Bengamide Analogues Show A Potent Antitumor Activity against Colon Cancer Cells: A Preliminary Study

 , , , ,
, , , ,  and
and
Abstract
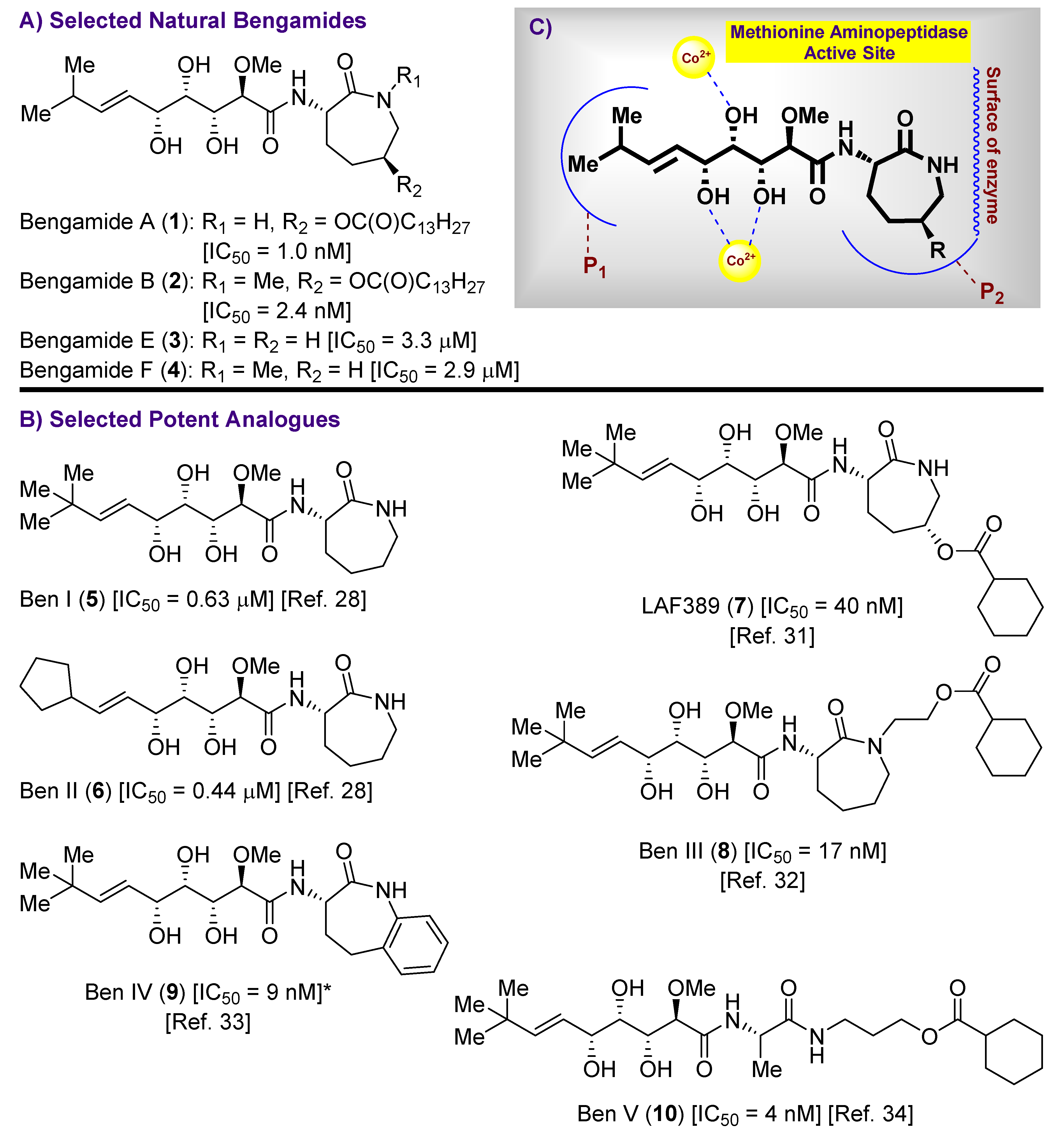
1. Introduction
2. Results and Discussion
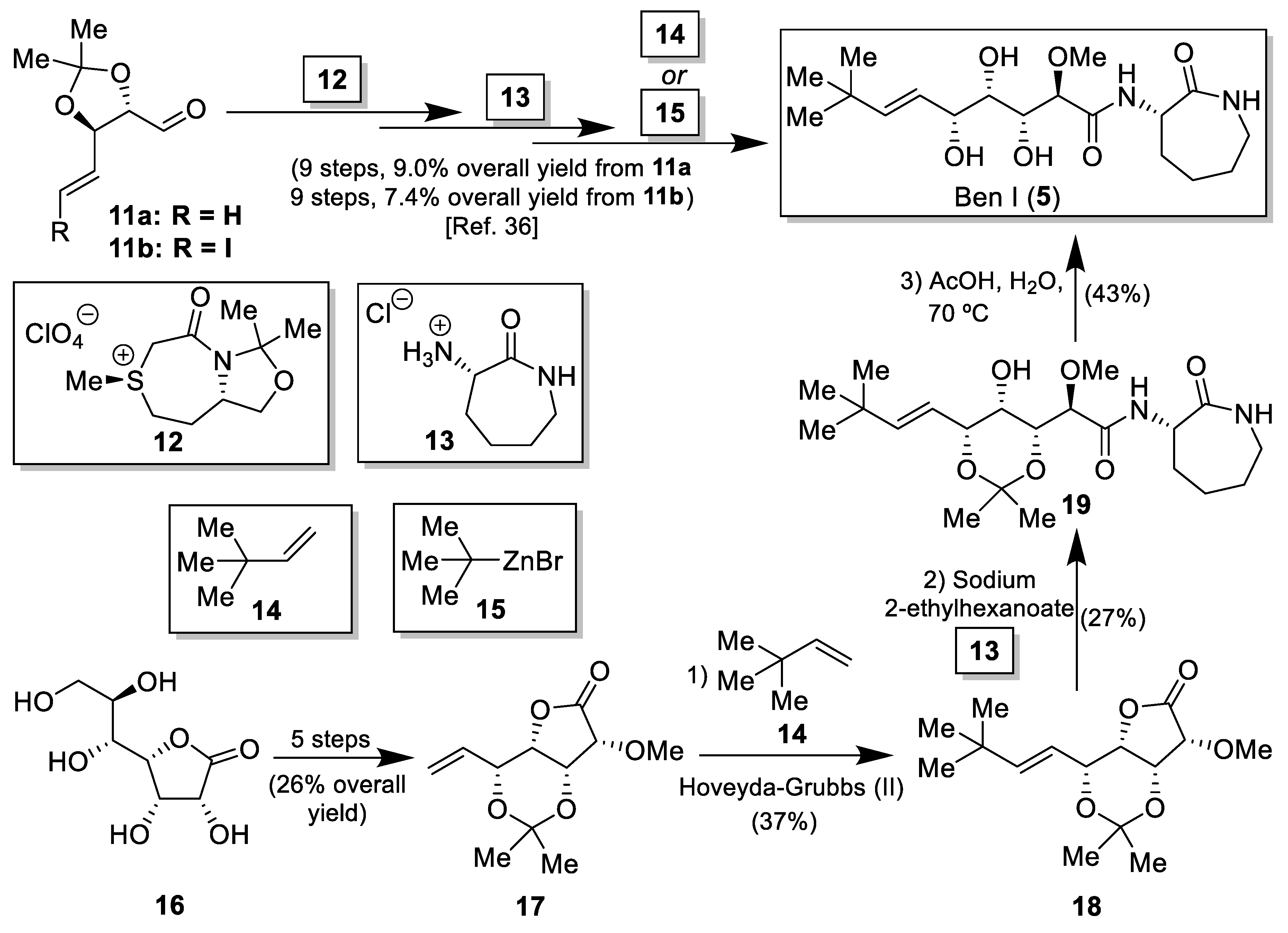
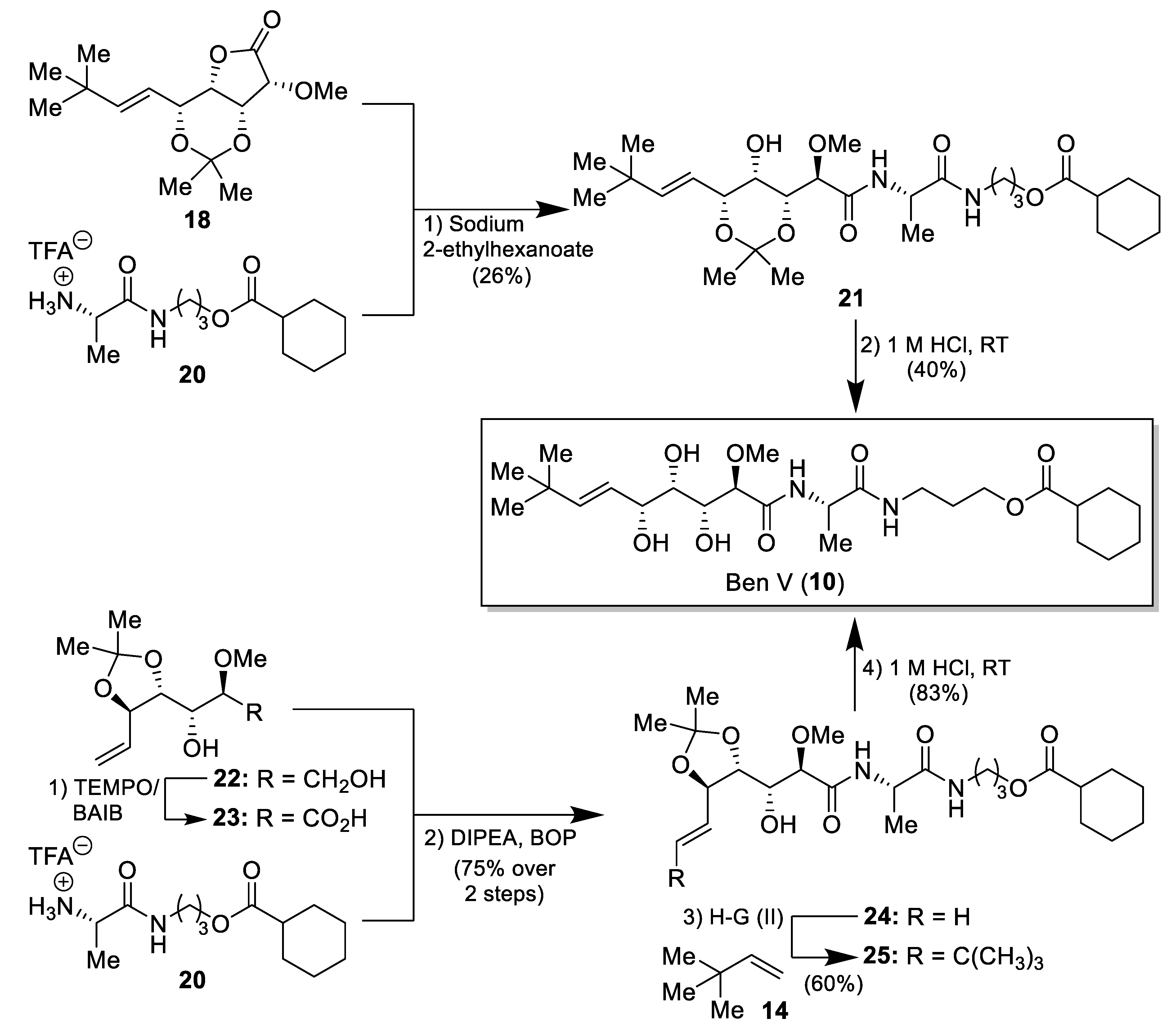
2.1. Synthesis of the Bengamide Analogues
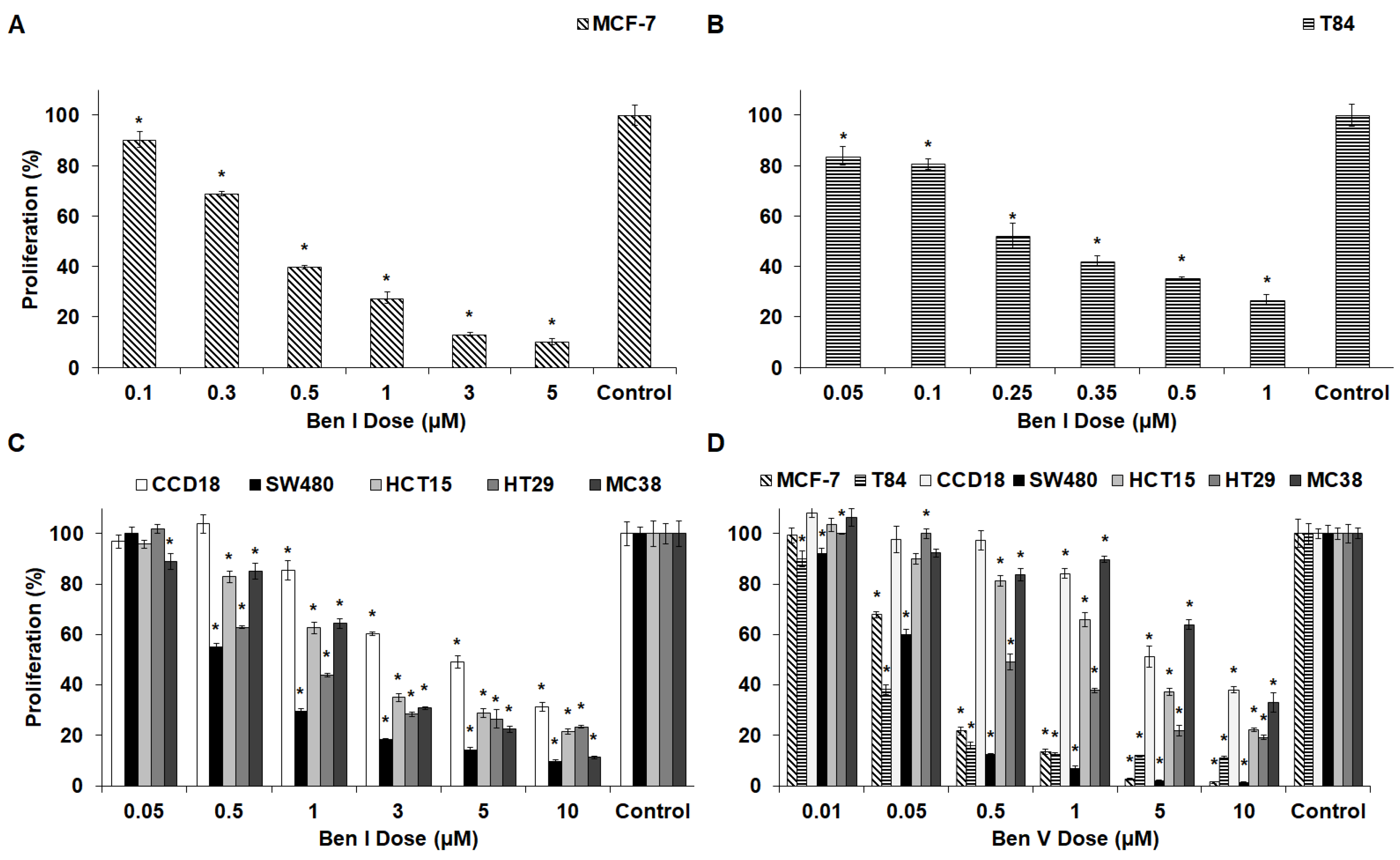
2.2. In Vitro Antiproliferative Assays
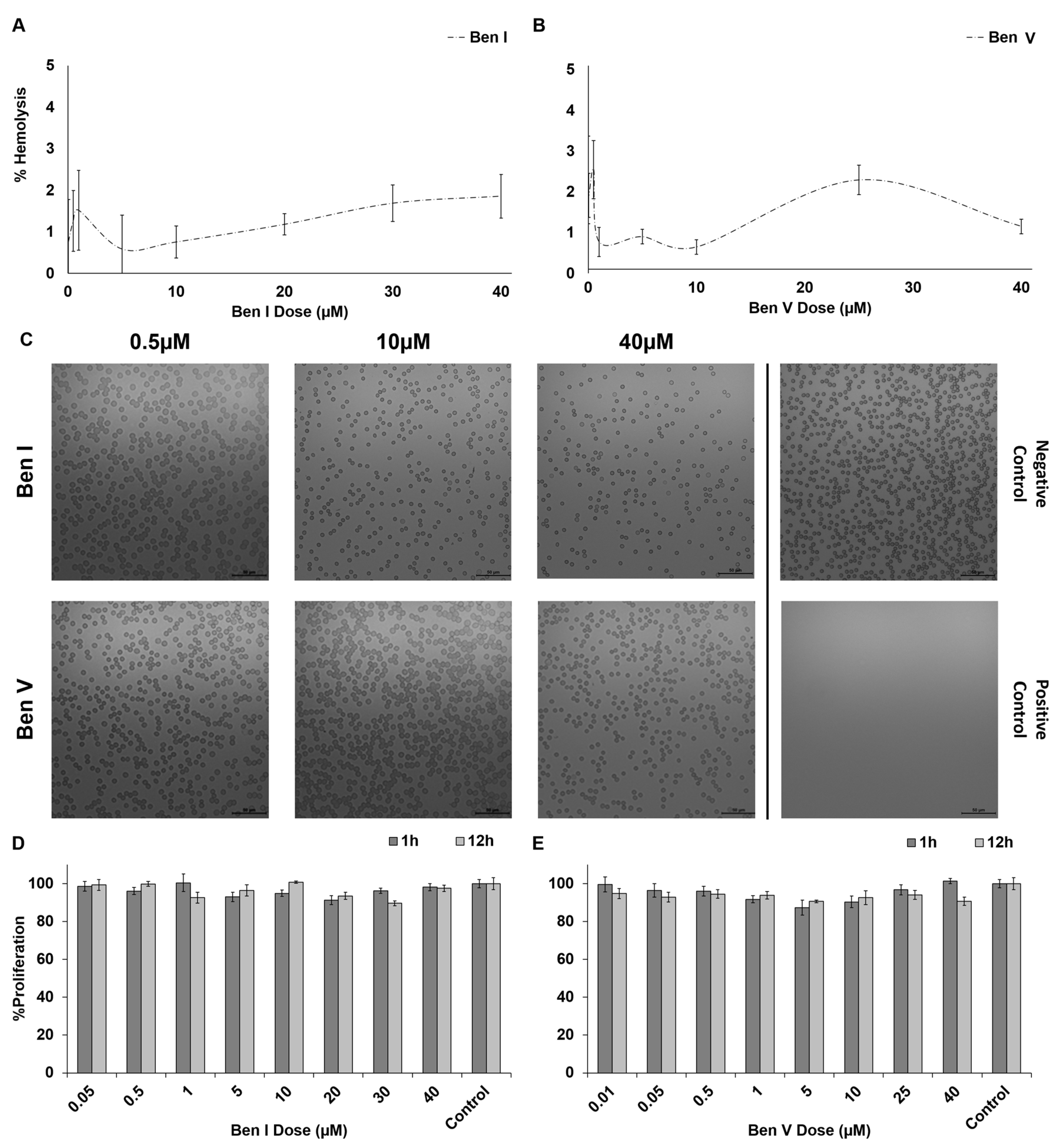
2.3. Blood Cell Cytotoxicity
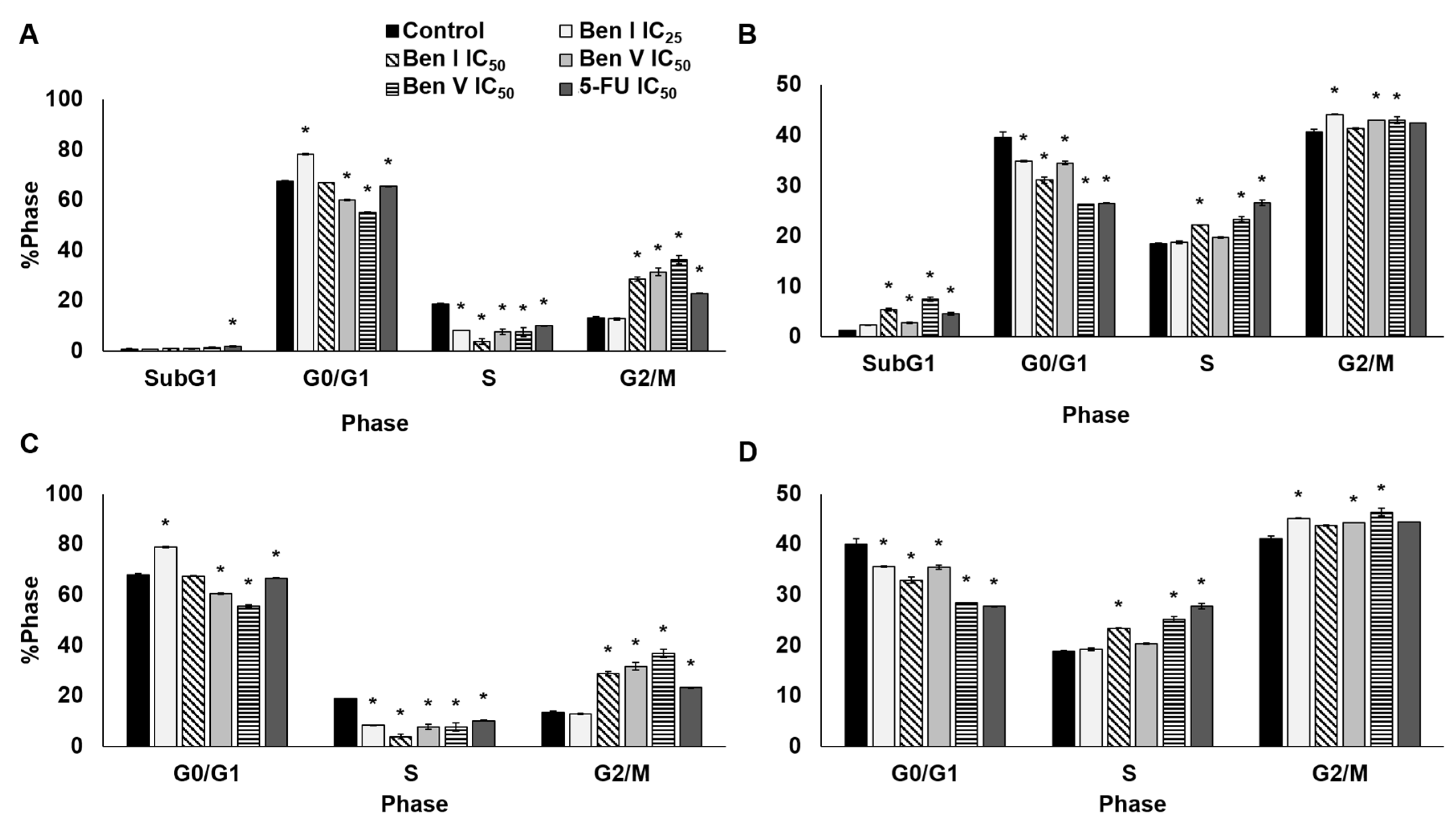
2.4. Cell Cycle Analysis
3. Experimental Section
3.1. General Techniques
3.2. Synthesis of the Bengamide Analogues
3.3. Cell Culture
3.4. Proliferation Assay
3.5. Hemocytotoxicity Assays
3.6. Cell Cycle Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- The, L. GLOBOCAN 2018: Counting the toll of cancer. Lancet 2018, 392, 985. [Google Scholar]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef]
- Seeber, A.; Gastl, G. Targeted Therapy of Colorectal Cancer. Oncol. Res. Treat. 2016, 39, 796–802. [Google Scholar] [CrossRef]
- Fernandez Montes, A.; Martinez Lago, N.; Covela Rua, M.; de la Camara Gomez, J.; Gonzalez Villaroel, P.; Mendez Mendez, J.C.; Jorge Fernandez, M.; Salgado Fernandez, M.; Reboredo Lopez, M.; Quintero Aldana, G.; et al. Efficacy and safety of FOLFIRI/aflibercept in second-line treatment of metastatic