Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy




Abstract
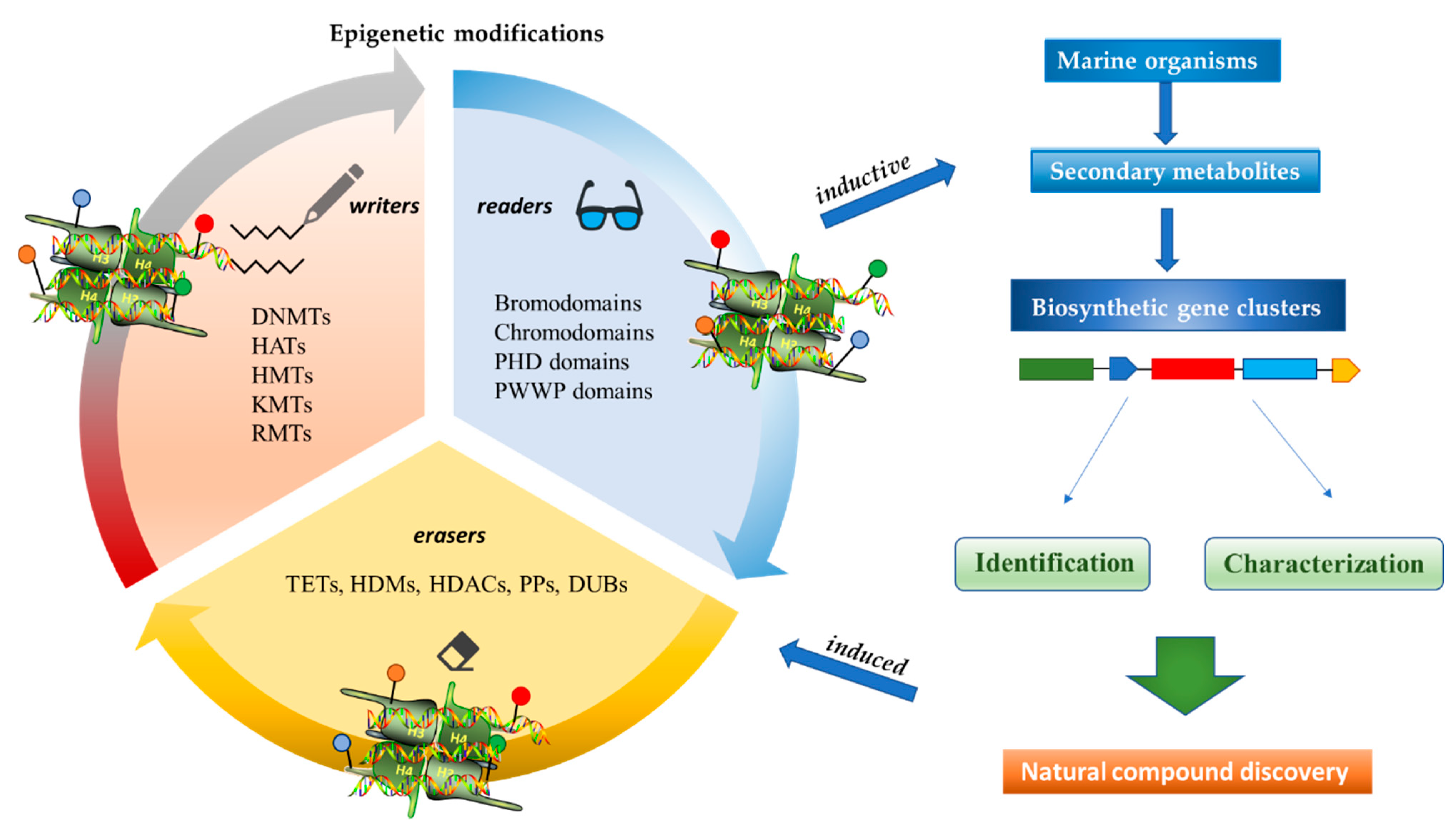
1. Introduction
2. Anticancer Activities of Marine-Derived Secondary Metabolites with Inductive and Induced Epigenetic Modifications
3. Sustainability and Health
4. (Poly)phenolic Compounds
4.1. Psammaplin A
4.2. Indole-Derived Psammaplin a Analogues
4.2.1. UVI5008
4.2.2. Panobinostat
4.2.3. NVP-LAQ824
4.2.4. Trichostatin A
4.2.5. Vorinostat
5. Cyclic Peptides
5.1. Romidepsin
5.2. Plitidepsin
5.3. Largazole
5.4. Azumamides
5.5. Trapoxins
5.6. Apicidin
6. Alkaloids
6.1. Brominated Alkaloids: Isofistularin-3
6.2. Bispyridinium Alkaloids: Cyclostellettamines
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Poli, A.; Finore, I.; Romano, I.; Gioiello, A.; Lama, L.; Nicolaus, B. Microbial Diversity in Extreme Marine Habitats and Their Biomolecules. Microorganisms 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Firn, R.D.; Jones, C.G. The evolution of secondary metabolism-a unifying model. Mol. Microbiol. 2000, 37, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Coppola, D.; Russo, R.; Denaro, R.; Giuliano, L.; Lauro, F.M.; di Prisco, G.; Verde, C. Marine Microbial Secondary Metabolites: Pathways, Evolution and Physiological Roles. Adv. Microb. Physiol. 2015, 66, 357–428. [Google Scholar] [PubMed]
- Carneiro, V.C.; Lyko, F. Rapid Epigenetic Adaptation in Animals and Its Role in Invasiveness. Integr. Comp. Biol. 2020, 60, 267–274. [Google Scholar] [CrossRef]
- Mirbahai, L.; Chipman, J.K. Epigenetic memory of environmental organisms: A reflection of lifetime stressor exposures. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 10–17. [Google Scholar] [CrossRef]
- Jeremias, G.; Barbosa, J.; Marques, S.M.; Asselman, J.; Goncalves, F.J.M.; Pereira, J.L. Synthesizing the role of epigenetics in the response and adaptation of species to climate change in freshwater ecosystems. Mol. Ecol. 2018, 27, 2790–2806. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Pinto, D. Plant Secondary Metabolites as Anticancer Agents: Successes in Clinical Trials and Therapeutic Application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef]
- Kiuru, P.; D’Auria, M.V.; Muller, C.D.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring marine resources for bioactive compounds. Planta Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-Derived Pharmaceuticals-Challenges and Opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef]
- Sun, W.; Wu, W.; Liu, X.; Zaleta-Pinet, D.A.; Clark, B.R. Bioactive Compounds Isolated from Marine-Derived Microbes in China: 2009–2018. Mar. Drugs 2019, 17, 339. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Brickner, J.H. Mechanisms of epigenetic memory. Trends Genet. 2014, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.L.; Marcato, P. Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.Y.; Kim, H.; Li, W.; Kong, A.N. Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr. Top. Med. Chem. 2016, 16, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Torres, I.O.; Fujimori, D.G. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr. Opin. Struct. Biol. 2015, 35, 68–75. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Bosch, T.C.; Adamska, M.; Augustin, R.; Domazet-Loso, T.; Foret, S.; Fraune, S.; Funayama, N.; Grasis, J.; Hamada, M.; Hatta, M.; et al. How do environmental factors influence life cycles and development? An experimental framework for early-diverging metazoans. Bioessays 2014, 36, 1185–1194. [Google Scholar] [CrossRef]
