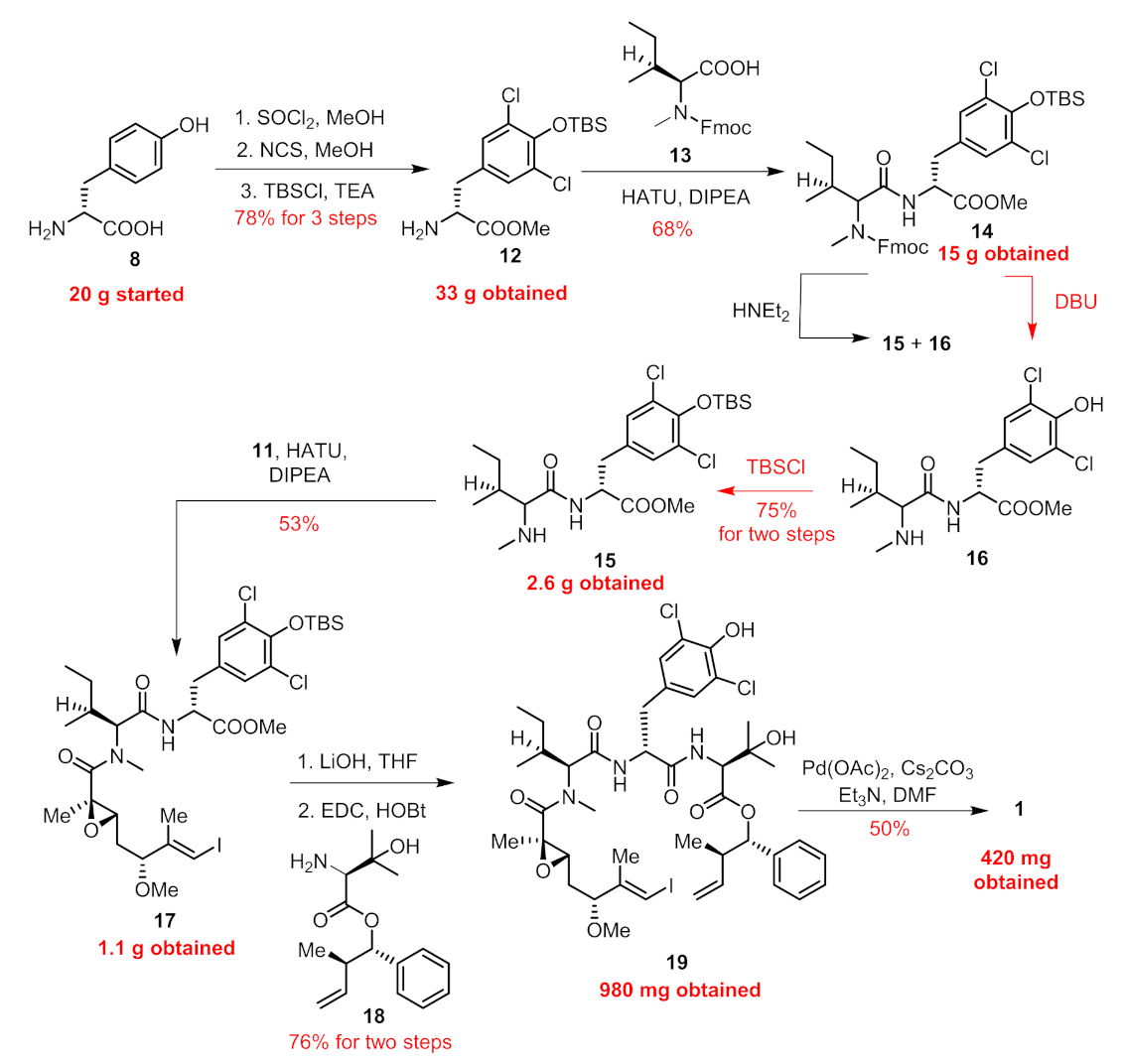
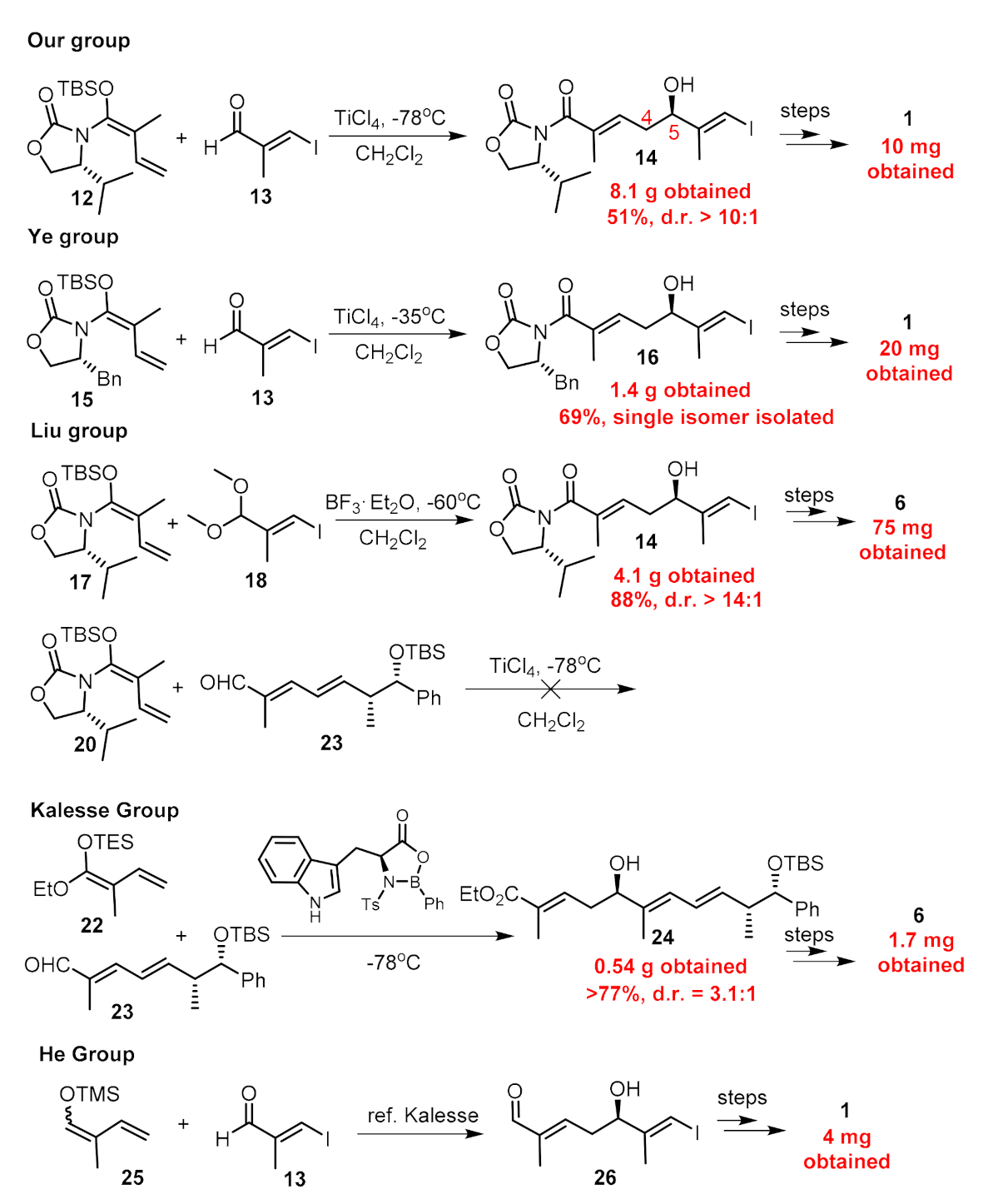
2. Results
Chemistry
3. Materials and Methods
3.1. General Information
3.2. Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schäberle, T.; Goralski, E.; Neu, E.; Erol, Ö.; Hölzl, G.; Dörmann, P.; Bierbaum, G.; König, G. Marine Myxobacteria as a Source of Antibiotics-Comparison of Physiology, Polyketide-Type Genes and Antibiotic Production of Three New Isolates of Enhygromyxa salina. Mar. Drugs 2010, 8, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Albataineh, H.; Stevens, D. Marine Myxobacteria: A Few Good Halophiles. Mar. Drugs 2018, 16, 209. [Google Scholar] [CrossRef]
- Höfle, G.; Bedorf, N.; Gerth, K.; Reichenbach, H. (GBF). German Patent DE 91-4138042, 1993. Chem. Abstr. 1993, 120, 52841. [Google Scholar]
- Lee, F.; Borzilleri, R.; Fairchild, C.; Kim, S.; Long, B.H.; Reventos-Suarez, C.; Vite, G.; Rose, W.; Kramer, R. BMS-247550: A Novel Epothilone Analog with a Mode of Action Similar to Paclitaxel but Possessing Superior Antitumor Efficacy. Clin. Cancer Res. 2001, 7, 1429–1437. [Google Scholar] [PubMed]
- Borzilleri, R.; Zheng, X.; Schmidt, R.; Johnson, J.; Kim, S.; DiMarco, J.; Fairchild, C.R.; Gougoutas, J.Z.; Lee, F.Y.; Long, B.H.; et al. A Novel Application of a Pd(0)-Catalyzed Nucleophilic Substitution Reaction to the Regio- and Stereoselective Synthesis of Lactam Analogues of the Epothilone Natural Products. J. Am. Chem. Soc. 2000, 122, 8890–8897. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Krastel, P.; Roggo, S.; Schirle, M.; Ross, N.T.; Perruccio, F.; Aspesi, P.; Aust, T.; Buntin, K.; Estoppey, D.; Liechty, B.; et al. Nannocystin A: An Elongation Factor 1 Inhibitor from Myxobacteria with Differential Anti-Cancer Properties. Angew. Chem. Int. Ed. 2015, 54, 10149–10154. [Google Scholar] [CrossRef]
