Abstract
Bioactivity-guided fractionation of a methanolic extract of the Red Sea cucumber Holothuria spinifera and LC-HRESIMS-assisted dereplication resulted in the isolation of four compounds, three new cerebrosides, spiniferosides A (1), B (2), and C (3), and cholesterol sulfate (4). The chemical structures of the isolated compounds were established on the basis of their 1D NMR and HRMS spectral data. Metabolic profiling of the H. spinifera extract indicated the presence of diverse secondary metabolites, mostly hydroxy fatty acids, diterpenes, triterpenes, and cerebrosides. The isolated compounds were tested for their in vitro cytotoxicities against the breast adenocarcinoma MCF-7 cell line. Compounds 1, 2, 3, and 4 displayed promising cytotoxic activities against MCF-7 cells, with IC50 values of 13.83, 8.13, 8.27, and 35.56 µM, respectively, compared to that of the standard drug doxorubicin (IC50 8.64 µM). Additionally, docking studies were performed for compounds 1, 2, 3, and 4 to elucidate their binding interactions with the active site of the SET protein, an inhibitor of protein phosphatase 2A (PP2A), which could explain their cytotoxic activity. This study highlights the important role of these metabolites in the defense mechanism of the sea cucumber against fouling organisms and the potential uses of these active molecules in the design of new anticancer agents.
1. Introduction
Natural products have been a prime source of compounds with substantial structural diversity and numerous therapeutic activities [1], including plants, marine organisms, animals, and minerals [2]. Marine organisms can be considered as a great reservoir of new bioactive compounds that can aid in the prevention and treatment of different ailments, including cancer [3]. Research on marine-derived natural products emerged as early as the 19th century, and since 1980, the study of marine-derived natural products has led to the discovery of potential applications in drug development [4,5]. As a result of this intense research, more than 10,000 bioactive metabolites have so far been discovered [6]. The marine ecosystem is recognized as a treasure trove of unprecedented bioactive natural products and nutraceuticals with exceptional structural and chemical properties [7,8]. Marine organisms are commonly soft-bodied, and thus, tend to produce poisonous secondary metabolites or acquire them from microorganisms to protect themselves from predators [8]. Today, there is rising interest in exploiting marine diversity and complexity for drug discovery [9]. Many bioactive compounds have been isolated from different groups of marine organisms; among them are corals, tunicates, sponges, and sea cucumbers [6]. The Egyptian Red Sea coast and the Gulf of Aqaba host numerous marine communities, and one of the most abundant marine taxa in that area are the holothurians (sea cucumbers) [10]. They belong to the phylum Echinodermata, class Holothuroidea, and are commonly known as holothurians, comprising about 1200 known species worldwide [6]. Sea cucumbers constitute a huge group of organisms, from which a vast range of secondary metabolites have been isolated—among them, cerebrosides [11,12]. These compounds possess several pharmacologically relevant bioactivities, such as antiviral, antibacterial, antifungal, or anticancer effects [6]. It has also been reported that sea cucumber extracts show multiple biological activities, such as wound healing, antioxidant, and immunomodulatory effects [6]. Metabolomics encompass comprehensive analysis of the total number of metabolites within a biological sample using a specific set of parameters; metabolomics could serve as a reliable method to provide a picture of the central information of a cell, tissue, or whole organism [13]. In this paper, we report on research including bioactivity-guided fractionation complemented by LC-HRESIMS-assisted chemical investigation of the Red Sea cucumber Holothuria spinifera, leading to the isolation and structural characterization of three new cerebrosides, named spiniferoside A (1)—which is a mixture of three cerebrosides A1 (1a), A2 (1b), and A3 (1c)—spiniferoside B (2), and spiniferoside C (3), along with a fourth, a known sterol, cholesterol sulfate (4). The SET [Su(var)3-9, enhancer-of-zeste and Trithorax] is a protein domain that plays a key role in the methylation of histones, hence regulating gene expression. It is also a potent inhibitor of protein phosphatase 2A (PP2A), a serine/threonine enzyme with a tumor suppressing effect. In breast cancer, the SET protein was found to be overexpressed, and its knockdown decreased tumorigenesis. SET inhibits PP2A activity through binding to both N-terminus and C-terminus regions of PP2A. Given that PP2A maintains the activation of some oncogenic survival signals, SET is therefore an attractive and powerful therapeutic target for breast cancer therapy. In the same context, a recent topic that has gained interest in breast cancer research is the important role of the ceramide metabolism in this disease. Sphingolipid ceramide has long been described to activate PP2A through direct binding to SET oncoprotein [14,15].Our research efforts were oriented to discover bioactive drug candidates from the Red Sea marine organism, and supported by in silico studies, we have investigated a promising lead offered by sea cucumber Holothuria spinifera from the Red Sea, and three new cerebrosides were isolated. In vitro studies on the breast carcinoma cell line MCF-7 revealed that the new compounds displayed promisingantitumor activity comparable to the control drug doxorubicin. In the same context, and, to better understand the reasons for this cytotoxicity and the possible underlying mechanisms, investigations using molecular modeling research tools were carried out on the SET oncoprotein.
2. Results and Discussion
2.1. Structure Elucidation of the Isolated Compounds
Compound 1 (Figure 1) was isolated as an amorphous white substance. It was identified as a cerebroside mixture 1 of three similar metabolites, compounds, 1a, 1b, and 1c, all with a sphingosine-type moiety having the molecular formulae C37H72NO9, C38H74NO9, and C39H76NO9, respectively; two degrees of unsaturation in all three cases. The LC-HRESIMS displayed three peaks at m/z 674.5207, 688.5369, and 702.5513 [M + H]+ (Figure S1). The 1H and 13C NMR spectral data of compound 1 are listed in Table 1 (Figures S2–S5). The 1H NMR spectrum (measured in C5D5N, 400 MHz) of 1 indicated a sphingolipid skeleton, by the presence of an exchangeable proton signal of NH group at δH 8.37 (1H, d, J = 8.0 Hz) along with the typical aliphatic chain resonances, overlapping methyls at δH 0.85, and the long methylene chain protons at δH 1.25. Further characteristic NMR resonances were observed at δH 4.82 (1H, m, H-2), 4.82 (2H, m, H-3), 4.79 (2H, m, H-2’), 5.48 (1H, m, H-4), and 5.96 (1H, m, H-5), besides the signal for an anomeric proton at δH 4.53 (1H, d, J = 8.0 Hz, H-1’’), indicative of a sphingosine-type cerebroside. The 13C NMR spectrum (C5D5N, 100 MHz) showed signals at δC 14.1 for the methyl groups and at δC 173.4 for an amide carbonyl function. The resonances of the 2-amino-1,3,2′-triol part of the hydrocarbon chain were observed at δC 54.9 (C-2), 70.5 (C-1), 72.6 (C-3), and 72.6 (C-2’). The two methine groups resonated at δC 132.2 and 132.6 for (C-4) and (C-5), respectively. The position of the double bond was assigned at C-4 based on the typical biosynthetic pathway of ceramides as in [16]. The 13C NMR spectrum displayed characteristic signal corresponding to an anomeric carbon at δC 105.9 in addition to related downfield-shifted resonances at δC 75.2, 78.5, 71.5, 78.5, and 62.6, revealing the presence of a sugar moiety. The glucopyranosyl moiety was assigned to be β-configured based on the observed coupling constant of the anomeric proton at δH 4.53 (1H, d, J = 8.0 Hz, H-1’’) characterizing the di-axial interactions between H-1′’ and H-2′ and the chemical shift of the anomeric carbon δC 105.9 ppm (in the case of an α-configuration, the coupling constant should be 3.7 Hz, and the 13C NMR signal of the anomeric carbon should resonate at δC 98.5 ppm) [17,18]. In order to determine the length of the fatty acid chain, compound 1 was subjected to methanolysis followed by peak detection by LC-HRESIMS according to the method adopted by Sun and coworkers [19]. In brief, compound 1 was treated with aqueous HCl/MeOH for methanolysis, by which hydroxy acid methyl esters, sphingosine, and a sugar moiety were produced. LC-HRESIMS analysis of the fatty acid methyl esters of compound 1 molecular species mixture gave three molecular-ion peaks at m/z 273.2461, 287.2573, and 301.2729 [M]+, corresponding to the molecular formulas C16H32O3, C17H34O3, and C18H36O3 of the fatty acid methyl esters, methyl-2-hydroxypentadecanoate, methyl-2-hydroxyhexadecanoate, and methyl-2-hydroxyheptadecanoate, respectively. The relative configuration of the cerebroside moieties was deduced to be (2S, 3R, 4E, 2’R), as evidenced from the optical rotation +17.20 (c =1.00, MeOH) and the aforementioned 13C NMR signals (C-1, C-2, C-3, C-4, C-5, and C-2’) and their corresponding protons in the 1H NMR (measured in C5D5N) spectrum, which were in accordance with those of the known, previously reported sphingosine-type cerebrosides HLG-2 [20] and HPC-2 [12] possessing the same 2S,3R,4E,2’R-configuration. The sugar produced during the hydrolysis exhibited a comparable specific rotation +17.2 (c = 0.1%, H2O) as the standard, d-glucose. Moreover, the acetylated thiazolidine derivative of the obtained sugar was analyzed using a Cosmosil-5C18-AR-II column and it displayed the same tR as the standard, d-glucose (19.7 min). From the aforementioned data, the structure of compound 1 was deduced to be a molecular species of the sphingosine-type glucocerebrosides possessing three different 2-hydroxy fatty acids with the molecular formulas C37H72NO9, C38H74NO9, and C39H76NO9. To the best of our knowledge, compound 1 (henceforth named spiniferoside A) is a new molecular species. Rooted in the considerable interest and importance of determining the molecular compositions of species of sphingolipids, the isolation and the structural elucidation of the cerebrosides in the molecular species 1 were conducted. Using a reversed phase HPLC adsorbent, 1 was separated into its three peaks and they were recovered to yield three cerebrosides spiniferoside, A1 (1a), spiniferoside A2 (1b), and spiniferoside A3 (1c) following the method described in [21]. Spiniferoside A2 was the major metabolite in the molecular species mixture with a molecular mass of m/z 688.5363 [M + H]+, while spiniferosides A1 and A3, with molecular masses of m/z 674.5207 [M + H]+ and 702.5513 [M + H]+ respectively, occurred in smaller quantities (Figures S9–S15).