colorectal cancer in a real-world population: Prognostic and predictive markers. Cancer Med. 2019, 8, 882–889. [Google Scholar] [CrossRef]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Welt, S.; Ritter, G.; Williams, C., Jr.; Cohen, L.S.; John, M.; Jungbluth, A.; Richards, E.A.; Old, L.J.; Kemeny , N.E. Phase I study of anticolon cancer humanized antibody A33. Clin. Cancer Res. 2003, 9, 1338–1346. [Google Scholar] [PubMed]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Colon cancer: Cancer stem cells markers, drug resistance and treatment. Biomed. Pharmacother. 2014, 68, 911–916. [Google Scholar] [CrossRef]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, N.; Kruger, C.A.; Abrahamse, H. Targeted photodynamic therapy as potential treatment modality for the eradication of colon cancer and colon cancer stem cells. Tumor Biol. 2017, 39, 101042831773469. [Google Scholar] [CrossRef] [PubMed]
- Quiñoà, E.; Adamczeski, M.; Crews, P. Bengamides, heterocyclic anthelminthics from a Jaspidae. marine sponge. J. Org. Chem. 1986, 51, 4494–4497. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Sarabia, F. Chemistry and biology of bengamides and bengazoles, bioactive natural products from Jaspis. Sponges. Mar. Drugs 2014, 12, 1580–1622. [Google Scholar] [CrossRef]
