1. Introduction
There is little doubt that UV-radiation affects all people in their daily life. Harmful effects of solar radiation cause skin damage and skin aging, but the body counters these harmful influences through the production of the pigment, melanin [
1,
2]. Despite the considerable virtue of melanin, irregular production of this pigment underlies several skin diseases and conditions, including melasma, leukoplakia, lentigo, albinism, moles, and freckles [
3]. To address the consequences of abnormal melanogenesis processes, numerous researchers have investigated melanogenesis mechanisms. The shared mechanism of melanin production is initiated as an oxidative progress in which tyrosine is converted first to 1-3,4-dihydroxyphenylalanine (DOPA), and then to dopaquinone, dopachrome and finally the pigment melanin, primarily by tyrosinase and related enzyme systems [
4]. Given that catalysis is central to the oxidative action of tyrosinase, the capacity to suppress tyrosinase activity and expression is important for anti-melanogenesis activity. A number of medications that possess this capacity, such as kojic acid, 4-n-butylresorcinol, hydroquinone and tretinoin, are currently available [
5]. Although these synthetic agents are very effective, they are accompanied by undesirable side-effects. For example, hydroquinone causes blistering, skin cracking, and dryness in individuals with sensitive skin. Therefore, recent research on anti-melanogenesis in the cosmetic industry has focused on the development of novel, natural sources-derived agents that are free from adverse effects.