- Dias, B.G.; Maddox, S.; Klengel, T.; Ressler, K.J. Epigenetic mechanisms underlying learning and the inheritance of learned behaviors. Trends Neurosci. 2015, 38, 96–107. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Pinto, D. Biological Potential and Medical Use of Secondary Metabolites. Medicines 2019, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, I.; Mahajan, S.; Tyagi, M.; Hoffmann, L.; Sanejouand, Y.H.; Srinivasan, N.; de Brevern, A.G.; Cadet, F.; Offmann, B. Knowledge-based prediction of protein backbone conformation using a structural alphabet. PLoS ONE 2017, 12, e0186215. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Xiao, M.; Zhao, J.; Li, Z.; Xing, B.; Li, X.; Kong, M.; Li, L.; Zhang, Q.; Liu, Y.; et al. An Overview of Plant Phenolic Compounds and Their Importance in Human Nutrition and Management of Type 2 Diabetes. Molecules 2016, 21, 1374. [Google Scholar] [CrossRef] [PubMed]
- Baral, B.; Akhgari, A.; Metsa-Ketela, M. Activation of microbial secondary metabolic pathways: Avenues and challenges. Synth. Syst. Biotechnol. 2018, 3, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, D.; Tanaka, S.; Hasegawa, D.; Iwamoto, S.; Hiramatsu, H.; Kiyokawa, N.; Miyachi, H.; Horibe, K.; Saito, A.M.; Taga, T.; et al. Evaluation of high-dose cytarabine in induction therapy for children with de novo acute myeloid leukemia: A study protocol of the Japan Children’s Cancer Group Multi-Center Seamless Phase II-III Randomized Trial (JPLSG AML-12). Jpn. J. Clin. Oncol. 2018, 48, 587–593. [Google Scholar] [CrossRef]
- Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules 2020, 25, 1223. [Google Scholar] [CrossRef]
- McDowell, G.C., 2nd; Pope, J.E. Intrathecal Ziconotide: Dosing and Administration Strategies in Patients with Refractory Chronic Pain. Neuromodulation 2016, 19, 522–532. [Google Scholar] [CrossRef]
- Czyz, K.; Sokola-Wysoczanska, E.; Bodkowski, R.; Cholewinska, P.; Wyrostek, A. Dietary Omega-3 Source Effect on the Fatty Acid Profile of Intramuscular and Perimuscular Fat-Preliminary Study on a Rat Model. Nutrients 2020, 12, 3382. [Google Scholar] [CrossRef]
- Nakano, K.; Hayakawa, K.; Funauchi, Y.; Tanizawa, T.; Ae, K.; Matsumoto, S.; Tomomatsu, J.; Ono, M.; Taira, S.; Nishizawa, M.; et al. Differences in the efficacy and safety of eribulin in patients with soft tissue sarcoma by histological subtype and treatment line. Mol. Clin. Oncol. 2020, 14, 13. [Google Scholar] [CrossRef]
- Oberic, L.; Delzor, F.; Protin, C.; Perriat, S.; Laurent, C.; Grand, A.; Canonge, J.M.; Borel, C.; Gauthier, M.; Ysebaert, L.; et al. Brentuximab vedotin in real life, a seven year experience in patients with refractory/relapsed CD30+ T cell lymphoma. J. Oncol. Pharm. Pract. 2020, 1078155220968615. [Google Scholar] [CrossRef] [PubMed]
- Eccles, R.; Winther, B.; Johnston, S.L.; Robinson, P.; Trampisch, M.; Koelsch, S. Efficacy and safety of iota-carrageenan nasal spray versus placebo in early treatment of the common cold in adults: The ICICC trial. Respir. Res. 2015, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Lee, J.Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.M.; Grandjenette, C.; Kim, J.G.; Yoon, A.Y.; et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Martins, N.; Barros, L. Phenolic Compounds and Its Bioavailability: In Vitro Bioactive Compounds or Health Promoters? Adv. Food Nutr. Res. 2017, 82, 1–44. [Google Scholar] [PubMed]