- Phillips, P.E.; Bair, K.W.; Bontempo, J.; Crews, P.; Czuchta, M.; Kinder, F.R.; Versace, R.W.; Wang, B.; Wang, J.; Wood, A.; et al. Bengamide E arrests cells at the G1/S restriction point and within the G2/M phase of the cell cycle. Proc. Am. Assoc. Cancer Res. 2000, 41, 59. [Google Scholar]
- Towbin, H.; Bair, K.W.; DeCaprio, J.A.; Eck, M.J.; Kim, S.; Kinder, F.R.; Morollo, A.; Mueller, D.R.; Schindler, P.; Song, H.K.; et al. Proteomics-based target identification: Bengamides as a new class of methionine aminopeptidase inhibitors. J. Biol. Chem. 2003, 278, 52964–52971. [Google Scholar] [CrossRef]
- Lowther, W.T.; Orville, A.M.; Madded, D.T.; Lim, S.; Rich, D.H.; Matthews, B.W. Escherichia coli methionine aminopeptidase: Implications of crystallographic analyses of the native, mutant and inhibited enzymes for the mechanism of catalysis. Biochemistry 1999, 38, 7678–7688. [Google Scholar] [CrossRef]
- Xu, W.; Lu, J.P.; Ye, Q.Z. Structural analysis of bengamide derivatives as inhibitors of methionine aminopeptidases. J. Med. Chem. 2012, 55, 8021–8027. [Google Scholar] [CrossRef]
- Ingber, D.; Fujita, T.; Kishimoto, S.; Sudo, K.; Kanamaru, T.; Brem, H.; Folkman, J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 1990, 348, 555–557. [Google Scholar] [CrossRef]