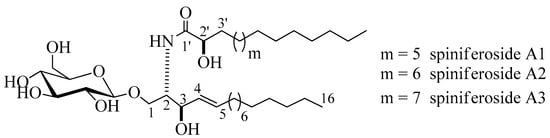
Figure 1.
Chemical structures of spiniferoside A1–A3.

Table 1.
1H (400 MHz) and 13C NMR (100 MHz) data for the new compounds 1, 2, and 3 in C5D5N.
Compound 2 (Figure 2) was isolated as a white powder with a molecular formula of C49H94NO9, representing three degrees of unsaturation, as deduced from its 1H and 13C NMR spectral data and further confirmed by LC-HRESIMS analysis, which displayed a molecular ion peak at m/z 840.6923 [M + H]+ (Figure S16). The 1H and 13C NMR data of 2 were listed in Table 1 (Figures S17–S21). The 1H NMR spectrum (C5D5N, 400 MHz) displayed resonances typical of long methylene chain protons at δH 1.25, overlapped signals of methyl groups at δH 0.85, and a peak for an amide proton at δH 8.37 (1H, d, J = 8.0 Hz) indicative of a sphingolipid skeleton. Another signal characteristic of a sphingosine-type cerebroside appeared at δH 4.79 (1H, m, H-2), 4.79 (2H, m, H-3), 4.56 (2H, m, H-2’), and 5.96 (2H, m, H-4 and H-5), and the anomeric proton resonated at δH 4.98 (1H, d, J = 8.0 Hz, H-1’’). The 13C NMR spectrum in (C5D5N, 100 MHz) displayed an amide carbonyl at δC 175.7, two terminal methyl groups at δC 14.1, and characteristic resonances at δC 54.6 (C-2), 70.0 (C-1), 72.4 (C-2’), 72.5 (C-3), 131.9 (C-4), and 131.1 (C-5). The 13C NMR spectrum displayed the characteristic signal of an anomeric carbon δC 105.6, together with other downfield-shifted signals at δC 75.0, 78.4, 71.4, 78.5, and 62.5 indicating the presence of a sugar part. The observed coupling constant of the anomeric proton at δH 4.98 (1H, d, J = 8.0 Hz, H-1’’) characterizing the di-axial interaction between H-1′’ and H-2′, together with the chemical shift for the anomeric carbon δC 105.6, confirmed the β-configuration of the glucopyranosyl moiety (α glucopyranoside: J = 3.7 Hz; δC 98.5) [17,18]. The length of the fatty acid chain was determined by its methanolysis, followed by peak detection using LC-HRESIMS analysis for the obtained methyl ester of the fatty acid portion of 2. As described in [19], compound 2 reacted with 5% methanolic HCl to produce the respective hydroxy acid methyl ester, sphingosine, and a sugar. LC-HRESIMS analysis of the fatty acid methyl ester of 2 exhibited a molecular ion peak at m/z 383.3547 [M]+ (Figure S25), corresponding to a C24H46O3 fatty acid methyl ester, (E)-methyl 2-hydroxytricos-15-enoate. Further confirmation of the double bond in the α-hydroxy fatty acid methyl ester was provided by GC–MS analysis. The α-hydroxy fatty acid methyl ester resulting after hydrolysis C24H46O3 with m/z 383.3547 [M]+ was subjected to oxidation by KmnO4, as described in [19], to yield fatty acid methyl ester C23H44O2 with m/z 352 that was subjected to GC–MS analysis; resulting fragments confirmed the position of the double bond by the presence of mass fragments at m/z 267 corresponding to a [C17H31O2•] fragment, m/z 253 corresponding to a [C16H29O2•] fragment, and m/z 125 corresponding to a [C9H17•] fragment (Figure S26). Therefore, the position of the double bond was authenticated in 15’ at (E) methyl 2-hydroxytricos-15-enoate. The sugar obtained from the hydrolysis displayed a specific rotation of +17.2 (c = 0.1%, H2O), which was identical to that of the standard, d-glucose. Furthermore, the tR value of the acetylated thiazolidine derivative of the liberated sugar was similar to that of the standard, acetylated d-glucose (19.7 min). Like for compound 1, the relative stereochemical details in the cerebroside moieties were determined to be (2S,3R,4E,2’R), by comparing its optical rotation and 1H NMR and 13C NMR spectral data (measured in C5D5N) with the correlatives reported previously. The optical rotation of +17.20 (c 1.00, MeOH) and the chemical shifts of C-1 (δC 70.0), C-2 (δC 54.6), C-3 (δC 72.5), C-4 (δC 131.9), C-5 (δC 131.1), and C-2’ (δC 72.4), along with the chemical shifts of their corresponding protons were similar to those of the known reported sphingosine-type glucocerebrosides HLG-2 [20] and HPC-2 [12], which possess 2S,3R,4E,2’R-configuration. From the above data, the structure of compound 2 was assigned as a cerebroside and it was given the name spiniferoside B, which is, to the best of our knowledge, a new compound.
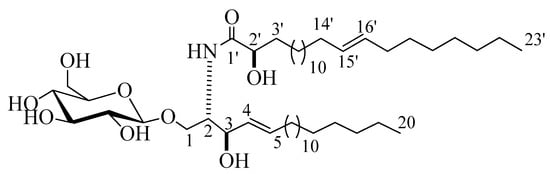
Figure 2.
Chemical structure of the newly discovered compound 2, spiniferoside B.