- Godert, A.M.; Angelino, N.; Woloszynska-Read, A.; Morey, S.R.; James, S.R.; Karpf, A.R.; Sufrin, J.R. An improved synthesis of psammaplin A. Bioorg. Med. Chem. Lett. 2006, 16, 3330–3333. [Google Scholar] [CrossRef]
- Jing, Q.; Hu, X.; Ma, Y.; Mu, J.; Liu, W.; Xu, F.; Li, Z.; Bai, J.; Hua, H.; Li, D. Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification. Mar. Drugs 2019, 17, 384. [Google Scholar] [CrossRef]
- Kim, D.; Lee, I.S.; Jung, J.H.; Yang, S.I. Psammaplin A, a natural bromotyrosine derivative from a sponge, possesses the antibacterial activity against methicillin-resistant Staphylococcus aureus and the DNA gyrase-inhibitory activity. Arch. Pharm. Res. 1999, 22, 25–29. [Google Scholar] [CrossRef]
- Jiang, Y.; Ahn, E.Y.; Ryu, S.H.; Kim, D.K.; Park, J.S.; Yoon, H.J.; You, S.; Lee, B.J.; Lee, D.S.; Jung, J.H. Cytotoxicity of psammaplin A from a two-sponge association may correlate with the inhibition of DNA replication. BMC Cancer 2004, 4, 70. [Google Scholar] [CrossRef]
- Shim, J.S.; Lee, H.S.; Shin, J.; Kwon, H.J. Psammaplin A, a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004, 203, 163–169. [Google Scholar] [CrossRef]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tsubuki, M.; Tani, H.; et al. Psammaplin A inhibits hepatitis C virus NS3 helicase. J. Nat. Med. 2013, 67, 765–772. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Kang, J.K.; Lee, E.K.; Lee, J.C.; Lee, S.H.; Jeon, Y.J.; Koh, D.H.; Ahn, S.H.; Seo, D.W.; Lee, H.Y.; et al. Histone deacetylase inhibitor apicidin downregulates DNA methyltransferase 1 expression and induces repressive histone modifications via recruitment of corepressor complex to promoter region in human cervix cancer cells. Oncogene 2008, 27, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.D.; Li, J.; Du, L.; Mahdi, F.; Le, T.P.; Chen, W.L.; Swanson, S.M.; Watabe, K.; Nagle, D.G. Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins. Mar. Drugs 2018, 16, 442. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.H.; Chie, E.K.; Young, P.D.; Kim, I.A.; Kim, I.H. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol. 2012, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef]
- Baud, M.G.; Leiser, T.; Petrucci, V.; Gunaratnam, M.; Neidle, S.; Meyer-Almes, F.J.; Fuchter, M.J. Thioester derivatives of the natural product psammaplin A as potent histone deacetylase inhibitors. Beilstein J. Org. Chem. 2013, 9, 81–88. [Google Scholar] [CrossRef]
- Pereira, R.; Benedetti, R.; Pérez-Rodríguez, S.; Nebbioso, A.; García-Rodríguez, J.; Carafa, V.; Stuhldreier, M.; Conte, M.; Rodríguez-Barrios, F.; Stunnenberg, H.G.; et al. Indole-derived psammaplin A analogues as epigenetic modulators with multiple inhibitory activities. J. Med. Chem. 2012, 55, 9467–9491. [Google Scholar] [CrossRef]
- Mujumdar, P.; Teruya, K.; Tonissen, K.F.; Vullo, D.; Supuran, C.T.; Peat, T.S.; Poulsen, S.A. An Unusual Natural Product Primary Sulfonamide: Synthesis, Carbonic Anhydrase Inhibition, and Protein X-ray Structures of Psammaplin C. J. Med. Chem. 2016, 59, 5462–5470. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, D.; Sun, D.; Yang, S.; Hu, G.; Wu, Z.; Zhao, L. Synthesis of the marine bromotyrosine psammaplin F and crystal structure of a psammaplin A analogue. Molecules 2010, 15, 8784–8795. [Google Scholar] [CrossRef]
- Piña, I.C.; Gautschi, J.T.; Wang, G.Y.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the sponge Pseudoceratina purpurea: Inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef]