- Liu, S. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 1998, 282, 1324–1327. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dang, Y.; Tenney, K.; Crews, P.; Tsai, C.W.; Sixt, K.M.; Cole, P.A.; Liu, J.O. Regulation of c-Src nonreceptor tyrosine kinase activity by bengamide A through inhibition of methionine aminopeptidases. Chem. Biol. 2007, 14, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Sohn, J.; Vaske, Y.M.; White, K.N.; Cohen, T.L.; Vervoort, H.C.; Tenney, K.; Valeriote, F.A.; Bjeldanes, L.F.; Crews, P. Myxobacteria versus sponge-derived alkaloids: The bengamide family identified as potent immune modulating agents by scrutiny of LC-MS/ELSD libraries. Bioorg. Med. Chem. 2012, 20, 4348–4355. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Williams, D.E.; Read, S.; Kuang, X.T.; Mwimanzi, P.; Wilhelm, E.; Markle, T.; Kinloch, N.N.; Naphen, C.N.; Tenney, K.; et al. Inhibition of NF-κB-dependent HIV-1 replication by the marine natural product bengamide A. Antivir. Res. 2018, 152, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Yuan, X.H.; Yuan, H.; Wang, W.L.; Wan, B.; Franzblau, S.G.; Ye, Q.Z. Inhibition of Mycobacterium tuberculosis methionine aminopeptidases by bengamide derivatives. ChemMedChem 2011, 6, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Quan, D.H.; Nagalingam, G.; Luck, I.; Proschogo, N.; Pillalamarri, V.; Addlagatta, A.; Martinez, E.; Sintchenko, V.; Rutledge, P.J.; Triccas, J.A. Bengamides display potent activity against drug-resistant Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 14396. [Google Scholar] [CrossRef]