Marine microorganisms are known to produce many bioactive molecules, with additional secondary metabolites exhibiting significant activities in various molecular and biochemical pathways [
6]. In our continuing research effort to discover anti-melanogenic agents from marine microorganisms for use as constituents of cosmetic products, we have focused on screening crude extracts from libraries of cultures produced by dozens of marine microorganisms. In the process, we discovered that crude extracts of
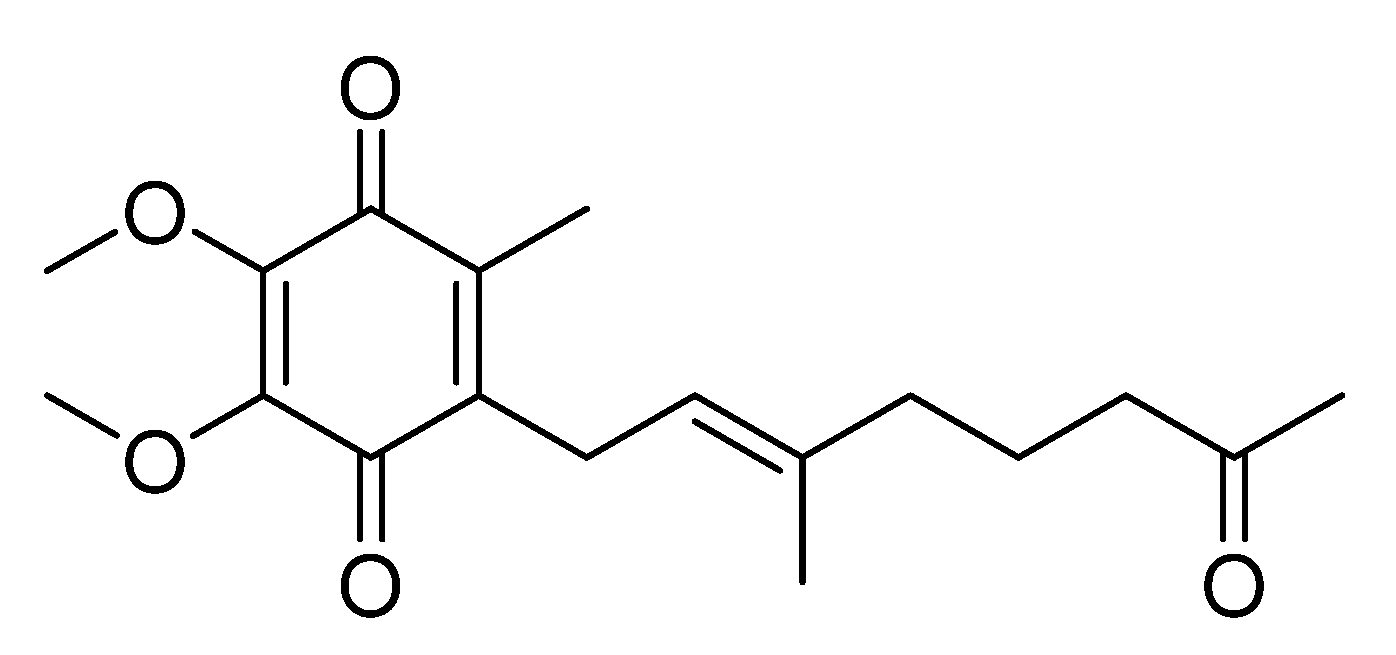
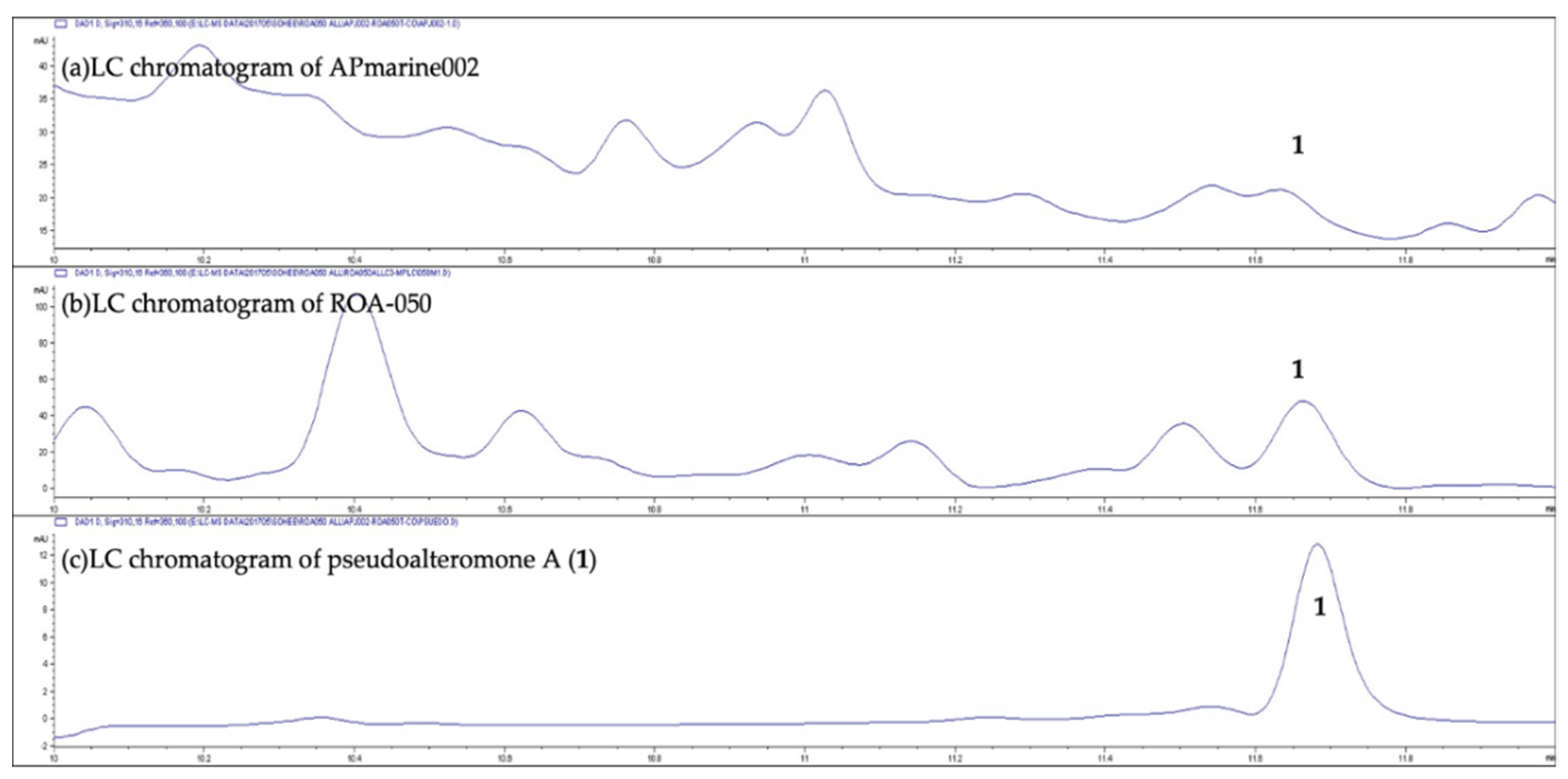
Pseudoalteromonas spp., APmarine002 and ROA-050, exhibit moderate inhibitory activity on melanogenesis in α-melanocyte-stimulating hormone (α-MSH) stimulated B16 melanoma cells. Using assay-guided purification of crude extracts of APmarine002 and ROA-050 based on anti-melanogenic activity, we identified the pseudoalteromone A (
1) and
p-coumaric acid as such anti-melanogenic agents (
Figure 1). Pseudoalteromone A, first isolated from
Pseudoalteromonas sp. CGH2XX and subsequently isolated from two other
Pseudoalteromonas spp.,
Pseudoalteromonas rubra QD1-2 and
Pseudoalteromonas sp. P1-9 [
7,
8], has been reported to exhibit cytotoxicity against MOLT-4 human acute lymphoblastic leukemia cells and inhibit the release of elastase by human neutrophils [
9]. However, anti-melanogenic effects of pseudoalteromone A have not been previously reported. Here, we describe the inhibitory effects of pseudoalteromone A on melanogenesis both in vitro and in a 3D pigmented-epidermis model (MelanoDerm).