- Park, Y.; Liu, Y.; Hong, J.; Lee, C.O.; Cho, H.; Kim, D.K.; Im, K.S.; Jung, J.H. New bromotyrosine derivatives from an association of two sponges, Jaspis wondoensis and Poecillastra wondoensis. J. Nat. Prod. 2003, 66, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Pereira, R.; Khanwalkar, H.; Matarese, F.; García-Rodríguez, J.; Miceli, M.; Logie, C.; Kedinger, V.; Ferrara, F.; Stunnenberg, H.G.; et al. Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Mol. Cancer Ther. 2011, 10, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Mayr, C.; Kiesslich, T.; Illig, R.; Neureiter, D.; Alinger, B.; Ganslmayer, M.; Wissniowski, T.; Fazio, P.D.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat affects angiogenesis in hepatocellular carcinoma models via modulation of CTGF expression. Int. J. Oncol. 2015, 47, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Helland, O.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjorge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Remiszewski, S.W. The discovery of NVP-LAQ824: From concept to clinic. Curr. Med. Chem. 2003, 10, 2393–2402. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Fu, A.; Osusky, K.; Huamani, J.; Hallahan, D.E.; Geng, L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs 2007, 18, 793–800. [Google Scholar] [CrossRef]
- Fuino, L.; Bali, P.; Wittmann, S.; Donapaty, S.; Guo, F.; Yamaguchi, H.; Wang, H.G.; Atadja, P.; Bhalla, K. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol. Cancer Ther. 2003, 2, 971–984. [Google Scholar]
- Atadja, P.; Gao, L.; Kwon, P.; Trogani, N.; Walker, H.; Hsu, M.; Yeleswarapu, L.; Chandramouli, N.; Perez, L.; Versace, R.; et al. Selective growth inhibition of tumor cells by a novel histone deacetylase inhibitor, NVP-LAQ824. Cancer Res. 2004, 64, 689–695. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, F.; Woan, K.; Sahakian, E.; Merino, O.; Rock-Klotz, J.; Vicente-Suarez, I.; Pinilla-Ibarz, J.; Wright, K.L.; Seto, E.; et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J. Immunol. 2011, 186, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.A.; Soliman, K.F.A. Whole-transcriptomic Profile of SK-MEL-3 Melanoma Cells Treated with the Histone Deacetylase Inhibitor: Trichostatin, A. Cancer Genom. Proteom. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.; Rymarchyk, S.; Zheng, S.; Cen, Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 2018, 638, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Hirokawa, T.; Takagi, M.; Shin-ya, K. Trichostatin analogues JBIR-109, JBIR-110, and JBIR-111 from the marine sponge-derived Streptomyces sp. RM72. J. Nat. Prod. 2012, 75, 285–289. [Google Scholar] [CrossRef]
- Ueda, J.Y.; Hwang, J.H.; Maeda, S.; Kato, T.; Ochiai, A.; Isshiki, K.; Yoshida, M.; Takagi, M.; Shin-ya, K. JBIR-17, a novel trichostatin analog from Streptomyces sp. 26634. J. Antibiot. 2009, 62, 283–285. [Google Scholar] [CrossRef]
- Codd, R.; Braich, N.; Liu, J.; Soe, C.Z.; Pakchung, A.A. Zn(II)-dependent histone deacetylase inhibitors: Suberoylanilide hydroxamic acid and trichostatin A. Int. J. Biochem. Cell Biol. 2009, 41, 736–739. [Google Scholar] [CrossRef]
- Makena, M.R.; Nguyen, T.H.; Koneru, B.; Hindle, A.; Chen, W.H.; Verlekar, D.U.; Kang, M.H.; Reynolds, C.P. Vorinostat and fenretinide synergize in preclinical models of T-cell lymphoid malignancies. Anticancer Drugs 2020, 32, 34–43. [Google Scholar] [CrossRef]
