Immunomodulatory Effects of a Low-Molecular Weight Polysaccharide from Enteromorpha prolifera on RAW 264.7 Macrophages and Cyclophosphamide- Induced Immunosuppression Mouse Models


Abstract
1. Introduction
2. Results
2.1. Physicochemical Properties of Polysaccharides Isolated from E. prolifera
2.2. Effects of EP1-2 on Cell Viability and NO Production in RAW 264.7 Cells
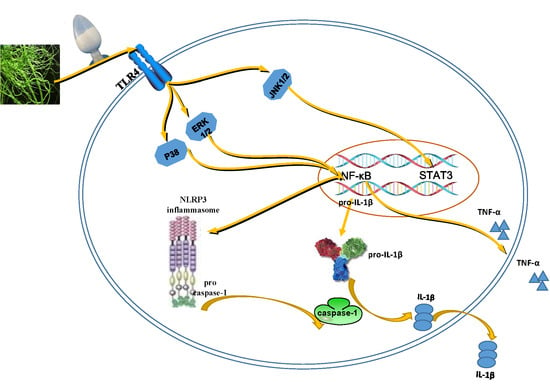
2.3. Effects of EP2 on Cytokines and Proinflammatory Proteins in RAW 264.7 Cells
2.4. EP2 Promoted the Activation of NLRP3 Inflammasome in RAW 264.7 Cells
2.5. EP2 Promoted the Activation of MAPK Signaling Pathway in RAW 264.7 Cells
2.6. EP2 Promoted the Activation of NF-κB and AP-1 in RAW 264.7 Cells
2.7. EP2 Improved Visceral Index and Inflammatory Cell Counts in CYP-induced Immunosuppression Mouse Models
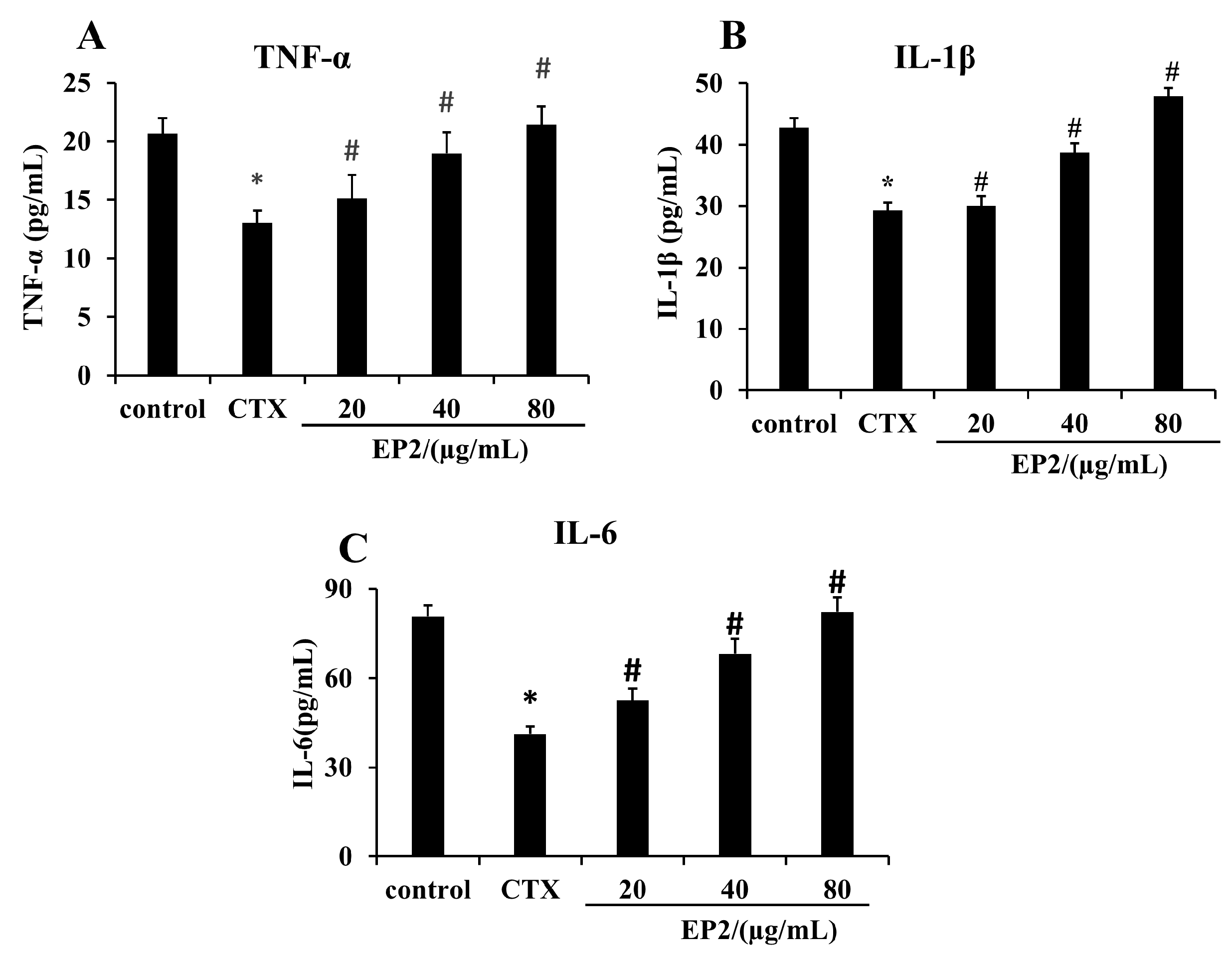
2.8. EP2 Enhanced the Production of Cytokines in CYP-induced Immunosuppression Mouse Models
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Extraction and Purification of the Polysaccharide
4.3. Composition Analysis
4.4. Endotoxin Test
4.5. Cell Culture and Cell Viability Assay
4.6. Nitric Oxide Production
4.7. Immunofluorescence Staining Analysis
4.8. Western Blot
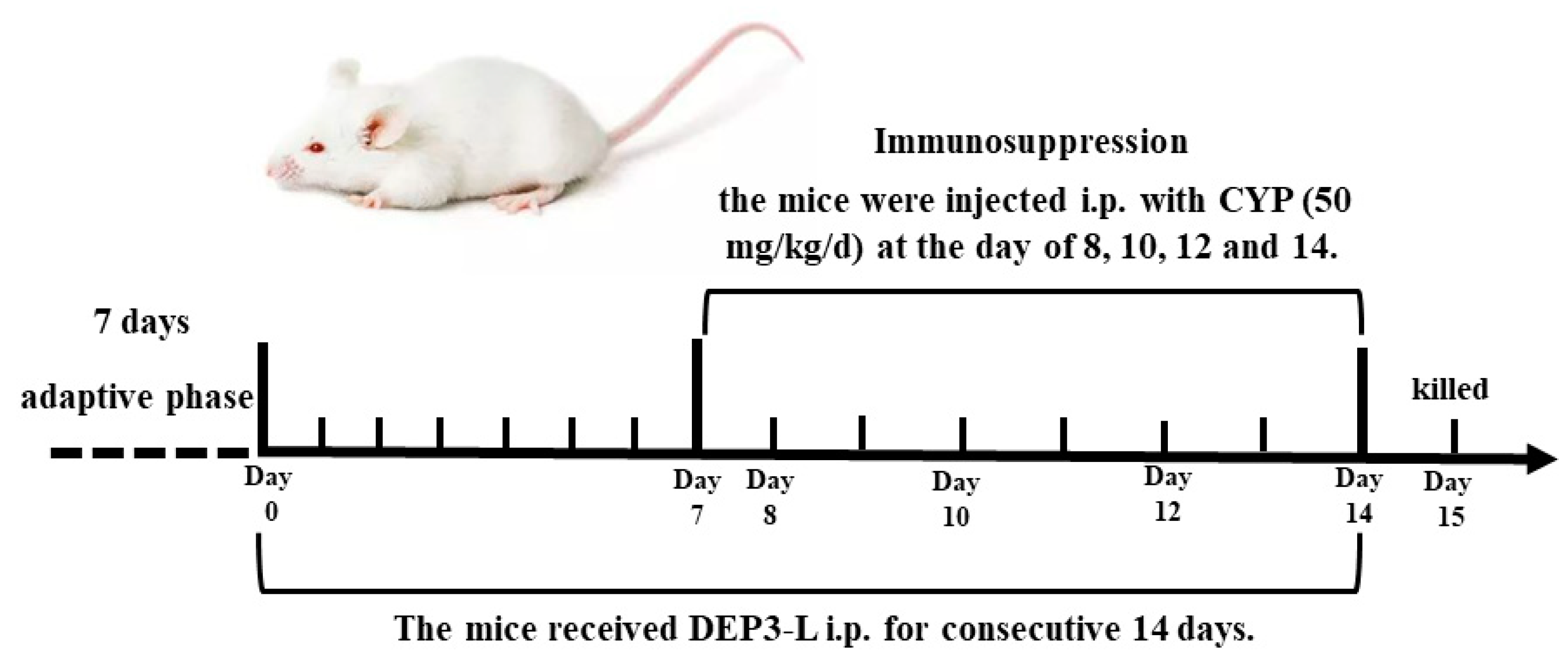
4.9. Animals
4.10. Inflammatory Cell Counts
4.11. Enzyme Linked Immunosorbent Assay (ELISA)
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hooper, L.; Manzella, S.; Baenziger, J. From legumes to leukocytes: Biological roles for sulfated carbohydrates. FASEB J. 1996, 10, 1137–1146. [Google Scholar] [CrossRef]
- Pomin, V.H.; Mourao, P.A. Structure, biology, evolution, and medical importance of sulfated fucans and galactans. Glycobiology 2008, 18, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Di, L.; Li, J.; Hu, L.; Cheng, J.; Wu, H. Elaboration in type, primary structure, and bioactivity of polysaccharides derived from mollusks. Crit. Rev. Food. Sci. Nutr. 2019, 59, 1091–1114. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Cho, M.L.; Karnjanapratum, S.; Shin, I.S.; You, S.G. In vitro and in vivo immunomodulatory activity of sulfated polysaccharides from Enteromorpha prolifera. Int. J. Biol. Macromol. 2011, 49, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Ray, B. Polysaccharides from Enteromorpha compressa: Isolation, purification and structural features. Carbohydr. Polym. 2006, 66, 408–416. [Google Scholar] [CrossRef]