3. Discussion
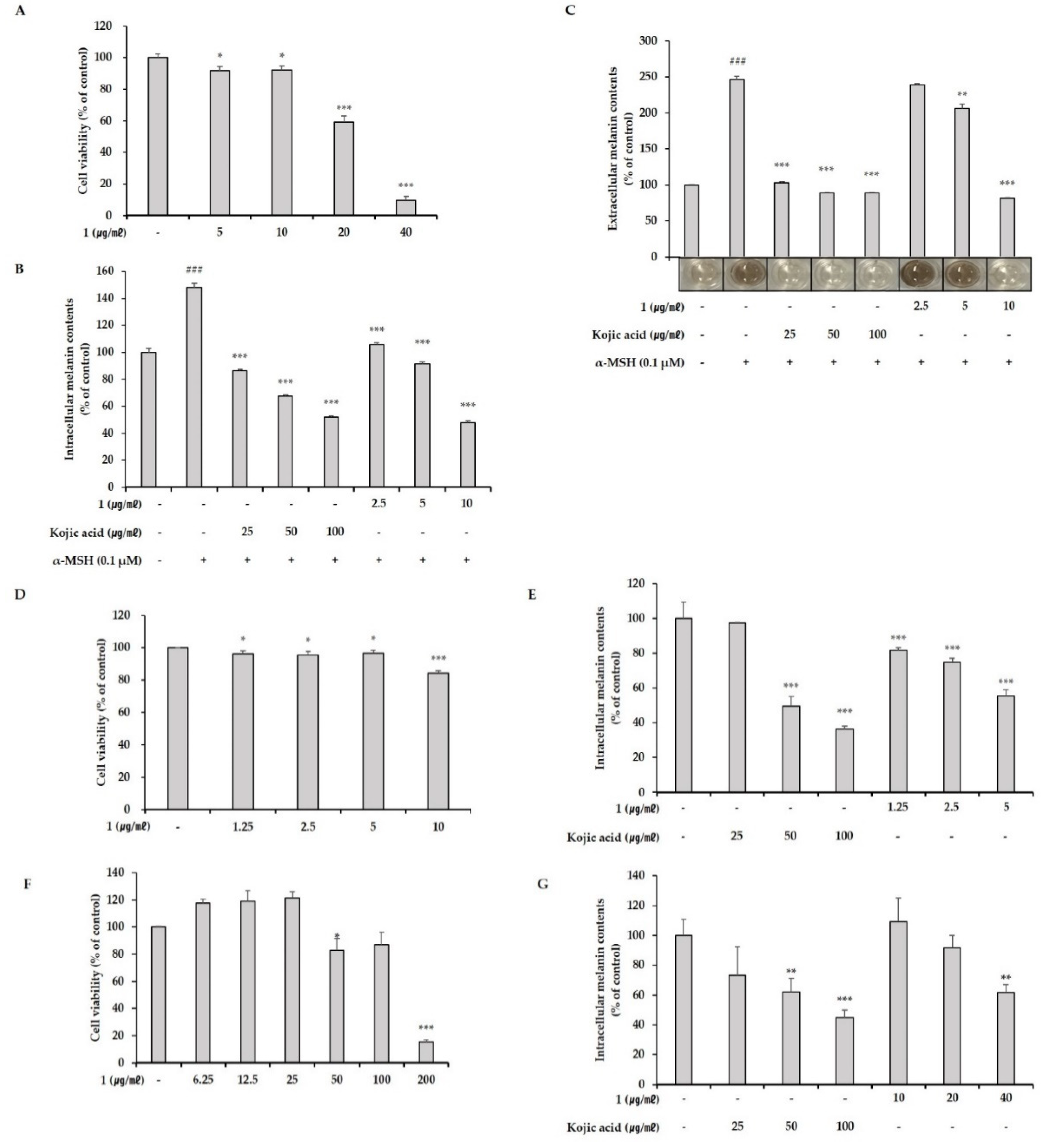
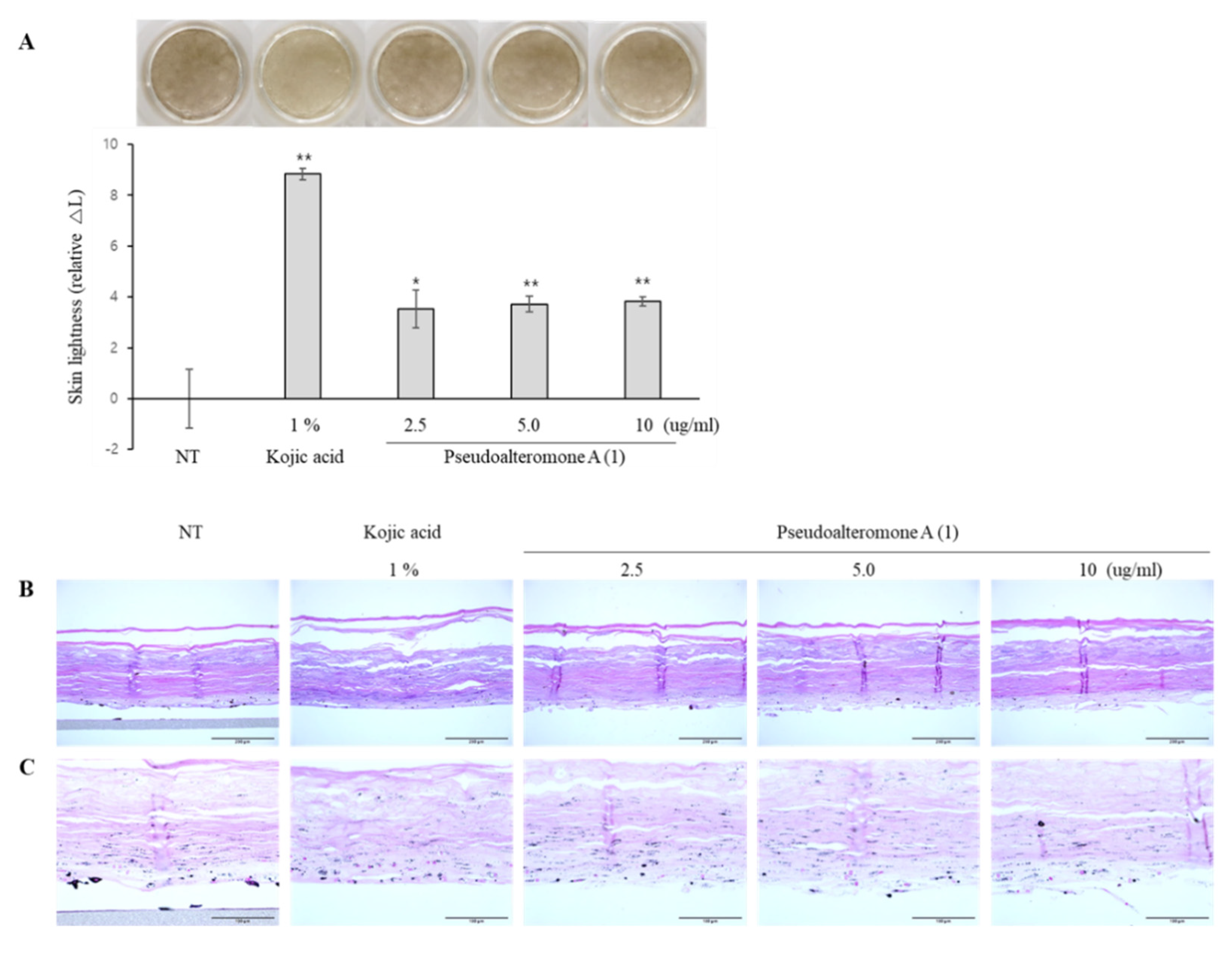
We showed that two different marine-derived Pseudoalteromonas strains, APmarine002 (isolated from soft coral in Jeju island) and ROA-050 (isolated from marine sediments of Pohang, South Korea), produced the same anti-melanogenic agents, pseudoalteromone A (1) and p-coumaric acid. We also demonstrated that pseudoalteromone A clearly inhibits melanin synthesis in different cell lines (B16, Melan-a and MNT-1 cells) and exerts a whitening effect in a human skin equivalent.