- Janku, F.; Park, H.; Call, S.G.; Madwani, K.; Oki, Y.; Subbiah, V.; Hong, D.S.; Naing, A.; Velez-Bravo, V.M.; Barnes, T.G.; et al. Safety and Efficacy of Vorinostat Plus Sirolimus or Everolimus in Patients with Relapsed Refractory Hodgkin Lymphoma. Clin. Cancer Res. 2020, 26, 5579–5587. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.; Raslan, S.; Tombuloglu, H.; Shamseddin, A.; Cevik, E.; Said, O.A.; Madyan, E.F.; Senel, M.; Bozkurt, A.; Rehman, S.; et al. Vorinostat-loaded titanium oxide nanoparticles (anatase) induce G2/M cell cycle arrest in breast cancer cells via PALB2 upregulation. 3 Biotech. 2020, 10, 407. [Google Scholar] [CrossRef]
- Kang, D.W.; Hwang, W.C.; Noh, Y.N.; Kang, Y.; Jang, Y.; Kim, J.A.; Min, D.S. Phospholipase D1 is upregulated by vorinostat and confers resistance to vorinostat in glioblastoma. J. Cell Physiol. 2020, 236, 549–560. [Google Scholar] [CrossRef]
- Skelton, W.P., 4th; Turba, E.; Sokol, L. Durable Complete Response to AMG 655 (Conatumumab) and Vorinostat in a Patient with Relapsed Classical Hodgkin Lymphoma: Extraordinary Response from a Phase 1b Clinical Protocol. Clin. Lymphoma Myeloma Leuk. 2020, 20, e944–e946. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Mina, R.; Shah, J.J.; Laubach, J.P.; Nooka, A.K.; Lewis, C.; Gleason, C.; Sharp, C.; Harvey, R.D.; Heffner, L.T.; et al. Phase 1 Trial Evaluating Vorinostat Plus Bortezomib, Lenalidomide, and Dexamethasone in Patients with Newly Diagnosed Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2020, 20, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Shinojima, N.; Hayashi, M.; Nakano, T.; Ichimura, K.; Mukasa, A. Histone deacetylase inhibition enhances the therapeutic effects of methotrexate on primary central nervous system lymphoma. Neurooncol. Adv. 2020, 2, vdaa084. [Google Scholar] [PubMed]
- Terranova-Barberio, M.; Pawlowska, N.; Dhawan, M.; Moasser, M.; Chien, A.J.; Melisko, M.E.; Rugo, H.; Rahimi, R.; Deal, T.; Daud, A.; et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 2020, 11, 3584. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61, 11169–11182. [Google Scholar] [CrossRef]
- Sarojini, V.; Cameron, A.J.; Varnava, K.G.; Denny, W.A.; Sanjayan, G. Cyclic Tetrapeptides from Nature and Design: A Review of Synthetic Methodologies, Structure, and Function. Chem. Rev. 2019, 119, 10318–10359. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Narita, K.; Kikuchi, T.; Watanabe, K.; Takizawa, T.; Oguchi, T.; Kudo, K.; Matsuhara, K.; Abe, H.; Yamori, T.; Yoshida, M.; et al. Total synthesis of the bicyclic depsipeptide HDAC inhibitors spiruchostatins A and B, 5″-epi-spiruchostatin B, FK228 (FR901228) and preliminary evaluation of their biological activity. Chemistry 2009, 15, 11174–11186. [Google Scholar] [CrossRef]
- Narita, K.; Fukui, Y.; Sano, Y.; Yamori, T.; Ito, A.; Yoshida, M.; Katoh, T. Total synthesis of bicyclic depsipeptides spiruchostatins C and D and investigation of their histone deacetylase inhibitory and antiproliferative activities. Eur. J. Med. Chem. 2013, 60, 295–304. [Google Scholar] [CrossRef]
- Fukui, Y.; Narita, K.; Dan, S.; Yamori, T.; Ito, A.; Yoshida, M.; Katoh, T. Total synthesis of burkholdacs A and B and 5,6,20-tri-epi-burkholdac A: HDAC inhibition and antiproliferative activity. Eur. J. Med. Chem. 2014, 76, 301–313. [Google Scholar] [CrossRef]