- Huang, W.; Wang, F.; Qiu, N.; Wu, X.; Zang, C.; Li, A.; Xu, L. Enteromorpha prolifera-derived Fe3C/C composite as advanced catalyst for hydroxyl radical generation and efficient removal for organic dye and antibiotic. J. Hazard. Mater. 2019, 378, 120728. [Google Scholar] [CrossRef]
- Lin, W.; Wang, W.; Liao, D.; Chen, D.; Zhu, P.; Cai, G.; Kiyoshi, A. Polysaccharides from Enteromorpha prolifera improve glucose metabolism in diabetic rats. J. Diabetes. Res. 2015, 2015, 675201. [Google Scholar] [CrossRef]
- Teng, Z.; Qian, L.; Zhou, Y. Hypolipidemic activity of the polysaccharides from Enteromorpha prolifera. Int. J. Biol. Macromol. 2013, 62, 254–256. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Z.; Zhao, J.; Yong, C.; Mao, Y. Polysaccharides from Enteromorpha Prolifera ameliorate acute myocardial infarction in vitro and in vivo via up-regulating HIF-1α. Int. Heart. J. 2019, 60, 964–973. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, C.; Zhou, D.; Ou, S.; Liu, Z.; Huang, H. Immune-enhancing activities of chondroitin sulfate in murine macrophage RAW 264.7 cells. Carbohydr. Polym. 2018, 198, 611–619. [Google Scholar] [CrossRef]
- Zhu, F.; Du, B.; Xu, B. Anti-inflammatory effects of phytochemicals from fruits, vegetables, and food legumes: A review. Crit. Rev. Food. Sci. Nutr. 2018, 58, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Yu, Q.; Zheng, B.; Wang, H.; Wang, J.; Chen, S.; Nie, S.; Xie, M. Cultured Cordyceps sinensis polysaccharides modulate intestinal mucosal immunity and gut microbiota in cyclophosphamide-treated mice. Carbohydr. Polym. 2020, 235, 115957. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Dong, J.; Jiang, H.; Wang, J.; Liu, Z.; Ma, C.; Kang, W. Effects of polysaccharide from Malus halliana Koehne flowers in cyclophosphamide-induced immunosuppression and oxidative stress on mice. Oxid. Med. Cell. Longev. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, H.; Jin, W.; Zhang, H.; Zhang, Q. Structure-activity relationship of sulfated hetero/galactofucan polysaccharides on dopaminergic neuron. Int. J. Biol. Macromol. 2016, 82, 878–883. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Zhang, Q.; Zhang, Z.; Shi, X.; Li, P. Synthesized different derivatives of low molecular fucoidan extracted from Laminaria japonica and their potential antioxidant activity in vitro. Int. J. Biol. Macromol. 2009, 44, 379–384. [Google Scholar] [CrossRef]
- Schroter, D.; Neugart, S.; Schreiner, M.; Grune, T.; Rohn, S.; Ott, C. Amaranth’s 2-caffeoylisocitric acid—An anti-inflammatory caffeic acid derivative that impairs NF-kappaB signaling in LPS-challenged RAW 264.7 macrophages. Nutrients 2019, 11, 571. [Google Scholar] [CrossRef]