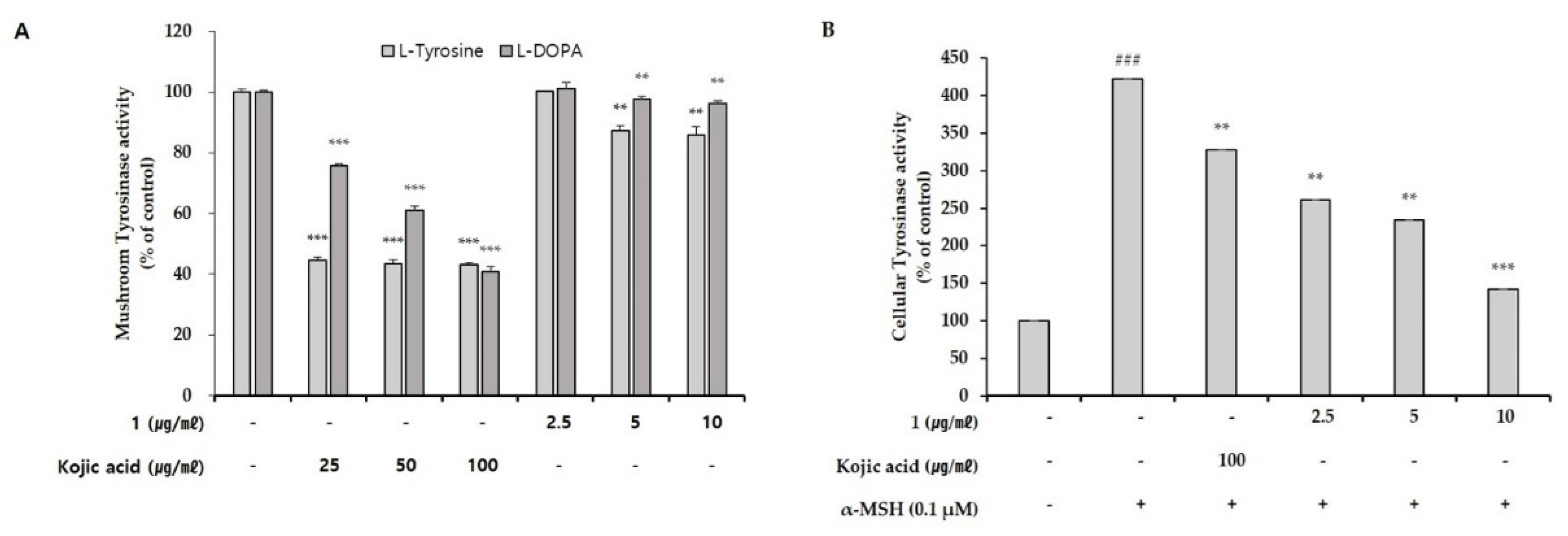
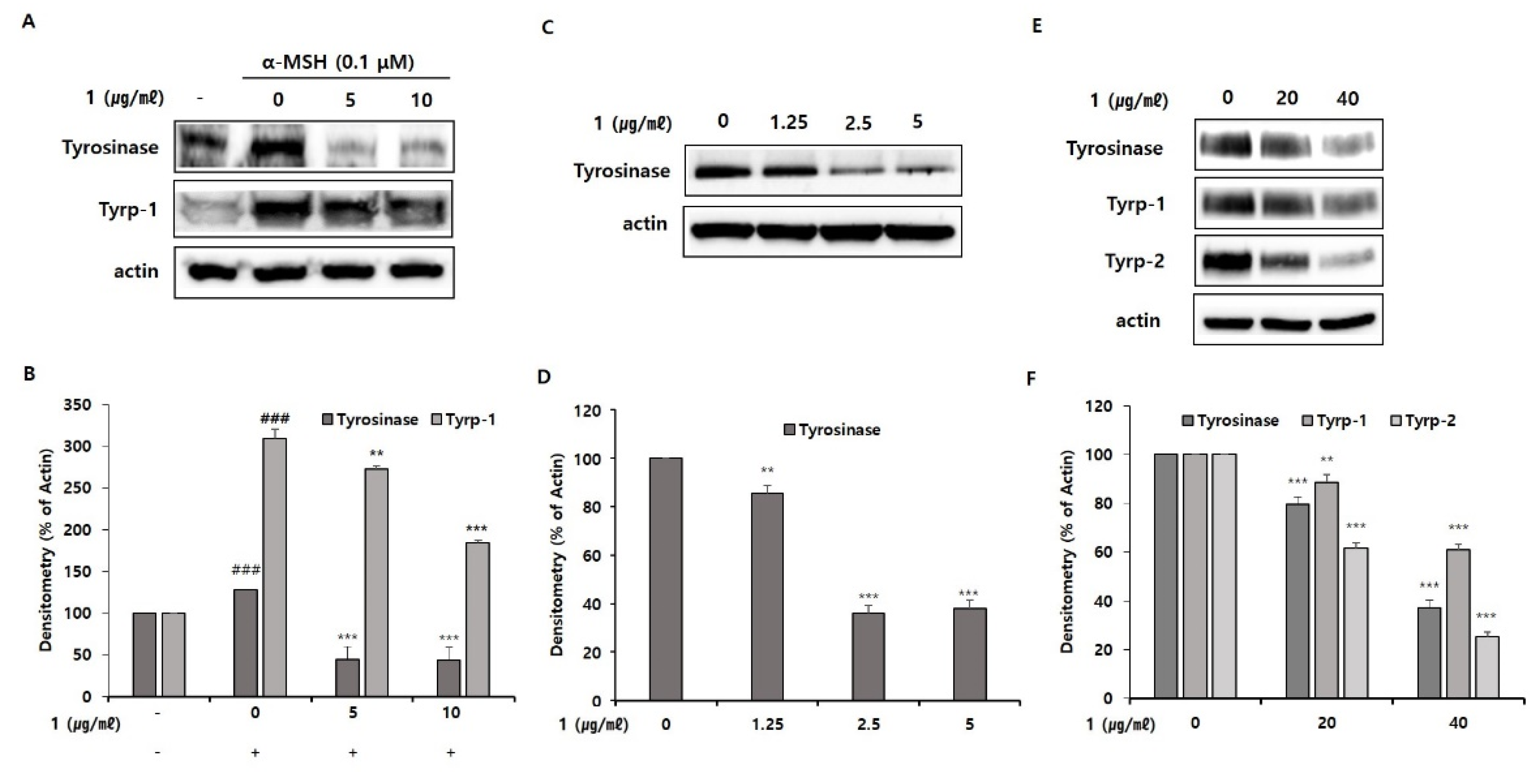
To begin defining the action mechanism of pseudoalteromone A, we tested the inhibitory effect of pseudoalteromone A on tyrosinase activity. First, we determined whether pseudoalteromone A directly inhibits tyrosinase activity. Simultaneously, we performed the total cellular tyrosinase activity after pseudoalteromone A treatment in α-MSH-treated B16 cells. In
Figure 5A, we found that the de-pigmentary effect of pseudoalteromone A on melanocytes mainly depends on reduced tyrosinase activity, as supported by the significant (albeit weak) direct inhibition of mushroom tyrosinase activity and robust inhibition of cellular tyrosinase activity by pseudoalteromone A (
Figure 5). Based on above data, we supposed that the decreased total cellular tyrosinase activity by pseudoalteromone A treatment may be mainly caused by the decreased level of tyrosinase expression rather than the direct inhibition of tyrosinase activity. In addition, further support is provided by the observation that pseudoalteromone A reduced expression of tyrosinase as well as that of the tyrosine-related enzymes, Tyrp-1 and Tyrp-2 (
Figure 6). Collectively, these findings suggest that pseudoalteromone A inhibits melanogenesis by suppressing both tyrosinase activity and expression.
Despite finding that pseudoalteromone A suppressed tyrosinase, Tyrp-1 and Tyrp-2 expression, our evidence for an underlying molecular action mechanism of pseudoalteromone A is incomplete. To establish a detailed molecular mechanism, we plan future studies to determine