Compound 3 (Figure 3) was isolated as a white powder. Its molecular formula was deduced to be C48H94NO10, representing two degrees of unsaturation on the basis of its NMR analysis and further confirmed by the mass spectra with the appearance of a molecular-ion peak at m/z 844.6872 [M + H]+ (Figure S27). The 1H and 13C NMR spectral data of compound 3 are listed in Table 1 (Figures S28–S30). The 1H NMR spectrum (C5D5N, 400 MHz) of 3 displayed resonances typical of long methylene chain protons at δH 1.26, overlapped peaks of methyl groups at δH 0.87, and an exchangeable proton of an NH group at δH 8.60 (1H, d, J = 8.0 Hz), indicating the presence of a sphingolipid skeleton. In addition, several multiplets at δH 5.30 (1H, br,m, H-2), 4.20 (1H, m, H-4), 4.35 (1H, m, H-3), 4.34 (1H, m, H-1), and 4.74 (1H, t, J = 8.0, H-2’) were observed, along with an anomeric proton at δH 4.97 (1H, d, J = 8.0 Hz, H-1’’), exhibiting a phytosphingosine-type cerebroside. The 13C NMR spectrum (measured in C5D5N, at 100 MHz), displayed an amide carbonyl resonating at δC 175.7, two terminal methyl groups at δC 14.1, and the resonances characteristic of the 2-amino-1,3,4,2′-tetrol part of the hydrocarbon chain at δC 51.4 (C-2), 70.2 (C-1), 72.1 (C-4), 72.2 (C-2’), and 74.9 (C-3). The 13C NMR spectrum displayed the characteristic signal of the anomeric carbon at δC 105.1, together with downfield-shifted signals at δC 75.0, 78.4, 71.4, 78.5, and 62.5 of the carbon skeleton of the sugar part. The chemical shift of the anomeric carbon at δC 105.1, along with the coupling constant of the anomeric proton at δH 4.97 (1H, d, J = 8.0 Hz, H-1’’) characterizing the di-axial interaction between H-1′’ and H-2′, confirmed the β-configuration of the glucopyranosyl moiety (for an α-glucopyranoside: J = 3.7 Hz; δC 98.5 ppm) [17,18]. As for compound 2, the length of the fatty acid chain was determined by its methanolysis, while ensuing peak detection using LC-HRESIMS analysis for the fatty acid methyl ester obtained from 3. According to the method described in [19], compound 3 reacted with 5% methanolic HCl, giving the corresponding hydroxy acid methyl ester, phytosphingosine, and a sugar molecule. The LC-HRESIMS spectrum of the fatty acid methyl ester of 3 exhibited a peak with a molecular ion of m/z 369.3358 [M]+ (Figure S32) corresponding to a C23H44O3 fatty acid methyl ester, (E)-methyl 2-hydroxydocos-14-enoate. Further confirmation of the double bond in α-hydroxy fatty acid methyl ester was provided by GC–MS analysis. The α-hydroxy fatty acid methyl ester resulted after hydrolysis C23H44O3 with m/z 369.3358 [M]+ was subjected to oxidation by KmnO4, as described in [19], to yield fatty acid methyl ester C22H42O2 with m/z 338 that was subjected to GC–MS analysis; resulting fragments confirmed the position of the double bond by the presence of mass fragments at m/z 239 corresponding to a [C15H27O2•] fragment, m/z 139 corresponding to a [C10H19•] fragment, and m/z 125 corresponding to a [C9H17•] fragment (Figure S33). Therefore, the position of the double bond was authenticated in 14’ at (E) methyl 2-hydroxydocos-14-enoate. The sugar liberated from the hydrolysis displayed a specific rotation of +17.2 (c = 0.1%, H2O), which was identical to that of the standard, d-glucose. Furthermore, the tR of the acetylated thiazolidine derivative of the liberated sugar was similar to that of the standard, d-glucose (19.7 min). The relative configurations at the stereocenters of the cerebroside moieties were assigned to be (2S, 3S, 4R, and 2’R), as evidenced by the comparison of their physical properties and NMR data (measured in C5D5N) with those of analogs reported in the literature [20]. The recorded optical rotation +17.40 (c 1.00, MeOH) and the aforementioned 13C NMR signals (C-1, C-2, C-3, C-4, and C-2’), along with the chemical shifts of their corresponding protons in the 1H NMR (measured in C5D5N) spectrum, were in line with those reported for cerebroside HLG-1 [20] and HPC-3 [12]. From the above data, the structure of compound 3 was assigned as presented in Figure 3. To the best of our knowledge, it was a new compound, henceforth named spiniferoside C.
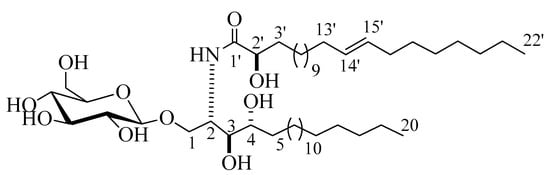
Figure 3.
Chemical structure of compound 3, spiniferoside C.
Compound 4 (Figure 4) was isolated as a white powder. Derived from its NMR spectral data, the molecular formula of 4 was C27H46O4S, representing five double bond equivalents. The interpretation of the 1H NMR spectrum of compound 4 (Figure S34) indicated the presence of two methyl singlets at δH 0.62 and 0.75 assigned to CH3-19 and CH3-18, respectively, along with three methyl doublets at δH 0.85, 0.84, and 0.91 assigned to CH3-21, CH3-26, and CH3-27, respectively, which was in agreement with the presence of a steroidal nucleus [22]. Moreover, a trisubstituted olefinic proton at δH 5.28 (m) assigned to H-6 was observed in addition to an oxygenated methine at δH 3.93 (m), corresponding to an axial proton next to the O-sulfate group at C-3. The 13C NMR spectrum (Figure S35) indicated the presence of five methyl groups (CH3), eleven methylenes (CH2), eight methines (CH), and three quaternary carbons (C), besides an oxymethine carbon at δC 78.4 assigned to be C-3, to which the sulfate group was attached. Comparing the chemical shift values of H-3/C-3 with the data reported in [23] suggested the presence of a cholesterol sulfate structure. The afore-mentioned 1H NMR and 13C NMR spectral data came into line with those reported for cholesterol sulfate [23], so compound 4 was identified as cholesterol-3-O-sulfate.
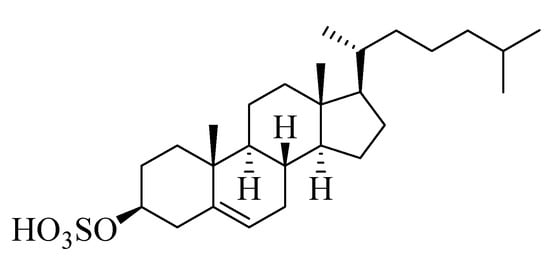
Figure 4.
Chemical structure of compound 4, cholesterol-3-O-sulfate.
2.2. Metabolic Profiling
The crude extract of H. spinifera was tested for its in vitro cytotoxicity on five cancerous cell lines: HepG2 (liver cancer cell line), MCF7 (breast cancer cell line), PC3 (prostate cancer cell line), HCT 116 (colon cancer cell line), and HeLa (cervix cancer cell line). This test was performed by applying the sulforhodamine B (SRB) assay using the method adapted by Skehan et al. [24], which follows the protocol described by Vichai and Kirtikara [25]. An ELISA reader was used to measure the color intensity, and the corresponding IC50 values (extract concentration which reduces survival of cancer cells to 50%) were calculated.
The crude extract of H. spinifera displayed potent cytotoxic activity on MCF7 with an IC50 value of 4.58 µg/mL and weak activity on HCT 116 with an IC50 of 19.4 µg/mL. On the contrary, the extract exhibited no significant cytotoxicity on PC3, HepG2, and HeLa cells, achieving IC50 values of 44.7, 24.5, and 32.3 µg/mL respectively, as established by the U.S. National Cancer Institute (NCI; the criterion of cytotoxicity is an IC50 < 20 µg/mL), in a preliminary evaluation [26]. Upon treatment with the crude extract of H. spinifera, the MCF-7 cells exhibited an enhanced rate of cell death with a lower concentration of the extract compared to that in other tested cell lines.
Based on the aforementioned results and motivated by the potent cytotoxicity of the H. spinifera extract on MCF-7 cells, we identified the secondary metabolites in the extract using the LC-HRESIMS technique.
LC-HRESIMS metabolic analysis is a new strategy for the comprehensive phytochemical characterization of complex crude extracts, along with targeting specific metabolites that can be associated with certain biological activities prior to sometimes tedious and time-consuming purification procedures. By using combinations of different analytical methods, the bioassay-guided isolation route is shortened and putative identification of potentially new natural products is rapidly achieved. Since the metabolic profile in each extract varies by its physical nature and ionization potential, both the positive and negative ionization modes were combined, so that detection of the maximum possible metabolites was accomplished. A portion of 57% of the metabolites were detected in positive mode and 39% of the metabolites were found in negative mode, while only 4% were detected in both modes. Prior to dereplication, molecular formula prediction was done utilizing the MZmine algorithm, which employs a combination of empirical techniques that include isotope pattern matching. Using the positive and negative mode electrospray ionization, with spectral data at a MW tolerance within 5 ppm, known compounds were tentatively identified. In the data processing step, two parameters were used, the “retention time” and the “m/z tolerance range.” If the variance was deemed to be too high (i.e., 20% for LC-HRESIMS data), the feature was removed from the analysis. This processing step was performed at the start of the data analysis process.
The metabolomic profiling of Holothuria spinifera using LC-HRESIMS led to the identification of secondary metabolites belonging to the following chemical groups: fatty acids, phenolic diterpenes, and triterpenes. The metabolites in the extract (Table 2 and Figure 5) were tentatively identified by searching in databases, e.g., in the Dictionary of Natural Products (DNP), and in (METLIN).
Table 2.
Dereplicated metabolites reported to occur in Holothuria spinifera.