- Biggins, J.B.; Gleber, C.D.; Brady, S.F. Acyldepsipeptide HDAC inhibitor production induced in Burkholderia thailandensis. Org. Lett. 2011, 13, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Katoh, T. Total Synthesis of Thailandepsin B, a Potent HDAC Inhibitor Isolated from a Microorganism. Chem. Pharm. Bull. 2016, 64, 913–917. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brosowsky, J.; Lutterbeck, M.; Liebich, A.; Keller, M.; Herp, D.; Vogelmann, A.; Jung, M.; Breit, B. Syntheses of Thailandepsin B Pseudo-Natural Products: Access to New Highly Potent HDAC Inhibitors via Late-Stage Modification. Chemistry 2020, 26, 16241–16245. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gambs, C.; Abe, Y.; Wentworth, P., Jr.; Janda, K.D. Total synthesis of the depsipeptide FR-901375. J. Org. Chem. 2003, 68, 8902–8905. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef]
- Yao, L. Aplidin PharmaMar. IDrugs 2003, 6, 246–250. [Google Scholar]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Kurihara, N.; Atkinson, E.G.; Nelson, J.; Miyagawa, K.; Galmarini, C.M.; Roodman, G.D.; Bellido, T. Aplidin (plitidepsin) is a novel anti-myeloma agent with potent anti-resorptive activity mediated by direct effects on osteoclasts. Oncotarget 2019, 10, 2709–2721. [Google Scholar] [CrossRef]
- Borjan, B.; Steiner, N.; Karbon, S.; Kern, J.; Francesch, A.; Hermann, M.; Willenbacher, W.; Gunsilius, E.; Untergasser, G. The Aplidin analogs PM01215 and PM02781 inhibit angiogenesis in vitro and in vivo. BMC Cancer 2015, 15, 738. [Google Scholar] [CrossRef]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic modulators from “The Big Blue”: A treasure to fight against cancer. Cancer Lett. 2014, 351, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Izzo, I.; Maulucci, N.; Bifulco, G.; De Riccardis, F. Total synthesis of azumamides A and E. Angew. Chem. Int. Ed. Engl. 2006, 45, 7557–7560. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Florean, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Chromatin-modifying agents in anti-cancer therapy. Biochimie 2012, 94, 2264–2279. [Google Scholar] [CrossRef]
- Nakao, Y.; Narazaki, G.; Hoshino, T.; Maeda, S.; Yoshida, M.; Maejima, H.; Yamashita, J.K. Evaluation of antiangiogenic activity of azumamides by the in vitro vascular organization model using mouse induced pluripotent stem (iPS) cells. Bioorg. Med. Chem. Lett. 2008, 18, 2982–2984. [Google Scholar] [CrossRef]
- Porter, N.J.; Christianson, D.W. Binding of the Microbial Cyclic Tetrapeptide Trapoxin A to the Class I Histone Deacetylase HDAC8. ACS Chem. Biol. 2017, 12, 2281–2286. [Google Scholar] [CrossRef]
- Yoshida, M.; Furumai, R.; Nishiyama, M.; Komatsu, Y.; Nishino, N.; Horinouchi, S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol. 2001, 48 (Suppl. S1), S20–S26. [Google Scholar] [CrossRef]
- Ahn, M.Y. HDAC inhibitor apicidin suppresses murine oral squamous cell carcinoma cell growth in vitro and in vivo via inhibiting HDAC8 expression. Oncol. Lett. 2018, 16, 6552–6560. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Lee, J.; Na, Y.J.; Choi, W.S.; Lee, B.M.; Kang, K.W.; Kim, H.S. Mechanism of apicidin-induced cell cycle arrest and apoptosis in Ishikawa human endometrial cancer cells. Chem. Biol. Interact. 2009, 179, 169–177. [Google Scholar] [CrossRef]
- Hwang, W.C.; Kang, D.W.; Kang, Y.; Jang, Y.; Kim, J.A.; Min, D.S. Inhibition of phospholipase D2 augments histone deacetylase inhibitor-induced cell death in breast cancer cells. Biol. Res. 2020, 53, 34. [Google Scholar] [CrossRef]