whether pseudoalteromone A suppresses expression of melanocyte-inducing transcription factor (MiTF) through inhibition of mitogen-activated protein kinase (MAPK) or cAMP-depend on protein kinase (PKA) signaling pathways in melanocytes, reflecting the fact that MAPK and PKA signaling are intimately linked with MiTF-induced tyrosinase expression during melanogenesis [
11,
12,
13,
14,
15,
16,
17]. Collectively, the data from this study provide preliminary evidence supporting the possibility of pseudoalteromone A as a potent and effective whitening agent that could be used in cosmetics and medicinal formulations.
4. Materials and Methods
4.1. General Experimental Procedures
UV spectra were recorded in methanol (MeOH) on a Cinco UVS-2100 spectrophotometer. 1D and 2D NMR spectra were recorded on a Varian Unity-Enova at 400 MHz using signals of the residual solvent as internal references (ph. 3.31 ppm and act 49.00 ppm for methanol-d4 (CD3OD-d4). Low-resolution LC/MS measurement was performed at the National Research Facilities and Equipment Center (Nano Bioenergy Materials Center) at Elwha Woman’s University using an Agilent Technologies 1260 quadrupole and Waters micromassZQ LC/MS system with a reversed-phase column (Phenomenex Luna C18 (2) 100 Å, 50 mm × 4.6 mm, 5 µm) at a flow rate 1.0 mL/min. Column chromatography separation was performed using a C18 column eluting with a gradient of MeOH and H2O. Fractions were purified using a WATERS 1525 binary HPLC pump with a reversed-phase C18 column (Phenomenex Luna C18 (2), 250 mm × 10 mm, 5 μm) eluting with 48% CH3CN in H2O at flow rate of 2.0 mL/min.