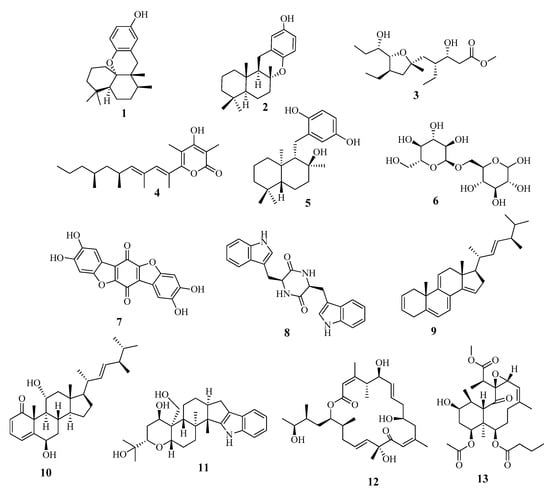
Figure 5.
Dereplicated metabolites from Holothuria spinifera.
According to the literature, the majority of the identified secondary metabolites displayed cytotoxic and anticancer effects. For instance, Aureol in vitro cytotoxic activities were tested against human liver carcinoma (Hepa59T/VGH), human oral epidermoid carcinoma (KB), and human cervical epithelioid carcinoma (Hela) tumor cell lines and it showed promising cytotoxic activities. [27]. Assays on cytotoxic activity were performed for epichromazonarol against P-388, A-549, HT-29, and MEL-28 cells, and in all cases an IC50 = 15.9 μM was obtained for epichromazonarol [28], while Plakortether E was evaluated for cytotoxic activity against two different cell lines: WEHI 164 (murine fibrosarcoma) and RAW 264-7 (murine macrophage). Plakortether E proved to be selectively active against the second cell line [29]. Moreover, stoloniferone O, imposed significant antitumoral effects on HT-29 and P-388 cell lines [30]. Furthermore, the antiangiogenesis activity of thelephoric acid was reported, as it was evaluated using a tyrosine kinase assay kit with recombinant human VEGFR2. Since VEGFR2, a principal regulator of angiogenesis, is highly expressed in umbilical vein endothelial cells, thelephoric acid was examined on cell growth of HUVECs by a WST-8 assay. As a result, it exhibited antiproliferative activity in HUVECs. It also markedly inhibited cell invasion [31]. In addition, terpendole F inhibited cholesterol acyl-transferase (ACAT), a crucial enzyme in cancer progression [32].
All the above-mentioned data supported the displayed cytotoxicity of the H. spinifera extract.
2.3. In Vitro Evaluation of the Antitumor Activities of the Isolated Compounds
The cytotoxicity levels of ceramides and cerebrosides on wide array of cancer cell lines have previously been reported [42,43,44]. SJG-2 is one example of a ganglioside molecular species recently isolated from Stichopus japonicas possessing neurogenic activity against the rat pheochromocytoma cell line PC12 [45]. Jia and coworkers proved the effectiveness of a glucocerebroside mixture isolated from the sea cucumber Cucumaria frondosa on the HepG2 liver cancer cell line [46]. The ophidiacerebrosides, which are cerebroside molecular species isolated from the African starfish Narcissia canariensis, displayed promising cytotoxicity on multiple myeloma (KMS-11), glioblastoma (GBM), and colorectal adenocarcinoma (HCT-116) cell lines [47]. Therefore, compounds 2, 3, and 4 and the spiniferoside A molecular mixture 1 isolated from the sea cucumber Holothuria spinifera, were assessed for their cytotoxic effect on MCF-7 cancer cell line by applying the sulforhodamine B (SRB) assay using the method adapted by Skehan et al. [24], which follows the protocol described by Vichai and Kirtikara [25]. The MCF-7 cancer cells were chosen particularly for the cytotoxicity evaluation rather than any cancerous cell lines based on our results, which revealed the potent cytotoxicity of H. spinifera on MCF-7 cells. As presented in Table 3, all four compounds showed promising anticancer activities against cells of the MCF-7 breast cancer cell line. Compounds 2 and 3 demonstrated promising cytotoxic activities against MCF-7 cancer cells, with IC50 values of 8.13 and 8.27 µM respectively compared to the standard drug doxorubicin with IC50 values of 8.64 µM. Compound 1 showed a slightly lower effect, with an IC50 value of 13.83µM. The exhibited cytotoxicity of compound 1 could be attributed to the major component spiniferoside A2, as reported in the literature [47,48,49]. Cholesterol sulfate (4) exhibited the least cytotoxicity, with an IC50 value of 35.65 µM.
Table 3.
IC50 values of compounds 1, 2, 3, and 4 on the MCF-7 breast cancer cell line.
Ceramide acts as a cytotoxic agent and tumor-suppressor by induction of apoptosis. It was observed that treatment with exogenous ceramide activates a cascade of caspases, protein kinases, and phosphatases and leads to apoptosis [50]. On the other hand, glucosylceramide exerts its cytotoxic effect by arresting cell renewal capacity instead of induction of apoptosis. This could be explained in the light of the fact that sphingoid bases were reported to induce cell cycle arrest at G2/M phase. The presence of a glucose moiety increases the water-solubility of the compound, enhancing its membrane permeability. As a consequence, it is possible that a small amount of ceramide penetrates into cells, and then hydrolyzes to its sphingoid base, which in turn affects the cell physiology [51].
Doxorubicin was used as a standard drug in our cytotoxic assay based on the fact that it represents a pillar of various cancer treatment protocols, including breast cancer [52]. It is known that doxorubicin increases endogenous ceramide in drug-sensitive MCF-7, resulting in apoptosis [53] by induction of ceramide de novo synthesis [50].
Impairment of ceramide metabolism in cancer cells is an escape pathway necessary for their survival which leads to drug resistance. As a consequence, tumor progression and metastasis occur [54]. To overcome chemotherapy resistance, drug combinations were used. A recent study revealed that combining exogenous C6-ceramide sensitizes multiple cancer cell lines to doxorubicin or etoposide. In addition, combining sorafenib with nano liposomal ceramide enhances sensitivity to sorafenib in other cancer cell types, including breast carcinoma and melanoma [50].
Therefore, the isolated cerebrosides 2 and 3 could be used as an adjuvant therapy with doxorubicin to overcome drug resistance and increase the sensitivity to doxorubicin. In the future, a comparative pharmacological study will be conducted to evaluate the in vivo efficacy of the isolated cerebroside as an anticancer agent individually and in combination of doxorubicin to verify our assumption.
2.4. Molecular Docking Studies
A literature survey revealed that the SET oncoprotein plays a role in cancer progression by affecting multiple cellular processes through the inhibition of tumor suppressor protein phosphatase 2A (PP2A) and metastasis suppressor nm23-H1, or through alterations of inhibitory ceramide synthesis [55,56]. Sphingolipid ceramide has been reported to activate PP2A via direct interaction between them. Recently it has been revealed that an additional mechanism which involves the direct binding of ceramide with the SET protein may contribute to the activation of ceramide to PP2A [57]. Molecular docking studies suggested that the SET domain possesses a hydrophobic ceramide-binding pocket characteristic of the lipid binding pocket.
For this reason, targeting the inhibition of SET would have a significant and effective role in inhibiting the proliferation of cancer cells. The ability of the cerebrosides 1, 2, and 3, and the sterol sulfate 4 to bind to the SET oncoprotein was examined by molecular docking. To further estimate a predictive picture concerning the site where the natural compounds are binding to the SET oncoprotein, docking scores for the natural compounds with the ligand are provided (Table 4).
Table 4.
Molecular docking studies.
In this study, the docking pose of spiniferoside A (1) showed the hydrophobic skeleton, which was stabilized by the hydrophobic interactions with the amino acids Phe 68 and Pro 214. Per the predicated binding pose of spiniferoside C (3), two hydrogen bond donors were formed between the hydroxy group of 3 and the carbonyl oxygens of Asn 61 and Glu 57 with distances of 3.22 and 2.64 Å, respectively. Additionally, an extra hydrogen bond acceptor was found to be formed with the Glu 116 residue (Figure 6). This may explain the potent inhibitory activity of spiniferoside C (3) against the breast cancer cell line with an IC50 value of 8.27 µM. Spiniferoside B (2), on the other hand, formed two hydrogen bonds with Asn 61 and Glu 57 (Figure 6). These interactions may be missing, with relatively low binding energy scores, in cholesterol sulfate (4), which showed the least inhibitory activity against the MCF-7 cancer cell line. Based on molecular modeling, the compounds exhibited their anti-tumor activity through targeting the PP2A inhibitor, SET. Overexpression of the SET oncoprotein was reported in breast cancer cell lines, including MCF-7 [58,59]. The use of SET antagonists and PP2A activators was postulated as a relatively novel strategy for breast cancer therapy [59]. In a study that involved MDA-MB-231 breast adenocarcinoma cell line, targeting SET inhibited cellular proliferation and decreased cellular migration and invasion via activation of the tumor suppressors PP2A and nm23-H1 [55]. Suppression of SET reversed the resistance in MCF-7/PTX human breast carcinoma [60]. The pronounced cytotoxic effect of SET inhibitors in breast cancer might be explained by the concomitant activation of PP2A, a suggested therapeutic target in resistant MCF-7 cells [61]. Inhibition of PP2A has been associated with poor prognosis of breast cancer [62]. PP2A regulates the expression of estrogen receptor (ER), a key molecular determinant of breast cancer status and survival [62,63]. PP2A activation was raised as an effective treatment target in MCF-7 breast cancer cell line through its mediation of ERα expression via modulating ER mRNA stability [64].