- Geng, L.; Hu, W.; Liu, Y.; Wang, J.; Zhang, Q. A heteropolysaccharide from Saccharina japonica with immunomodulatory effect on RAW 264.7 cells. Carbohydr. Polym. 2018, 201, 557–565. [Google Scholar] [CrossRef]
- Liu, H.; Yan, Y.; Zhang, F.; Wu, Q. The immuno-enhancement effects of Tubiechong (Eupolyphaga sinensis) Lyophilized powder in cyclophosphamide-induced immunosuppressed mice. Immunol. Invest. 2019, 48, 844–859. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef]
- Venkatesan, T.; Choi, Y.W.; Lee, J.; Kim, Y.K. Falcarindiol inhibits LPS-induced inflammation via attenuating MAPK and JAK-STAT signaling pathways in murine macrophage RAW 264.7 cells. Mol. Cell. Biochem. 2018, 445, 169–178. [Google Scholar] [CrossRef]
- Xing, B.; Chen, H.; Wang, L.; Weng, X.; Chen, Z.; Li, X. Ozone oxidative preconditioning protects the rat kidney from reperfusion injury via modulation of the TLR4-NF-kappaB pathway. Acta. Cir. Bras. 2015, 30, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z.; Zhang, X. A review of the green tides in the Yellow Sea, China. Mar. Environ. Res. 2016, 119, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, F.; Chi, Z.; Han, D.; Yu, L.; Liu, C. Development of Enteromorpha prolifera polysaccharide-based nanoparticles for delivery of curcumin to cancer cells. Int. J. Biol. Macromol. 2018, 112, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Weis, G.C.C.; Assmann, C.E.; Cadona, F.C.; Bonadiman, B.; Alves, A.O.; Machado, A.K.; Duarte, M.; da Cruz, I.B.M.; Costabeber, I.H. Immunomodulatory effect of mancozeb, chlorothalonil, and thiophanate methyl pesticides on macrophage cells. Ecotoxicol. Environ. Saf. 2019, 182, 109420. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, T.; Takahashi, M. Exogenous nanoparticles and endogenous crystalline molecules as danger signals for the NLRP3 inflammasomes. J. Cell. Physiol. 2019, 234, 5436–5450. [Google Scholar] [CrossRef]
- Uh, S.T.; Koo, S.M.; Kim, Y.; Kim, K.; Park, S.; Jang, A.S.; Kim, D.; Kim, Y.H.; Park, C.S. The activation of NLRP3-inflammsome by stimulation of diesel exhaust particles in lung tissues from emphysema model and RAW 264.7 cell line. Korean. J. Intern. Med. 2017, 32, 865–874. [Google Scholar] [CrossRef]
- Fu, S.; Liu, L.; Han, L.; Yu, Y. Leptin promotes IL18 secretion by activating the NLRP3 inflammasome in RAW 264.7 cells. Mol. Med. Rep. 2017, 16, 9770–9776. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Yuan, Y.H.; Chen, N.H.; Wang, H.B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef]