4.2. Strain Isolation
Pseudoalteromonas strain APmarine002 was isolated from coral collected at Jeju island. Pseudoalteromonas strain ROA-050 was isolated from the marine sediments of Pohang, South Korea. An NCBI blast analysis of 16S rRNA gene sequences identified Strain APmarine002 as Pseudoalteromonas rubra QD1-2 and strain ROA-050 as Pseudoalteromonas piscicida, with 100.0% identity in both cases.
4.3. Fermentation, Extraction and Purification
Strains APmarine002 and ROA-050 were independently cultured in 1 × 2.5 L Ultra Yield Flasks, each containing 1 L of seawater-based medium (10 g starch, 2 g yeast extract, 4 g peptone, 34.75 g artificial sea salt dissolved in distilled H2O), and at 27 °C with shaking (150 rpm). After 7 days of cultivation, each broth was extracted using ethyl acetate (EtOAc; 1 L overall), and the solvent was removed in vacuo to yield 23.1 mg and 84.2 mg of EtOAc crude extract, respectively. Both extracts were used to perform the LC-MS analysis. For the large cultivation to isolate compound 1, strain ROA-050 was cultured in 40 × 2.5 L Ultra Yield Flasks, each containing 1 L of seawater-based medium (10 g starch, 2 g yeast extract, 4 g peptone, 34.75 g artificial sea salt dissolved in distilled H2O), and at 27 °C with shaking (150 rpm). After 7 days of cultivation, the broth was extracted using ethyl acetate (EtOAc; 40 L overall), and the solvent was removed in vacuo to yield 3.5 g of an EtOAc crude extract. The extract was fractionated by open column chromatography on a C-18 resin using a step gradient elution of H2O and MeOH, yielding eight fractions. Fraction 4 and 5, eluted with 60~80% of MeOH in H2O (770.4 mg), was further purified by reversed-phased HPLC under isocratic conditions with 48% aqueous CH3CN to yield 1 (17.3 mg).
Pseudoalteromone A (1): orange oil, UV (MeOH) λmax (log ε) 200 (2.15), 281 (2.09) and 262 (1.24); LRESIMS m/z 343.1 [M + Na]+.
4.4. Cell Culture
B16 cells, mouse melanoma cell line, were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Welgen, Korea) containing 5% fatal bovine serum (FBS; ATCC, Manassas, VA, USA) and 1% penicillin-streptomycin mixture (Lonza, Basel, Switzerland). Melan-a cells, an immortalized mouse melanocyte cell line, were grown in RPMI-1640 medium (Lonza, Walkersvile, MD, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA), 1% penicillin-streptomycin mixture and 200 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, St. Louis, MO, USA). MNT-1 cells, a human melanoma cell line, were cultured in Minimum Essential Medium (MEM; Gibco, Grand Island, NY, USA) containing 1% DMEM (Welgen, Korea), 20% FBS, 1% penicillin-streptomycin mixture and 1% N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid) (HEPES 1 M; Gibco, Taiwan). All cells were cultured at 37 °C in a humidified environment with 5% CO2/95% air environment.
4.5. Cell Viability
B16 cells, Melan-a cells and MNT-1 cells were seeded on 96-well plates (1 × 104 cells/well) and cultured for 24 h, after which the medium was replaced with the medium containing pseudoalteromone A, diluted to the indicated concentrations, and plates were incubated for 72 h. The medium was then removed and the fresh culture medium containing 10% CCK-8 solution (DOJINDO, Tokyo, Japan) was added. After incubating 1 h, supernatants of each cell were transferred to 96-well plates, and the absorbance was measured at 450 nm using a Synergy™ HTX Multi-Mode Microplate Reader (Bioteck, Winooski, VT, USA).
4.6. Measurement of Melanin Content
B16 cells were seeded overnight in 48-well plates (2 × 104 cells/well) and then treated for 72 h with increasing concentrations of pseudoalteromone A. The extracellular melanin content in the phenol red-free cell culture medium was measured at 405 nm with Synergy™ HTX Multi-Mode Microplate Reader (Bioteck, Winooski, VT, USA). After removing the medium, 200 μL of 1 N NaOH was added, and plates were heated to 60 °C for 30 min to solubilize intracellular melanin. Next, 100 μL aliquots of lysate were transferred to a 96-well plate and absorbance was measured at 405 nm. Melan-a cells and MNT-1 cells were seeded in 24-well plates (10 × 104 cells/well) for 24 h, and treated with pseudoalteromone A. Then, the intracellular melanin contents were measured. Results were normalized to total protein content, determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Intracellular melanin levels are presented relative to those in the control group, expressed as a percentage.