- Hoffmann, H.; Kogler, H.; Heyse, W.; Matter, H.; Caspers, M.; Schummer, D.; Klemke-Jahn, C.; Bauer, A.; Penarier, G.; Debussche, L.; et al. Discovery, Structure Elucidation, and Biological Characterization of Nannocystin A, a Macrocyclic Myxobacterial Metabolite with Potent Antiproliferative Properties. Angew. Chem. Int. Ed. 2015, 54, 10145–10148. [Google Scholar] [CrossRef]
- Schoffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopez-Martin, J.A.; Pollard, P.; Dumez, H.; del Muro, X.G.; et al. Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma. Mar. Drugs 2009, 7, 57–70. [Google Scholar] [CrossRef]
- Eisen, T.; Thomas, J.; Miller, W.H., Jr.; Gore, M.; Wolter, P.; Kavan, P.; Martin, J.A.; Lardelli, P. Phase II Study of Biweekly Plitidepsin as Second-line Therapy in Patients with Advanced Malignant Melanoma. Melanoma Res. 2009, 19, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Concise Total Synthesis of Nannocystin A. Angew. Chem. Int. Ed. 2016, 55, 13263–13266. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, Z. Total Syntheses of Nannocystins A and A0, Two Elongation Factor 1 Inhibitors. Org. Lett. 2016, 18, 4702–4705. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, X.; Yang, C.-H.; Tian, Y.; Chen, X.; Lian, L.; Pan, W.; Su, X.; Zhang, W.; Chen, Y. Total Synthesis of Nannocystin A. Org. Lett. 2016, 18, 5768–5770. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Liu, B. Total synthesis of nannocystin Ax. Chem. Commun. 2017, 53, 5549–5552. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, P.; He, Y. Asymmetric Total Synthesis of Nannocystin A. J. Org. Chem. 2017, 82, 9217–9222. [Google Scholar] [CrossRef] [PubMed]
- Poock, C.; Kalesse, M. Total Synthesis of Nannocystin Ax. Org. Lett. 2017, 19, 4536–4539. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Souillart, L.; Monks, B.; Huwyler, N.; Herrmann, J.; Müller, R.; Fürstner, A. A “Motif-Oriented”Total Synthesis of Nannocystin Ax. Preparation and Biological Assessment of Analogues. J. Org. Chem. 2017, 83, 6977–6994. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. The Chemical Syntheses of Nannocystins. Synthesis 2019, 51, 2252–2260. [Google Scholar] [CrossRef]