- Gu, W.; Silverman, R.B. Synthesis of (S)-2-Boc-Amino-8-(R)-(tert-butyldimethylsilanyloxy) decanoic acid, a Precursor to the Unusual Amino Acid Residue of the Anticancer Agent Microsporin, B. Tetrahedron Lett. 2011, 52, 5438–5440. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Nagai, K.; Shindoh, N.; Terada, Y.; van Soest, R.W.; Matsunaga, S.; Fusetani, N. Three new cyclostellettamines, which inhibit histone deacetylase, from a marine sponge of the genus Xestospongia. Bioorg. Med. Chem. Lett. 2004, 14, 2617–2620. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Balado, C.; Nebbioso, A.; Rodríguez-Graña, P.; Minichiello, A.; Miceli, M.; Altucci, L.; de Lera, A.R. Bispyridinium dienes: Histone deacetylase inhibitors with selective activities. J. Med. Chem. 2007, 50, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Ratovitski, E.A. Anticancer Natural Compounds as Epigenetic Modulators of Gene Expression. Curr. Genom. 2017, 18, 175–205. [Google Scholar] [CrossRef]
- Howard, E.C.; Sun, S.; Reisch, C.R.; del Valle, D.A.; Bürgmann, H.; Kiene, R.P.; Moran, M.A. Changes in dimethylsulfoniopropionate demethylase gene assemblages in response to an induced phytoplankton bloom. Appl. Environ. Microbiol. 2011, 77, 524–531. [Google Scholar] [CrossRef]
- Bourne, D.G.; Morrow, K.M.; Webster, N.S. Insights into the Coral Microbiome: Underpinning the Health and Resilience of Reef Ecosystems. Annu. Rev. Microbiol. 2016, 70, 317–340. [Google Scholar] [CrossRef]
- Quina, A.S.; Buschbeck, M.; Di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef]
- Ishijima, J.; Iwabe, N.; Masuda, Y.; Watanabe, Y.; Matsuda, Y. Sponge cytogenetics-mitotic chromosomes of ten species of freshwater sponge. Zool. Sci. 2008, 25, 480–486. [Google Scholar] [CrossRef][Green Version]




{kind=link}
{kind=link}
{kind=link}
| Compound | Structural Formula | Chemical Class | Source | Species | Epigenetic Mechanism | Ref |
|---|---|---|---|---|---|---|
| Psammaplin A |  | Phenolic compound | Sponge | Psammaplin aplysilla, Thorectopsammaxana | DNMT inhibition (in vitro) | [36,44,47,50] |
| HDAC inhibition (in vitro) | [37,42,43,46,47,57,58,59,61,64,66] | |||||
| Topoisomerase II inhibition (in vitro) | [39] | |||||
| Inhibition of DNA regulation (in vitro) | [36] | |||||
| Aminopeptidase N inhibition (in vitro) | [40] | |||||
| SIRT1 induction (in vitro) | [47] | |||||
| HDAC inhibition (in vitro) | [46] | |||||
| Increased H3 acetylation (in vitro) | ||||||
| Psammaplin F |  | Phenolic compound | Sponge | Pseudoceratina purpurea | HDAC inhibition (in vitro) | [50] |
| Psammaplin G |  | Phenolic compound | Sponge | Pseudoceratina purpurea | DNMT inhibition (in vitro) | [50] |
| Bisaprasin |  | Phenolic compound | Sponge | Pseudoceratina purpurea | DNMT inhibition (in vitro) | [50] |
| UVI5008 |  | Phenolic compound | Psammaplin derivative | DNMT3a inhibition (in vitro) | [52] | |
| H3 hyperacetylation (ex vivo) | ||||||
| HDAC inhibition (in vitro) | ||||||
| HDAC1–4 inhibition (in vitro) | ||||||
| SIRT inhibition (in vitro) | ||||||
| NVP-LAQ824 (dacinostat) |  | Hydroxamic acid | Psammaplin derivative | HDAC inhibition (in vitro) | [57,59,60] | |
| HDAC inhibition (in vivo) | [57,58] | |||||
| Increased H3 acetylation (in vitro) | [59,60,61] | |||||
| Increased H4 acetylation (in vitro) | [60,61] | |||||
| Trichostatin A |  | Hydroxamic acid | Bacterium | Streptomyces platensis | HDAC inhibition (in vitro) | [62,66] |
| MAPK/MEK/BRAF downregulation (in vitro) | [62] | |||||