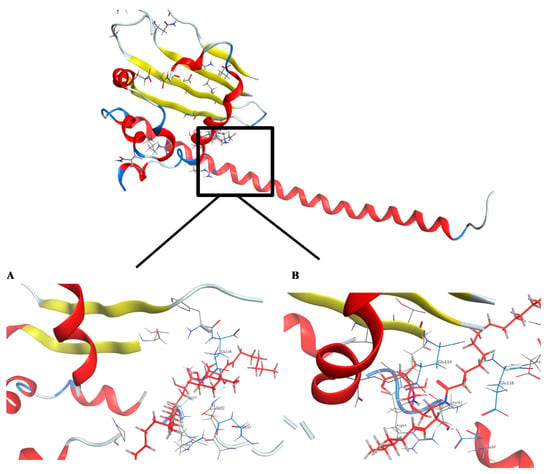
Figure 6.
Crystal structure of the predicted docking pose (in blue) of (A) compound 2 and (B) compound 3 with the SET oncoprotein (PDB code: 2E50).
3. Materials and Methods
3.1. General Experimental Procedures
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded using the residual solvent signal as an internal standard on a Varian AS 400 (Varian Inc., Palo Alto, CA, USA). High-resolution mass spectra were recorded using a Bruker BioApex (Bruker Corporation). Pre-coated silica gel G-25 UV254 plates were used for thin-layer chromatography (TLC) (20 cm × 20 cm) (E. Merck, Darmstadt, Germany). Silica gel (Purasil 60Å, 230–400 mesh) was used for flash column chromatography (Whatman, Sanford, ME, USA). Semi-preparative high-performance liquid chromatography (HPLC) was performed on a Cosmosil 5 C18–MS–II (150 × 4.6 mm) at a flow rate of 0.5 mL/min. The column was equipped with a TOSOH RI-8020 detector and a JASCO BIP-I HPLC pump [20].
3.2. Sea Cucumber Material
The sea cucumber Holothuria spinifera was collected from Sharm El Sheikh in the Egyptian Red Sea. The material was air-dried and stored at −24 °C until further processing. Identification of the sea cucumber was performed by Dr. Tarek Temraz, Marine Science Department, Faculty of Science, Suez Canal University, Ismailia, Egypt. Voucher specimens were deposited at the Herbarium Section of the Pharmacognosy Department, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt under the registration number SAA-129.
3.3. Extraction and Isolation
Material of the sea cucumber Holothuria spinifera (2 kg) was frozen and chopped into small pieces, and then extracted with a mixture of MeOH/CH2Cl2 (1:1) (3 × 2 L) at room temperature. The resulting crude extract was evaporated under vacuum to afford 100 g of a dark-greenish residue. The extract was slurred with a small portion of silica gel. The mixture was transferred to the top of a sintered-glass Büchner funnel (15 cm × 10 cm) packed with 300 g of silica gel and connected to a vacuum pump. Fractionation was performed by step gradient elution using a non-polar solvent (n-hexane) with increasing the polarity using ethyl acetate (EtOAc) and then MeOH to give nine fractions: (HS-1–HS-9). Fraction HS-5 (75:25 EtOAc:MeOH) (2.52 g) was placed on a silica gel column and eluted initially with 100% CHCl3, followed by gradient systems of CHCl3:MeOH to (65:35), which yielded eight subfractions (HS-5-S-1 to HS-5-S-8). Subfraction HS-5-S-3 (150 mg) was chromatographed on silica gel using gradient systems of CHCl3:MeOH starting with (90:10) to (65:35) followed by a Sephadex LH-20 column for final purification using CHCl3:MeOH (1:1) isocratic elution to obtain compound 1 (17 mg, white amorphous powder). Compound 1, as a molecular species mixture (3 mg) was finally resolved using semi-preparative HPLC (Cosmosil 5 C18, 100% MeOH) to afford spiniferoside A2 (1b, 0.9 mg), spiniferoside A1 (1a, 0.12 mg), and spiniferoside A3 (1c, 0.3 mg).
Subfraction HS-5-S-4 (200 mg) was re-chromatographed on a silica gel column using gradient systems of CHCl3:MeOH starting with (90:10) to (65:35) followed by a Sephadex LH-20 column twice using CHCl3:MeOH (1:1) isocratic elution to obtain compound 2 (15 mg, white amorphous powder). Subfraction HS-5-S-6 (250 mg) was subjected to silica gel column chromatography using gradient systems of CHCl3:MeOH starting with (90:10) to (65:35) followed by purification on Sephadex LH-20 twice using CHCl3:MeOH (1:1) isocratic elution to obtain compound 3 (19 mg, white amorphous powder). Fraction HS-4 (100% EtOAc) (0.83 g) was placed on a silica gel column and eluted initially with 100% CHCl3, and then with gradient systems of CHCl3:MeOH until (65:35); the effluent yielded eight subfractions (HS-4-H-1 to HS-4-H-8). Subfraction HS-4-H-6 (300 mg) was re-chromatographed on silica gel using gradient systems of CHCl3:MeOH starting with (90:10) to (65:35) and then subjected to a Sephadex LH-20 column chromatography for final purification using CHCl3:MeOH (1:1) isocratic elution to obtain compound 4 (40 mg, white amorphous powder).
3.4. Cerebroside Hydrolysis
Compounds 1, 2, and 3 were subjected to methanolysis, in which 2 mg each of the respective compounds was heated with 5% HCl in MeOH (0.5 mL) at 70 °C for 8 h in a sealed small-volume vial. The reaction mixture was extracted with n-hexane, and the hexane layer was evaporated under vacuum until dryness to give a mixture of FAM (fatty acid methyl esters) for LC-HRESIMS analysis. For further confirmation of the double bond in fatty acid side chain in compounds 2 and 3, α-hydroxy fatty acid methyl esters obtained from hydrolysis were subjected to Lemieux oxidation. Hence, 0.023 mol/L aqueous KMnO4 and 0.09 mol/L NaIO4 (2.0 mL) were slowly added to the α-hydroxy fatty acids/methyl esters mixture, t-BuOH (1.0 mL), and 0.04 mol/L aqueous K2CO3 (0.5 mL). Then, the mixture was stirred for 18 h at 37 °C, quenched with 2.5 mol/L H2SO4 (0.1–0.3 mL) and saturated aqueous Na2SO3, and then extracted with Et2O. The organic layer was dried over Na2SO4. Finally, the concentrated, dried residue was esterified with excess CH2N2 in Et2O overnight. The resulting fatty acid methyl esters were used for GC–MS analysis [19]. The identification of the fatty acids and methyl esters was de-convoluted using AMDIS software (www.amdis.net) and identification was done by retention indices (mass spectrum matching to authentic standards; Wiley spectral library collection and NSIT library database).
3.5. Identification of the Sugar Moiety in Compounds 1, 2, and 3
In a sealed small-volume vial, 5 mg of each compound 1, 2, and 3 was heated in 5% HCl/MeOH (0.5 mL) at 70 °C for 8 h. CHCl3 was used to extract the reaction mixture for the removal of the fatty acids released. The methanolic layer was then neutralized with Ag2CO3 to provide the methylated sugar followed by HPLC checking (Cosmosil-sugar-D, 4.6 ID X 250 mm, 1 mL/min, RI detector, 95% acetonitrile) against the standards, glucose and galactose. Glucose displayed a retention time of 4.62 min, which was found as being our sample retention time; meanwhile, the retention time of galactose was 4.76 min.