4.7. Cell-Free Enzymatic Assay for Tyrosinase Activity
The inhibitory effects of pseudoalteromone A on tyrosinase activity, tested using a mushroom tyrosinase enzyme, was assessed as a function of DOPA oxidase activity and t and tyrosine hydroxylase activity. For these experiments, a 5 μL sample was added to a 96-well plate to which was added 50 μL of 0.1 M sodium phosphate buffer (pH 6.8), 40 μL distilled water, and 5 μL of enzyme solution (2000-unit mushroom tyrosinase). After mixing, 50 μL of 20 mM L-DOPA or 0.03% L-tyrosine (tyrosinase substrate) was added to each well and the amount of products formed in the reaction mixture was determined by kinetically measuring absorbance values at 475 nm at 10 min intervals for at least 1 h at 37 °C using a spectrophotometer.
4.8. Assay of Cellular Tyrosinase Activity
The activity of tyrosinase enzyme was evaluated by measuring the oxidase rate of L-DOPA to dopachrome, an intermediate in melanin biosynthesis. B16 melanoma cells were plated in 6-well plates (20 × 104 cells/well) and incubated overnight, then treated with increasing concentrations of pseudoalteromone A for 3 days in a humidified incubator. Cells were then washed twice in ice-cold phosphate-buffered saline (PBS) and lysed in 1 mL of 0.1 M sodium phosphate buffer (pH 6.8) containing 1% (w/v) Triton X-100. The resulting whole-cell lysates were microcentrifuged at 13,000 rpm for 20 min at 4 °C, and the resulting supernatants were placed in a 96-well plate for reactions with L-DOPA (2 mg/10 mL). Tyrosinase activity was analyzed spectrophotometrically by kinetically measuring absorbance values at 475 nm at 10 min intervals for at least 1 h at 37 °C using a spectrophotometer.
4.9. Western Blotting
B16, Melan-A and MNT-1 cells were seeded onto 6-well plates (20 × 10⁴ cells/well) and incubated for 24 h. The medium was then removed and fresh culture medium containing different concentrations of pseudoalteromone A was added, after which plates were incubated for the appropriate time. After washing twice with ice-cold PBS, intracellular proteins were extracted from each cell type by lysing cells in 1 × RIPA buffer (Cell Signaling Technology, Danvers, MA, USA), diluted with distilled water containing protease inhibitor cocktail III and phosphatase inhibitor Cocktail set III (Merck, Darmstadt, Germany) at a ratio of 200:1. Cell lysates were centrifuged at 13,000 rpm at 4 °C, and protein concentration was measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Appropriate amounts of protein from each cell type were resolved by SDS-PAGE on 10% gels and then transferred onto a nitrocellulose membrane (Bio-Rd, Hercules, CA, USA). Membranes were blocked by incubating in 1 × Tris-buffered saline (TBS) containing 5% skim milk for at least 1 h, then incubated at 4 °C overnight with primary antibodies, diluted as recommended by the manufacturer. Membranes are washed three times in TBS containing Tween 20 and incubated with species-appropriate secondary antibodies at recommended dilutions for 1 h at room temperature. Immuno-reactive proteins were visualized using Clarity™ Western ECL substrate (Bio-Rad, Hercules, CA, USA). The densities of target protein bands were measured using iBright™ CL750 Imaging System (Invitrogen, Carlsbad, CA, USA).
4.10. Skin-Whitening Assay Using a Pigmented 3D Skin Model
MelanoDerm (MEL-300-B, MatTek Corp., Ashland, MA, USA), used for the skin-lightening study, was cultured in EPI-100-NMM-113-PRF medium (MatTek Corp., Ashland, MA, USA) at 37 °C in a humidified 5% CO2 incubator. Different concentrations of pseudoalteromone A were added to the culture medium every other day for 14 days. Thereafter, epidermal pigmentation was examined by performing optical and histological analyses. The epidermal pigmentation level was calculated by comparing variations in L* values (a lightness/darkness index) on days 1 and 14 and estimating the difference between them (ΔL). For the histological examination, tissues were fixed in 10% neutral buffered formalin (BBC Biochemical, Mount Vernon, WA, USA) and embedded in paraffin. Paraffin-embedded samples were then sliced into 5 μm sections, stained with hematoxylin and eosin (H and E) and Fontana Masson’s (F-M) staining (for melanin), and then examined histologically.
4.11. Two-Photon Excitation Fluorescence (TPEF) Imaging
TPEF imaging was used to visualize the three-dimensional distribution of melanin in the human skin equivalent, as described in our previous studies [
18,
19]. Intracellular melanin (green in
Figure 8) in the melanocyte-rich layer was measured by imaging the basement membrane side of MelanoDerm samples in a total volume of 400 (x) × 400 (y) × 40 (z) μm
3. Relative TPEF signal intensities and melanin volume were quantified using Image-Pro Premier 3D software (Media Cybernetics, Inc., Bethesda, MD, USA).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}