| Increased H3 acetylation (in vitro) | [63] | |||||
| JBIR-109 |  | Trichostatin analogue | Sponge | Streptomyces sp. strain RM72 | HDAC inhibition (in vitro) | [64] |
| JBIR-110 |  | Trichostatin analogue | Sponge | Streptomyces sp. strain RM72 | HDAC inhibition (in vitro) | [64] |
| JBIR-111 |  | Trichostatin analogue | Sponge | Streptomyces sp. strain RM72 | HDAC inhibition (in vitro) | [64] |
| JBIR-17 |  | Phenolic compound | Bacterium | Kerria japonica | HDAC inhibition (in vitro) | [65] |
| Panobinostat |  | Phenolic compound | Sponge | Psammaplin aplysilla | Pan-HDAC inhibition (in vitro) | [53,54] |
| HDAC inhibition (in vitro) | [55] | |||||
| Vorinostat |  | Hydroxamic acid | Trichostatin A derivative | Pan HDAC inhibition (in vitro) | [67] | |
| HDAC inhibition (in vitro) | [66,69,70,73] | |||||
| HDAC inhibition (in vivo) | [67,68] | |||||
| mTOR inhibition (in vivo) | [68] | |||||
| PLD-1 upregulation (in vitro) | [70] |
| Compound | Structural Formula | Chemical Class | Source | Species | Epigenetic Mechanism | Ref |
|---|---|---|---|---|---|---|
| Romidepsin |  | Depsipeptide | Bacterium | Chromobacterium violaceum | HDAC inhibition (in vitro) | [80,82,83,84] |
| Plitidepsin |  | Cyclic tetrapeptide | Tunicate | Aplidium albicans | Caspase-3 upregulation (in vitro) | [87] |
| Dephosphorylation of ERK1/2 and 5 (in vitro) | [88] | |||||
| PM01215 |  | Aplidin analogues | p16INK4A induction (in vitro) | [89] | ||
| VEGF downregulation (in vitro) | ||||||
| PM02781 |  | Aplidin analogues | p16INK4A induction (in vitro) | [89] | ||
| Largazole |  | Macrocyclic depsipeptide | Cyanobacterium | Symplocasp | HDAC inhibition (in vitro) | [85,91] |
| HDAC inhibition (in vivo) | [85] | |||||
| Azumamides |  | Cyclic tetrapeptide | Sponge | Mycale izuensis | HDAC inhibition (in vitro) | [92,93,95] |
| Increased H3 acetylation (in vitro) | [94] | |||||
| Trapoxins |  | Cyclic tetrapeptide | Fungus | Helicoma ambiens | Class I HDAC inhibition (in vitro) | [96] |
| HDAC inhibition (in vitro) | [97] | |||||
| Apicidin |  | Cyclic tetrapeptide | Fungus | Fusarium pallidoroseum | HDAC8 inhibition (in vivo) | [98] |
| p21 upregulation (in vitro) | [99] | |||||
| HDAC inhibition (in vitro) | [99,100] | |||||
| DNMT inhibition (in vitro) | [42] | |||||
| HDACs2/3 inhibition (in vitro) | [42] | |||||
| Microsporins A and B |  | Cyclic peptide | Fungus | Microsporum cf. gypseum | HDAC inhibition (in vitro) | [101] |
| HDAC8 inhibition (in vitro) | ||||||
| Isofistularin-3 |  | Alkaloid | Sponge | Aplysina aerophoba | DNMT1 inhibition (in vitro) | [33] |
| Cyclostellettamines |  | Alkaloid | Sponge | Xestospongia | HDAC inhibition (in vitro) | [102,103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, M.; Fontana, E.; Nebbioso, A.; Altucci, L. Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy. Mar. Drugs 2021, 19, 15. https://doi.org/10.3390/md19010015
Conte M, Fontana E, Nebbioso A, Altucci L. Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy. Marine Drugs. 2021; 19(1):15. https://doi.org/10.3390/md19010015
Chicago/Turabian StyleConte, Mariarosaria, Elisabetta Fontana, Angela Nebbioso, and Lucia Altucci. 2021. "Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy" Marine Drugs 19, no. 1: 15. https://doi.org/10.3390/md19010015
APA StyleConte, M., Fontana, E., Nebbioso, A., & Altucci, L. (2021). Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy. Marine Drugs, 19(1), 15. https://doi.org/10.3390/md19010015