3.6. Determination of the Configuration of the Sugar Moiety in 1, 2, and 3
For compounds 1, 2, or 3, 2 mg each was hydrolyzed by heating in 0.5 M HCl (0.1 mL) and then neutralized with Amberlite IRA400. After drying in vacuo, the residue was dissolved in pyridine (0.1 mL) containing L-cysteine methyl ester hydrochloride (0.5 mg) and heated at 60 °C for 1 h. A 0.1 mL solution of o-tolylisothiocyanate (0.5 mg) in pyridine was added to the mixture, which was heated to 60 °C for 1 h. The reaction mixture was directly analyzed by reversed-phase HPLC (Cosmosil 5C18-AR-II, 4.6 ID X 250 mm, 0.8 mL/min, λ = 250 nm, 25% acetonitrile in 50 mM H3PO4). The same derivatization procedures were done for 5 mg of standards, d-glucose (tR = 19.7 min), l-glucose (tR = 19.5 min), d-galactose (tR = 18.6 min), and l-galactose (tR = 19.3 min), evidencing the sugar portion to be d-glucose in all three cases.
3.7. Metabolomic Profiling
Metabolic profiling of the sea cucumber material was performed using the method described by Elsayed et al. [65]. These files were imported for peak picking, deconvolution, deisotoping, alignment, and formula prediction into the data mining program MZmine 2.10. Comparison with the MarinLit database and the Dictionary of Natural Products (DNP) 2015 achieved dereplication of a broad array of compounds.
3.8. Spectroscopic Data
Compounds 1, 2 and 3; were isolated as white powder; their 1H NMR (C5D5N, 400 MHz) and 13C NMR (C5D5N, 100 MHz) spectral data—see Table 1.
Compound 4; was isolated as white powder; 1H NMR (DMSO, 400 MHz) and 13C NMR (DMSO, 100 MHz) spectral data were similar to those reported in the literature [23].
1H NMR (DMSO, 400 MHz); δH 0.91–1.89* (2H, H-1), 1.23–1.97*(2H, H-2), 3.93 (1H, m, H-3), 2.35–2.38* (2H, H-4), 5.28 (1H, m, H-6), 1.94–2.33* (2H, H-7), 1.15–2.33* (1H, H-8), 1.15–2.33* (1H, H-9), 0.91–1.07* (2H, H-11), 1.15–2.38* (2H, H-12), 1.15–2.38* (1H, H-14), 1.15–2.38* (2H, H-15), 1.15–2.38* (2H, H-16), 1.15–2.38* (1H, H-17), 0.75 (3H, s, H-18), 0.99 (3H, s, H-19), 1.15–2.38* (1H, H-20), 0.85* (3H, H-21), 1.15–2.38* (2H, H-22), 1.15–2.38* (2H, H-23), 1.15–2.38* (2H, H-24), 1.15–2.38* (1H, H-25), 0.84–0.88* (3H, H-26), 0.84–0.88* (3H,H-27).
* The overlapped signals are reported without multiplicity
13C NMR (DMSO, 100 MHz); δC 38.2 (C-1), 31.9 (C-2), 78.4 (C-3), 42.1 (C-4), 143.8 (C-5), 124.2 (C-6), 40.0 (C-7), 34.5 (C-8), 52.7 (C-9), 38.2 (C-10), 22.1 (C-11), 25.5 (C-12), 38.4 (C-13), 56.8 (C-14), 25.5 (C-15), 21.7 (C-16), 56.8 (C-17), 14.8 (C-18), 21.7 (C-19), 34.8 (C-20), 21.7 (C-21), 34.5 (C-22), 23.9 (C-23), 40.0 (C-24), 27.0 (C-25), 22.9 (C-26), 22.9 (C-27).
3.9. Cytotoxicity Assays
The cyctotoxic activities of H. spinifra MeOH/CH2Cl2 (1:1) crude extract and the isolated compounds 1, 2, 3, and 4 were investigated by the sulforhodamine B (SRB) assay, as mentioned in Skehan et al. [24], according to the method described by Vichai and Kirtikara [25]. The cytotoxicity of the crude extract was tested on HepG2 (liver cancer cell line), MCF7 (breast cancer cell line), PC3 (prostate cancer cell line), HCT 116 (colon cancer cell line), and HeLa (cervix cancer cell line). For the evaluation of the cytotoxicity of the isolated compounds, MCF-7 cancer cells were chosen; 96-well microtiter plates were used to seed cells at an initial concentration of 3 × 103 cell/well in 150 µL of fresh medium and left for 24 h to get attached to the plates. The drugs were applied to the plates at serial different concentrations of 0, 5, 12.5, 25, and 50 µg/mL, and then all plates were incubated for 48 h. The cells were fixed with 50 μL of cold trichloroacetic acid 10% for 1 h at 4 °C, and then distilled water was used to wash the plates (automatic washer Tecan, Germany). SRB (50 μL, 0.4%) dissolved in 1% acetic acid used to stain the plates for 30 min at room temperature; then the plates were washed with 1% acetic acid and air-dried; 100 μL/well of 10 M tris base (pH 10.5) was used to solubilize the dye, and the optical density (OD) of each well was measured spectrophotometrically at 570 nm using an ELISA microplate reader (Sunrise Tecan reader, Germany). The mean background absorbance was automatically taken and each drug’s mean value of concentration was calculated. The experiment was done three times, and then the IC50 values were calculated.
3.10. Molecular Docking Studies
The crystallographic structure of the SET protein, inhibitor of PP2A, in complex with its ligand, was available from the Protein DataBank [66]. The structure arrangement process was used to revise the protein errors in addition to the creation of a reasonable protein structure that was set up on default rules on MOE (Molecular Operating Environment). Removal of water molecules and the ligands that were not involved in the binding was carried out using quick preparation protocol for the preparation of the protein. Finally, the Gasteiger methodology was applied to calculate the partial charges of the protein. Molecular docking studies were performed using Molecular Operating Environment 2019.0101 software [67]. The ligand coordinates of compounds 1b, 1b, 1c, 2, 3, and 4 were built using ChemDraw Ultra 11.0. Their protonation, the atom correction, and bond types were then defined; hydrogen atoms were added; protonation, and final minimization were performed (AMBER10, gradient: 0.01). The docking experiment on the SET gene protein was performed by redocking the ligand on in the PDB file 2E50, after which the ligand was deleted. The default Triangle Matcher placement method was selected for docking. GBVI/WSA dG scoring function, which determines the free energy of binding of the ligand from a given pose, was chosen to rank the final poses. The ligand complex with the protein having the lowest S-score was selected. The redocking of the ligand with its target revealed an RMSD (root mean square deviation) 0.606 Å, which confirms that the ligand binds to the same pocket and assures the dependability of parameters of docking.
4. Conclusions
The results presented in this paper demonstrate the beneficial impact of LC-HRESIMS profiling when combined with bioassay-guided drug research from marine invertebrates to accelerate the commonly long processes of identifying an active metabolites by successive isolation from crude extracts. Dereplication experiments focused on chemotaxonomic sorting helped identify putatively active metabolites, while structural elucidation of the isolated compounds, using both HRMS and NMR, verified the hits. The bioactive extract metabolomic profiling indicated the existence of various secondary metabolites, primarily fatty acids, phenolic diterpenes, and triterpenes. Thus, bioassay-guided isolation coupled to LC-HRESIMS metabolomic profiling of the Red Sea cucumber Holothuria spinifera led to the characterization of four compounds: three new cerebrosides (compounds 1, 2, and 3), and cholesterol sulfate (compound 4), marking the first report of it in the sea cucumber species Holothuria spinifera. Compounds 2 and 3 showed larger cytotoxic effects against MCF-7 cancer cells, with IC50 values of 8.13 and 8.27 µM, respectively. Compound 1 displayed a slightly lower activity, with an IC50 value of 13.83 µM, while the steroid 4 was the least active, displaying an IC50 value of 35.56 µM. The cytotoxicities of compounds 1, 2, 3, and 4 were compared to that of the standard drug doxorubicin with an IC50 value of 8.64 µM, which was used as a positive control. Finally, the obtained findings were confirmed by docking studies, which demonstrated the abilities of the four compounds to fit to the binding site of the SET oncoprotein and the inhibitor of PP2A. We assume that in vivo studies on these remarkable metabolites will be of impact to the potential development of new antineoplastic drugs.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/18/8/405/s1, Figures S1−S15: LC-HRESIMS, 1H NMR, 13C NMR and HPLC purification of compound 1, Figures S16–S24: LC-HRESIMS, 1H NMR, 13C NMR of compound 2, Figure S25: LC-HRESIMS for α-hydroxy fatty acid methyl ester after hydrolysis of compound 2, Figure S26: GC-MS analysis of fatty acid methyl ester carried out after oxidation of α-hydroxy fatty acid methyl ester compound 2, Figures S27–31: LC-HRESIMS, 1H NMR and 13C NMR of compound 3, Figure S32: LC-HRESIMS for α-hydroxy fatty acid methyl ester after hydrolysis of compound 3, Figure S33: GC-MS analysis of fatty acid methyl ester carried out after oxidation of α-hydroxy fatty acid methyl ester of compound 3, Figures S34–S35: 1H NMR and 13C NMR of compound 4 and Figures S36–S40: Cytotoxicity of compounds 1, 2, 3, 4 and doxorubicin on MCF-7.