- Zhang, W. From Target-Oriented to Motif-Oriented: A Case Study on Nannocystin Total Synthesis. Molecules 2020, 25, 5327. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xia, M.; Zhang, Y.; Fu, S.; Liu, B. The journey of total synthesis toward nannocystin Ax. Tetrahedron 2019, 75, 1781–1794. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, X.; Ding, Y.; Hao, X.; Bai, Y.; Tang, Y.; Zhang, X.; Li, Q.; Yang, Z.; Zhang, W.; et al. Synthesis and Biological Evaluation of Nannocystin Analogues toward Understanding the Binding Role of the (2R,3S)-Epoxide in Nannocystin A. Eur. J. Med. Chem. 2018, 150, 626–632. [Google Scholar] [CrossRef]
- Tian, Y.; Ding, Y.; Xu, X.; Bai, Y.; Tang, Y.; Hao, X.; Zhang, W.; Chen, Y. Total Synthesis and Biological Evaluation of Nannocystin Analogues Modified at the Polyketide Phenyl Moiety. Tetrahedron Lett. 2018, 59, 3206–3209. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.; Liu, W.; Yuan, X.; Tang, Y.; Li, J.; Chen, Y.; Zhang, W. Stereodivergent Total Synthesis of Br-Nannocystins Underpinning the Polyketide (10R,11S) Configuration as a Key Determinant of Potency. J. Mol. Struct. 2019, 1181, 568–578. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, X.; Ji, J.; Zhang, S.-L.; He, Y. Novel Nannocystin A Analogues as Anticancer Therapeutics: Synthesis, Biological Evaluations and Structure-activity Relationship Studies. Eur. J. Med. Chem. 2019, 170, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Shirokawa, S.-I.; Kamiyama, M.; Nakamura, T.; Okada, M.; Nakazaki, A.; Hosokawa, S.; Kobayashi, S. Remote Asymmetric Induction with Vinylketene Silyl N,O-Acetal. J. Am. Chem. Soc. 2004, 126, 13604–13605. [Google Scholar] [CrossRef] [PubMed]
- Keck, G.E.; Krishnamurthy, D. Pronounced Solvent and Concentration Effects in an Enantioselective Mukaiyama Aldol Condensation Using BINOL-Titanium(IV) Catalysts. J. Am. Chem. Soc. 1995, 117, 2363–2364. [Google Scholar] [CrossRef]