- Gong, A.G.; Zhang, L.M.; Lam, C.T.; Xu, M.L.; Wang, H.Y.; Lin, H.Q.; Dong, T.T.; Tsim, K.W. Polysaccharide of Danggui Buxue Tang, an ancient Chinese herbal decoction, induces expression of pro-inflammatory cytokines possibly via activation of NFkappaB signaling in cultured RAW 264.7 cells. Phytother. Res. 2017, 31, 274–283. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, J.; Hu, Y.; Lu, W.; Zhang, X.; Wang, R.; Chu, K. Rosmarinic acid mitigates lipopolysaccharide-induced neuroinflammatory responses through the inhibition of TLR4 and CD14 expression and NF-kappaB and NLRP3 inflammasome activation. Inflammation 2018, 41, 732–740. [Google Scholar] [CrossRef]
- Zhang, X.; Du, Q.; Yang, Y.; Wang, J.; Dou, S.; Liu, C.; Duan, J. The protective effect of Luteolin on myocardial ischemia/reperfusion (I/R) injury through TLR4/NF-kappaB/NLRP3 inflammasome pathway. Biomed. Pharmacother. 2017, 91, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zhang, Q.S.; Heng, Y.; Chen, Y.; Wang, S.; Yuan, Y.H.; Chen, N.H. NLRP3 inflammasome activation in the thymus of MPTP-induced Parkinsonian mouse model. Toxicol. Lett. 2018, 288, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mankhong, S.; Iawsipo, P.; Srisook, E.; Srisook, K. 4-methoxycinnamyl p-coumarate isolated from Etlingera pavieana rhizomes inhibits inflammatory response via suppression of NF-kappaB, Akt and AP-1 signaling in LPS-stimulated RAW 264.7 macrophages. Phytomedicine 2019, 54, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ngabire, D.; Seong, Y.A.; Patil, M.P.; Niyonizigiye, I.; Seo, Y.B.; Kim, G.D. Anti-inflammatory effects of Aster incisus through the inhibition of NF-kappaB, MAPK, and Akt pathways in LPS-stimulated RAW 264.7 macrophages. Mediators. Inflamm. 2018, 2018, 4675204. [Google Scholar] [CrossRef]
- Tanaka, M.; Kishimoto, Y.; Sasaki, M.; Sato, A.; Kamiya, T.; Kondo, K.; Iida, K. Terminalia bellirica (Gaertn.) Roxb. extract and gallic acid attenuate LPS-induced inflammation and oxidative stress via MAPK/NF-kappaB and Akt/AMPK/Nrf2 pathways. Oxid. Med. Cell. Longev. 2018, 2018, 9364364. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xing, R.; Liu, S.; Qin, Y.; Li, K.; Yu, H.; Li, P. Immunostimulatory effects of chitooligosaccharides on RAW 264.7 mouse macrophages via regulation of the MAPK and PI3K/Akt signaling pathways. Mar. Drugs 2019, 17, 36. [Google Scholar] [CrossRef]