Author Contributions
Conceptualization, U.R.A., S.A.A., R.F.A.A., G.B., H.A.H., and A.K.I.; methodology, D.M.H. and R.F.A.A.; data curation, R.F.A.A., D.M.H., E.E.E., A.M.A., S.F., A.M.A., U.R.A., and T.A.; writing—original draft preparation, D.M.H. and U.R.A.; writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This publication was funded by the German Research Foundation (DFG).
Acknowledgments
All authors are thankful to Dr. Tarek Temraz (Department of Marine Science, Faculty of Education, University of Suez Canal, Ismailia, Egypt). Many thanks and our gratitude go to the Egyptian Environmental Affairs Agency (EEAA) for promoting the collection of samples along the Red Sea coasts. Thanks is due to the Deutscher Akademischer Austauschdienst (DAAD) for the generous award of a scholarship to S.F. This publication was supported by the Open Access Publication Fund of the University of Würzburg.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eltahawy, N.A.; Ibrahim, A.K.; Radwan, M.M.; Zaitone, S.A.; Gomaa, M.; ElSohly, M.A.; Hassanean, H.A.; Ahmed, S.A. Mechanism of action of antiepileptic ceramide from Red Sea soft coral Sarcophyton auritum. Bioorg. Med. Chem. Lett. 2015, 25, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; El-Hossary, E.M.; Oelschlaeger, T.A.; Donia, M.S.; Quinn, R.J.; Abdelmohsen, U.R. Potential of marine natural products against drug-resistant bacterial infections. Lancet Infect. Dis. 2019, 19, 237–245. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs. 2019, 17, 491. [Google Scholar] [CrossRef]
- Castellano, I.; Seebeck, F.P. On ovothiol biosynthesis and biological roles: From life in the ocean to therapeutic potential. Nat. Prod. Rep. 2018, 35, 1241–1250. [Google Scholar] [CrossRef]
- Gerdol, M.; Sollitto, M.; Pallavicini, A.; Castellano, I. The complex evolutionary history of sulfoxide synthase in ovothiol biosynthesis. Proc. Biol. Sci. 2019, 286, 20191812. [Google Scholar] [CrossRef]
- Mokhlesi, A.; Saeidnia, S.; Gohari, A.R.; Shahverdi, A.R.; Nasrolahi, A.; Farahani, F.; Khoshnood, R.; Es’haghi, N. Biological activities of the Sea Cucumber Holothuria leucospilota. Asian J. Anim. Vet. Adv. 2012, 7, 243–249. [Google Scholar] [CrossRef]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Ebada, S.S.; Lin, W.H.; Proksch, P. Bioactive Sesquiterpenes and Triterpenes from Marine Sponges: Occurrence and Pharmacological Significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Aamer, M.A.; Lawrence, A.J. Identification of the Holothurian species of the Red Sea and Gulf of Aqaba using DNA barcoding technique. Egypt. J. Aquat. Biol. Fish. 2016, 20, 1–7. [Google Scholar] [CrossRef]
- Yamada, K.; Harada, Y.; Nagaregawa, Y.; Miyamoto, T.; Isobe, R.; Higuchi, R. Isolation and structure of biologically active gangliosides from the sea cucumber Holothuria pervicax. Org. Chem. 1998, 1, 2519–2525. [Google Scholar] [CrossRef]
- Yamada, K.; Sasaki, K.; Harada, Y.; Isobe, R.; Higuchi, R. Isolation and structure of glucocerebrosides from the sea cucumber Holothuria pervicax. Chem. Pharm. Bull. 2002, 50, 1467–1470. [Google Scholar] [CrossRef][Green Version]
- Abdelmohsen, U.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49. Mar. Drugs. 2014, 12, 1220–1244. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Huang, T.T.; Chen, Y.T.; Chen, J.L.; Chu, P.Y.; Huang, C.T.; Wang, W.L.; Lau, K.Y.; Dai, M.S.; Shiau, C.W.; et al. Targeting SET to restore PP2A activity disrupts an oncogenic CIP2Afeedforward loop and impairs triple negative breast cancer progression. EBioMedicine 2019, 40, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ruckhäberle, E.; Rody, A.; Engels, K.; Gaetje, R.; Minckwitz, G.V.; Schiffmann, S.; Grösch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 2008, 112, 41–52. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Pomorski, T.; Raggers, R.J.; Sprong, H.; Meer, G.V. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 2001, 81, 1689–1723. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 3rd ed.; John Wiley & Sons: New York, NY, USA,, 1974. [Google Scholar]
- Thomford, A.K.; Abdelhameed, R.F.A.; Yamada, K. Chemical studies on the parasitic plant Thonningia sanguinea Vahl. RSC Adv. 2018, 8, 21002–21011. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Y.; Liu, K.; Hua, H.; Zhu, H.; Pei, Y. Gracilarioside and Gracilamides from the Red Alga Gracilaria asiatica. J. Nat. Prod. 2006, 69, 1488–1491. [Google Scholar] [CrossRef]
- Yamada, K.; Matsubara, R.; Kaneko, M.; Miyamoto, T.; Higuchi, R. Isolation and structure of a biologically active ganglioside molecular species from the sea cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2001, 49, 447–452. [Google Scholar] [CrossRef]
- Abdelhameed, R.F.; Ibrahim, A.K.; Yamada, K.; Ahmed, S.A. Cytotoxic and anti-inflammatory compounds from Red Sea grass Thalassodendron ciliatum. Med. Chem. Res. 2018, 27, 1238–1244. [Google Scholar] [CrossRef]
- Goad, L.J.; Akihisa, T. Analysis of Sterols; Blackie Academic and Professional: London, UK, 1997; 1H NMR spectroscopy of sterols; pp. 197–234. [Google Scholar]
- Gallo, C.; D’Ippolito, G.; Nuzzo, G.; Sardo, A.; Fontana, A. Autoinhibitory sterol sulfates mediate programmed cell death in a bloom-forming marine diatom. Nat. Commun. 2017, 8, 1292. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahn, J.M.; Vistica, D.; Warren, J.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Nat. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Vijayarathna, S.; Sasidharan, S. Cytotoxicity of methanol extracts of Elaeis guineensis on MCF-7 and Vero cell lines. Asian Pac. J. Trop. Biomed. 2012, 2, 826–829. [Google Scholar] [CrossRef]
- Shen, Y.C.; Liaw, C.C.; Ho, J.R.; Khalil, A.T.; Kuo, Y.H. Isolation of aureol from Smenospongia sp. and cytotoxic activity of some aureol derivatives. Nat. Prod. Res. 2006, 20, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Cytotoxic Terpene Quinones from Marine Sponges. Mar. Drugs 2010, 8, 2849–2870. [Google Scholar] [CrossRef]
- Campagnuolo, C.; Fattorusso, E.; Taglialatela-Scafati, O.; Ianaro, A.; Pisano, B. Plakortethers A−G: A New Class of Cytotoxic Plakortin-Derived Metabolites. Eur. J. Org. Chem. 2002, 1, 61–69. [Google Scholar] [CrossRef]
- Duh, C.Y.; Lo, I.W.; Wang, S.K.; Dai, C.F. New cytotoxic steroids from the soft coral Clavularia viridis. Steroids 2007, 72, 573–579. [Google Scholar] [CrossRef]
- Nagasawa, I.; Kaneko, A.; Suzuki, T.; Nishio, K.; Kinoshita, K.; Shiro, M.; Koyama, k. Potential Anti-angiogenesis Effects of p-Terphenyl Compounds from Polyozellus multiplex. J. Nat. Prod. 2014, 77, 963–968. [Google Scholar] [CrossRef]
- Tomoda, H.; Tabata, N.; Yang, D.-J.; Takayanagi, H.; Omura, S. Terpendoles, novel ACAT inhibitors produced by Albophoma yamanashiensis. J. Antibiot. 1995, 48, 793–804. [Google Scholar] [CrossRef]
- Longeon, A.; Copp, B.R.; Quévrain, E.; Roué, M.; Kientz, B.; Cresteil, T.; Petek, S.; Debitus, C.; Bourguet-Kondracki, M.L. Bioactive indole derivatives from the South Pacific marine sponges Rhopaloeides odorabile and Hyrtios sp. Mar. Drugs 2011, 9, 879–888. [Google Scholar] [CrossRef]