- White, K.N.; Tenney, K.; Crews, P. The Bengamides: A mini-review of natural sources, analogues, biological properties, biosynthetic origins, and future prospects. J. Nat. Prod. 2017, 80, 740–755. [Google Scholar] [CrossRef]
- Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Valeriote, F.A.; Sarabia, F. An array of bengamide E analogues modified at the terminal olefinic position: Synthesis and antitumor properties. ChemMedChem 2013, 8, 819–831. [Google Scholar] [CrossRef]
- Sarabia, F.; Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Vivar-García, C. Epi-, epoxy-, and C2-modified bengamides: synthesis and biological evaluation. J. Org. Chem. 2013, 78, 5239–5253. [Google Scholar] [CrossRef]
- Zhang, W.; Liang, Q.; Li, H.; Meng, X.; Li, Z. Concise synthesis and antitumor activity of Bengamide E and its analogs. Tetrahedron 2013, 69, 664–672. [Google Scholar] [CrossRef]
- Kinder, F.R., Jr.; Versace, R.W.; Bair, K.W.; Bontempo, J.M.; Cesarz, D.; Chen, S.; Crews, P.; Czuchta, A.M.; Jagoe, C.T.; Mou, Y.; et al. Synthesis and antitumor activity of ester-modified analogues of bengamide B. J. Med. Chem. 2001, 44, 3692–3699. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ma, Y.M.; Tai, W.Y.; Xie, C.M.; Li, Y.L.