- Park, J.Y.; Park, S.D.; Koh, Y.J.; Kim, D.I.; Lee, J.H. Aqueous extract of Dipsacus asperoides suppresses lipopolysaccharide-stimulated inflammatory responses by inhibiting the ERK1/2 signaling pathway in RAW 264.7 macrophages. J. Ethnopharmacol. 2019, 231, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xing, R.; Liu, S.; Qin, Y.; Li, K.; Yu, H.; Li, P. Hydroxypropyltrimethyl ammonium chloride chitosan activates RAW 264.7 macrophages through the MAPK and JAK-STAT signaling pathways. Carbohydr. Polym. 2019, 205, 401–409. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. β-glucan recognition by the innate immune system. Immunol. Rev. 2009, 230, 38–50. [Google Scholar] [CrossRef]
- Li, P.; Wang, F. Polysaccharides: Candidates of promising vaccine adjuvants. Drug. Discov. Ther. 2015, 9, 88–93. [Google Scholar] [CrossRef]
- Noh, E.M.; Kim, J.M.; Lee, H.Y.; Song, H.K.; Joung, S.O.; Yang, H.J.; Kim, M.J.; Kim, K.S.; Lee, Y.R. Immuno-enhancement effects of Platycodon grandiflorum extracts in splenocytes and a cyclophosphamide-induced immunosuppressed rat model. BMC. Complement. Altern. Med. 2019, 19, 322. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Jiang, Y.; Yao, Y.; Wu, N.; Luo, J.; Hu, M.; Tu, Y.; Xu, M. Ovotransferrin ameliorates the dysbiosis of immunomodulatory function and intestinal microbiota induced by cyclophosphamide. Food. Funct. 2019, 10, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Shin, J.S.; Song, Y.R.; Rhee, Y.K.; Cho, C.W.; Ryu, J.H.; Inn, K.S.; Hong, H.D.; Lee, K.T. Immunostimulatory effects of polysaccharides isolated from young barley leaves (Hordeum vulgare L.) with dual activation of Th1 and Th2 in splenic T cells and cyclophosphamide-induced immunosuppressed mice. Int. J. Biol. Macromol. 2020, 147, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Anastyuk, S.D.; Imbs, T.I.; Shevchenko, N.M.; Dmitrenok, P.S.; Zvyagintseva, T.N. ESIMS analysis of fucoidan preparations from Costaria costata, extracted from alga at different life-stages. Carbohydr. Polym. 2012, 90, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, M.; Lamy, F. Determination of proteins and sulfobetaine with the folin-phenol reagent. Anal. Biochem. 1986, 157, 28–31. [Google Scholar] [CrossRef]
- Jin, W.; Wang, J.; Ren, S.; Song, N.; Zhang, Q. Structural analysis of a heteropolysaccharide from Saccharina japonica by electrospray mass spectrometry in tandem with collision-induced dissociation tandem mass spectrometry (ESI-CID-MS/MS). Mar. Drugs. 2012, 10, 2138–2152. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uranic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Yields (%) | UA (%) | SO42− (%) | Total Sugar (%) | Mw (kDa) |