- Kumagai, M.; Nishikawa, K.; Matsuura, H.; Umezawa, T.; Matsuda, F.; Okino, T. Antioxidants from the Brown Alga Dictyopteris undulata. Molecules 2018, 23, 1214. [Google Scholar] [CrossRef]
- Hochlowski, J.E.; Faulkner, D.J. Antibiotics from the marine pulmonate Siphonaria diemenensis. Tetrahedron Lett. 1983, 24, 1917–1920. [Google Scholar] [CrossRef]
- Ishibashi, F.; Sato, S.; Sakai, K.; Hirao, S.; Kuwano, K. Algicidal sesquiterpene hydroquinones from the brown alga Dictyopteris undulate. Biosci. Biotechnol. Biochem. 2013, 77, 1120–1122. [Google Scholar] [CrossRef]
- Chatterjee, S.; Vining, L.C. Nutrient utilization in actinomycetes. Induction of α-glucosidases in Streptomyces venezuelae. Can. J. Microbiol. 1981, 27, 639–645. [Google Scholar]
- Wang, X.; Li, Y.; Zhang, X.; Lai, D.; Zhou, L. Structural diversity and biological activities of the cyclodipeptides from fungi. Molecules 2017, 22, 2026. [Google Scholar] [CrossRef]
- Ivonne, J.N.; AVILA, C.; Iris, M. Determination of fatty acids and triterpenoid compounds from the fruiting body of Suillus luteus. Rev. Colomb. Quím. 2008, 37, 297–304. [Google Scholar]
- Tsuda, M.; Oguchi, K.; Iwamoto, R.; Okamoto, Y.; Fukushi, E.; Kawabata, J.; Ozawa, T.; Masuda, A. Iriomoteolides-1b and -1c, 20-Membered Macrolides from a Marine Dinoflagellate Amphidinium Species. J. Nat. Prod. 2007, 70, 1661–1663. [Google Scholar] [CrossRef]
- Mootoo, B.S.; Ramsewak, R.; Sharma, R.; Tinto, W.F.; Lough, A.J.; Mclean, S.; Reynolds, W.F.; Yang, P.J.; Yu, M. Further briareolate esters and briareolides from the Caribbean Gorgonian octocoral Briareum asbestinum. Tetrahedron 1996, 52, 9953–9962. [Google Scholar] [CrossRef]
- Eltamany, E.E.; Ibrahim, A.K.; Radwan, M.M.; ElSohly, M.A.; Hassanean, H.A.; Ahmed, S.A. Cytotoxic ceramides from the Red Sea sponge Spheciospongia vagabunda. Med. Chem. Res. 2015, 24, 3467–3473. [Google Scholar] [CrossRef]
- Raslan, A.E.; Radwan, M.M.; Ahmed, S.A.; Nafady, A.M.; Zaki, M.A.; Wanas, A.S.; Abou-Karam, M.; Shier, T.W.; Hassanean, H.A.; ElSohly, M.A. Monanchoramides A–D, ceramides from the marine sponge Monanchora clathrata with cytotoxic activity. Phytochem. Lett. 2018, 23, 83–89. [Google Scholar] [CrossRef]
- Abdelhameed, R.F.A.; Habib, E.S.; Eltahawy, N.A.; Hassanean, H.A.; Ibrahim, A.K.; Mohammed, A.F.; Fayez, S.; Hayallah, A.M.; Yamada, K.; Behery, F.A.; et al. New Cytotoxic Natural Products from the Red Sea Sponge Stylissa carteri. Mar. Drugs 2020, 18, 241. [Google Scholar] [CrossRef]
- Kaneko, M.; Kisa, F.; Yamada, K.; Miyamoto, T.; Higuchi, R. Structure of a New Neuritogenic-Active Ganglioside from the Sea Cucumber Stichopus japonicus. Eur. J. Org. Chem. 2003, 1004–1008. [Google Scholar] [CrossRef]
- Jia, Z.; Song, Y.; Tao, S.; Cong, P.; Wang, X.; Xue, C.; Xu, J. Structure of Sphingolipids From Sea Cucumber Cucumaria frondosa and Structure-Specific Cytotoxicity Against Human HepG2 Cells. Lipids 2016, 51, 321–334. [Google Scholar] [CrossRef]
- Farokhi, F.; Wielgosz-Collin, G.; Clement, M.; Kornprobst, J.M.; Barnathan, G. Cytotoxicity on human cancer cells of ophidiacerebrosides isolated from the African starfish Narcissia canariensis. Mar. Drug 2010, 8, 2988–2998. [Google Scholar] [CrossRef]
- Umegaki, K.; Taki, Y.; Endoh, K.; Taku, K.; Tanabe, H.; Shinozuka, K.; Sugiyama, T. Bilobalide in Ginkgo biloba Extract Is a Major Substance Inducing Hepatic CYPs. J. Pharm. Pharmacol. 2007, 59, 871–877. [Google Scholar] [CrossRef]
- Souza, P.; Bianchi, S.; Figueiró, F.; Heimfarth, L.; Moresco, K.; Gonçalves, R.; Hoppe, J.; Klein, C.; Salbego, C.; Gelain, D.; et al. Anticancer activity of flavonoids isolated from Achyrocline satureioides in gliomas cell lines. Toxicol. Vitro 2018, 51, 23–33. [Google Scholar] [CrossRef]
- Huang, W.-C.; Chen, C.-L.; Lin, Y.-S.; Lin, C.-F. Apoptotic sphingolipid ceramide in cancer therapy. J. Lipids 2011, 2011, 1–15. [Google Scholar] [CrossRef]
- Oku, H.; Wongtangtintharn, S.; Iwasaki, H.; Inafuku, M.; Shimatani, M.; Toda, T. Tumor specific cytotoxicity of glucosylceramide. Cancer Chemother. Pharm. 2007, 60, 767–775. [Google Scholar] [CrossRef]
- Wei, T.; Xiaojun, X.; Peilong, C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed. Pharmacother. 2020, 121, 109139. [Google Scholar] [CrossRef]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D. Drug Resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 137414. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.; Cheng, R.; Vitek, T.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I2PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef]
- Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011, 30, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hannun, Y.A.; et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEBJ 2009, 23, 751–763. [Google Scholar] [CrossRef]
- Bayarkhangai, B.; Noureldin, S.; Yu, L.; Zhao, N.; Gu, Y.; Xu, H.; Guo, C. A comprehensive and perspective view of oncoprotein SET in cancer. Cancer Med. 2018, 7, 3084–3094. [Google Scholar] [CrossRef] [PubMed]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef]
- Zhang, W.; Cai, J.; Chen, S.; Zheng, X.; Hu, S.; Dong, W.; Lu, J.; Xing, J.; Dong, Y. Paclitaxel resistance in MCF-7/PTX cells is reversed by paeonol through suppression of the SET/phosphatidylinositol 3-kinase/Akt pathway. Mol. Med. Rep. 2015, 12, 1506–1514. [Google Scholar] [CrossRef]
- Hayashi, T.; Hikichi, M.; Yukitake, J.; Harada, N.; Utsumi, T. Estradiol suppresses phosphorylation of ERα serine 167 through upregulation of PP2A in breast cancer cells. Oncol. Lett. 2017, 14, 8060–8065. [Google Scholar] [CrossRef][Green Version]
- Cristóbal, I.; Torrejón, B.; Martínez-Useros, J.; Madoz-Gurpide, J.; Rojo, F.; García-Foncillas, J. PP2A regulates signaling through hormonal receptors in breast cancer with important therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 435–438. [Google Scholar] [CrossRef]
- Zhao, H.; Li, D.; Zhang, B.; Qi, Y.; Diao, Y.; Zhen, Y.; Shu, X. PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer. Molecules 2017, 22, 2277. [Google Scholar] [CrossRef]
- Keen, J.C.; Zhou, Q.; Park, B.H.; Pettit, C.; Mack, K.M.; Blair, B.; Brenner, K.; Davidson, N.E. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. J. Biol. Chem. 2005, 280, 29519–29524. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, Y.; Refaat, J.; Abdelmohsen, U.R.; Othman, E.M.; Stopper, H.; Fouad, M.A. Metabolomic profiling and biological investigation of the marine sponge-derived bacterium Rhodococcus sp. UA13. Phytochem. Anal. 2018, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Senda, M.; Akai, Y.; Sato, L.; Suzuki, T.; Nagai, R.; Senda, T.; Horikoshi, M. Relationship between the structure of SET/TAF-Ibeta/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. USA 2007, 104, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2019.0101 of Chemical Computing Group. Inc. Available online: http://www. chemcomp.com (accessed on 15 April 2020).






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).