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of caprolactam-modified bengamide analogues. ChemMedChem 2008, 3, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Hoffmann, H.; Zhang, J.; Debussche, L.; Haag-Richter, S.; Kurz, M.; Nardi, F.; Lukat, P.; Kochems, I.; Tietgen, H.; et al. Production of the bengamide class of marine natural products in Myxobacteria: Biosynthesis and structure-activity relationships. Angew. Chem. Int. Ed. 2015, 54, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.Y.; Zhang, R.T.; Ma, Y.M.; Gu, M.; Liu, G.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of ring-opened bengamide analogues. ChemMedChem 2011, 6, 1555–1558. [Google Scholar] [CrossRef]
- Dumez, H.; Gall, H.; Capdeville, R.; Dutreix, C.; van Oosterom, A.T.; Giaccone, G. A phase I and pharmacokinetic study of LAF389 administered to patients with advanced cancer. Anti-Cancer Drugs 2007, 18, 219–225. [Google Scholar] [CrossRef]
- Sarabia, F.; Martín-Gálvez, F.; Chammaa, S.; Martín-Ortiz, L.; Sánchez-Ruiz, A. Chiral sulfur ylides for the synthesis of bengamide E and analogues. J. Org. Chem. 2010, 75, 5526–5532. [Google Scholar] [CrossRef]
- Xu, D.D.; Waykole, L.; Calienni, J.V.; Ciszewski, L.; Lee, G.T.; Liu, W.; Szewczyk, J.; Vargas, K.; Prasad, K.; Repič, O.; et al. An expedient synthesis of LAF389, a bengamide B analogue. Org. Process. Res. Dev. 2003, 7, 856–865. [Google Scholar] [CrossRef]