- Xiang, J.; Ding, Y.; Li, J.; Zhao, X.; Sun, Y.; Wang, D.; Wang, L.; Chen, Y. Ovatodiolides: Scalable Protection-Free Syntheses, Configuration Determination, and Biological Evaluation against Hepatic Cancer Stem Cells. Angew. Chem. Int. Ed. 2019, 58, 10587–10590. [Google Scholar] [CrossRef]
- Kakei, H.; Tsuji, R.; Ohshima, T.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. Catalytic Asymmetric Epoxidation of a,b-Unsaturated Esters with Chiral Yttrium–Biaryldiol Complexes. Chem. Asian J. 2007, 2, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Candu, N.; Rizescu, C.; Podolean, I.; Tudorache, M.; Parvulescu, V.; Coman, S. Efficient Magnetic and Recyclable SBILC (Supported Basic Ionic Liquid Catalyst)-Based Heterogeneous Organocatalysts for the Asymmetric Epoxidation of Trans-Methylcinnamate. Catal. Sci. Technol. 2015, 5, 729–737. [Google Scholar] [CrossRef]
- Wu, X.; She, X.; Shi, Y. Highly Enantioselective Epoxidation of α,β-Unsaturated Esters by Chiral Dioxirane. J. Am. Chem. Soc. 2002, 124, 8792–8793. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Miao, S.; Zhang, M.; Liu, W.; Wang, L.; Chen, Y. Optimization of Two Steps in Scale-Up Synthesis of Nannocystin A. Mar. Drugs 2021, 19, 198. https://doi.org/10.3390/md19040198
Zhang T, Miao S, Zhang M, Liu W, Wang L, Chen Y. Optimization of Two Steps in Scale-Up Synthesis of Nannocystin A. Marine Drugs. 2021; 19(4):198. https://doi.org/10.3390/md19040198
Chicago/Turabian StyleZhang, Tingrong, Shaojie Miao, Mingxiao Zhang, Wenjie Liu, Liang Wang, and Yue Chen. 2021. "Optimization of Two Steps in Scale-Up Synthesis of Nannocystin A" Marine Drugs 19, no. 4: 198. https://doi.org/10.3390/md19040198
APA StyleZhang, T., Miao, S., Zhang, M., Liu, W., Wang, L., & Chen, Y. (2021). Optimization of Two Steps in Scale-Up Synthesis of Nannocystin A. Marine Drugs, 19(4), 198. https://doi.org/10.3390/md19040198