|---|---|---|---|---|---|
| EP1 | 52.0 | 17.3 | 13.3 | 76.8 | 8 |
| EP2 | 36.0 | 15.4 | 19.0 | 82.8 | 4 |
| Samples | Monosaccharides (Molar Ratio) | |||||
|---|---|---|---|---|---|---|
| Man | Rha | Glc A | Glc | Gal | Xyl | |
| EP1 | - | 1 | 0.29 | - | 0.07 | 0.27 |
| EP2 | - | 1 | 0.2 | - | 0.01 | 0.56 |
| Sample | Body Weight (g) | Spleen Index (mg/g) | Thymus Index (mg/g) |
|---|---|---|---|
| control | 30.12 ± 1.28 | 4.08 ± 0.42 | 2.16 ± 0.21 |
| CYP | 25.03 ± 1.76 * | 2.91 ± 0.61 * | 1.95 ± 0.38 |
| EP2 (20 mg/kg/d) | 27.62 ± 1.40 | 3.31 ± 0.84 | 2.02 ± 0.41 |
| EP2 (40 mg/kg/d) | 29.64 ± 1.21 # | 4.11 ± 0.49 # | 2.13 ± 0.57 |
| EP2 (80 mg/kg/d) | 29.22 ± 1.65 # | 4.36 ± 0.47 # | 2.21 ± 0.63 |
| Sample | WBC (109/L) | NEUT (109/L) | LYMPH (109/L) | RBC (109/L) | PLT (1011/L) |
|---|---|---|---|---|---|
| control | 4.24 ± 0.58 | 0.24 ± 0.02 | 3.68 ± 0.42 | 7.65 ± 0.82 | 8.62 ± 0.75 |
| CYP | 1.96 ± 0.43 * | 0.11 ± 0.01 * | 0.97 ± 0.21 * | 6.03 ± 0.64 | 5.76 ± 0.82 * |
| EP2 (20 mg/kg/d) | 2.87 ± 0.71 # | 0.16 ± 0.03 | 2.14 ± 0.45 # | 7.42 ± 0.81 | 7.02 ± 0.54 # |
| EP2 (40 mg/kg/d) | 4.09 ± 0.45 # | 0.19 ± 0.03 # | 3.47 ± 0.63 # | 7.36 ± 0.65 | 7.63 ± 0.61 # |
| EP2 (80 mg/kg/d) | 5.11 ± 0.72 # | 0.22 ± 0.02 # | 3.82 ± 0.66 # | 7.67 ± 0.73 | 8.24 ± 0.68 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wu, X.; Jin, W.; Guo, Y. Immunomodulatory Effects of a Low-Molecular Weight Polysaccharide from Enteromorpha prolifera on RAW 264.7 Macrophages and Cyclophosphamide- Induced Immunosuppression Mouse Models. Mar. Drugs 2020, 18, 340. https://doi.org/10.3390/md18070340
Liu Y, Wu X, Jin W, Guo Y. Immunomodulatory Effects of a Low-Molecular Weight Polysaccharide from Enteromorpha prolifera on RAW 264.7 Macrophages and Cyclophosphamide- Induced Immunosuppression Mouse Models. Marine Drugs. 2020; 18(7):340. https://doi.org/10.3390/md18070340
Chicago/Turabian StyleLiu, Yingjuan, Xiaolin Wu, Weihua Jin, and Yunliang Guo. 2020. "Immunomodulatory Effects of a Low-Molecular Weight Polysaccharide from Enteromorpha prolifera on RAW 264.7 Macrophages and Cyclophosphamide- Induced Immunosuppression Mouse Models" Marine Drugs 18, no. 7: 340. https://doi.org/10.3390/md18070340
APA StyleLiu, Y., Wu, X., Jin, W., & Guo, Y. (2020). Immunomodulatory Effects of a Low-Molecular Weight Polysaccharide from Enteromorpha prolifera on RAW 264.7 Macrophages and Cyclophosphamide- Induced Immunosuppression Mouse Models. Marine Drugs, 18(7), 340. https://doi.org/10.3390/md18070340