- Alam, S.; Dhimane, H. A Concise Synthesis of Bengamide E and analogues via E-selective cross-metathesis olefination. Synlett 2010, 19, 2923–2927. [Google Scholar]
- Phi, T.D.; Mai, H.D.T.; Tran, V.H.; Truong, B.N.; Tran, T.A.; Vu, V.L.; Chau, V.M.; Pham, V.C. Design, synthesis and cytotoxicity of bengamide analogues and their epimers. Med. Chem. Commun. 2017, 8, 445–451. [Google Scholar] [CrossRef]
- Ortiz, R.; Cabeza, L.; Arias, J.L.; Melguizo, C.; Álvarez, P.J.; Vélez, C.; Clares, B.; Áranega, A.; Prados, J. Poly(butylcyanoacrylate) and Poly(ε-caprolactone) nanoparticles loaded with 5-fluorouracil increase the cytotoxic effect of the drug in experimental colon cancer. AAPS J. 2017, 17, 918–929. [Google Scholar] [CrossRef]
- Lorente, C.; Cabeza, L.; Clares, B.; Ortiz, R.; Halbaut, L.; Delgado, A.V.; Perazzoli, G.; Prados, J.; Arias, J.L.; Melguizo, C. Formulation and in vitro evaluation of magnetoliposomes as a potential nanotool in colorectal cancer therapy. Colloids Surf. 2018, 171, 553–565. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Ben I | Ben V | Reference Compound (b) |
|---|---|---|---|
| CCD18 | 4.70 ± 0.28 | 5.08 ± 0.39 | 7.35 ± 0.41 |
| T84 | 0.36 ± 0.03 | 0.07 ± 0.02 | 2.68 ± 0.16 |
| SW480 | 0.59 ± 0.07 | 0.08 ± 0.00 | 6.35 ± 0.54 |
| HCT15 | 1.90 ± 0.15 | 2.44 ± 0.25 | 6.58 ± 0.35 |
| HT29 | 1.02 ± 0.19 | 0.66 ± 0.18 | 6.14 ± 0.94 |
| MC38 | 1.60 ± 0.13 | 6.51 ± 1.12 | 0.33 ± 0.01 |
| MCF7 | 0.49 ± 0.05 | 0.13 ± 0.01 | 0.04 ± 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pinel, B.; Porras-Alcalá, C.; Cabeza, L.; Ortiz, R.; Prados, J.; Melguizo, C.; Cheng-Sánchez, I.; López-Romero, J.M.; Sarabia, F. Bengamide Analogues Show A Potent Antitumor Activity against Colon Cancer Cells: A Preliminary Study. Mar. Drugs 2020, 18, 240. https://doi.org/10.3390/md18050240
García-Pinel B, Porras-Alcalá C, Cabeza L, Ortiz R, Prados J, Melguizo C, Cheng-Sánchez I, López-Romero JM, Sarabia F. Bengamide Analogues Show A Potent Antitumor Activity against Colon Cancer Cells: A Preliminary Study. Marine Drugs. 2020; 18(5):240. https://doi.org/10.3390/md18050240
Chicago/Turabian StyleGarcía-Pinel, Beatriz, Cristina Porras-Alcalá, Laura Cabeza, Raul Ortiz, José Prados, Consolación Melguizo, Iván Cheng-Sánchez, Juan Manuel López-Romero, and Francisco Sarabia. 2020. "Bengamide Analogues Show A Potent Antitumor Activity against Colon Cancer Cells: A Preliminary Study" Marine Drugs 18, no. 5: 240. https://doi.org/10.3390/md18050240
APA StyleGarcía-Pinel, B., Porras-Alcalá, C., Cabeza, L., Ortiz, R., Prados, J., Melguizo, C., Cheng-Sánchez, I., López-Romero, J. M., & Sarabia, F. (2020). Bengamide Analogues Show A Potent Antitumor Activity against Colon Cancer Cells: A Preliminary Study. Marine Drugs, 18(5), 240. https://doi.org/10.3390/md18050240