Characterization and Antibiofilm Activity of Mannitol–Chitosan-Blended Paste for Local Antibiotic Delivery System



Abstract
1. Introduction
2. Results
2.1. Material Characteristics
2.1.1. Assessment of Injectability
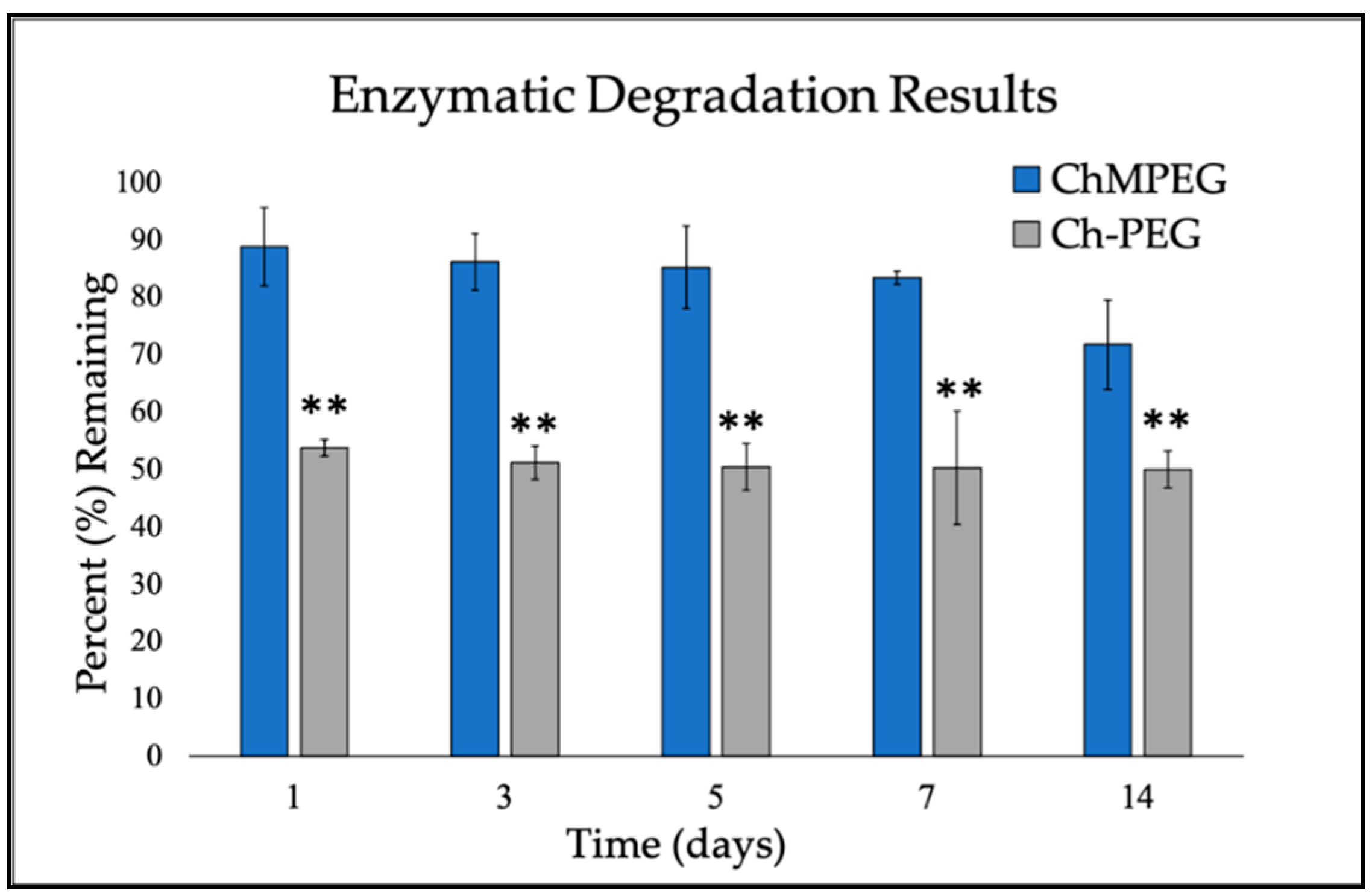
2.1.2. Enzymatic Degradation
2.2. Elution Profiles
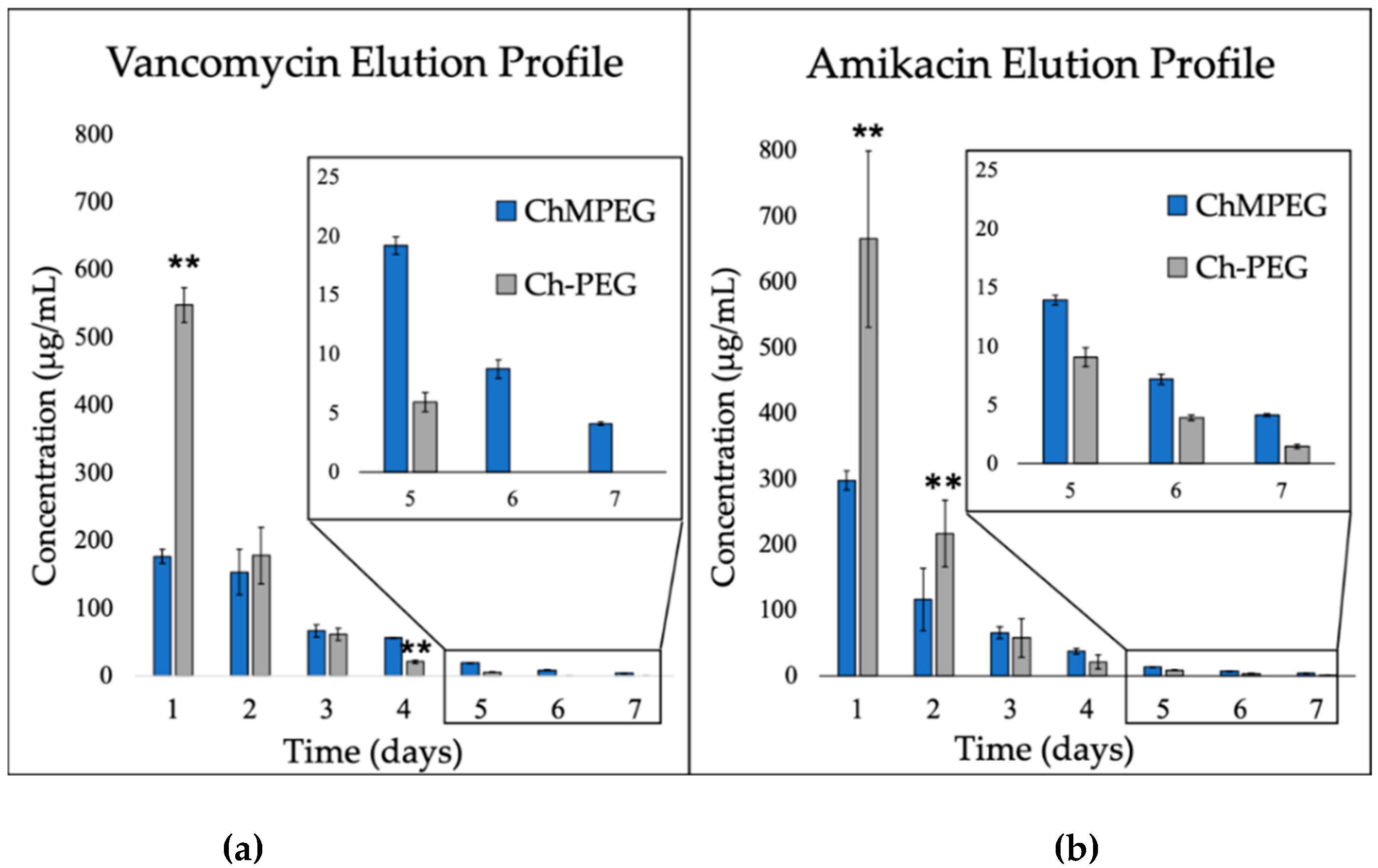
2.2.1. Antibiotic Elution: Vancomycin and Amikacin
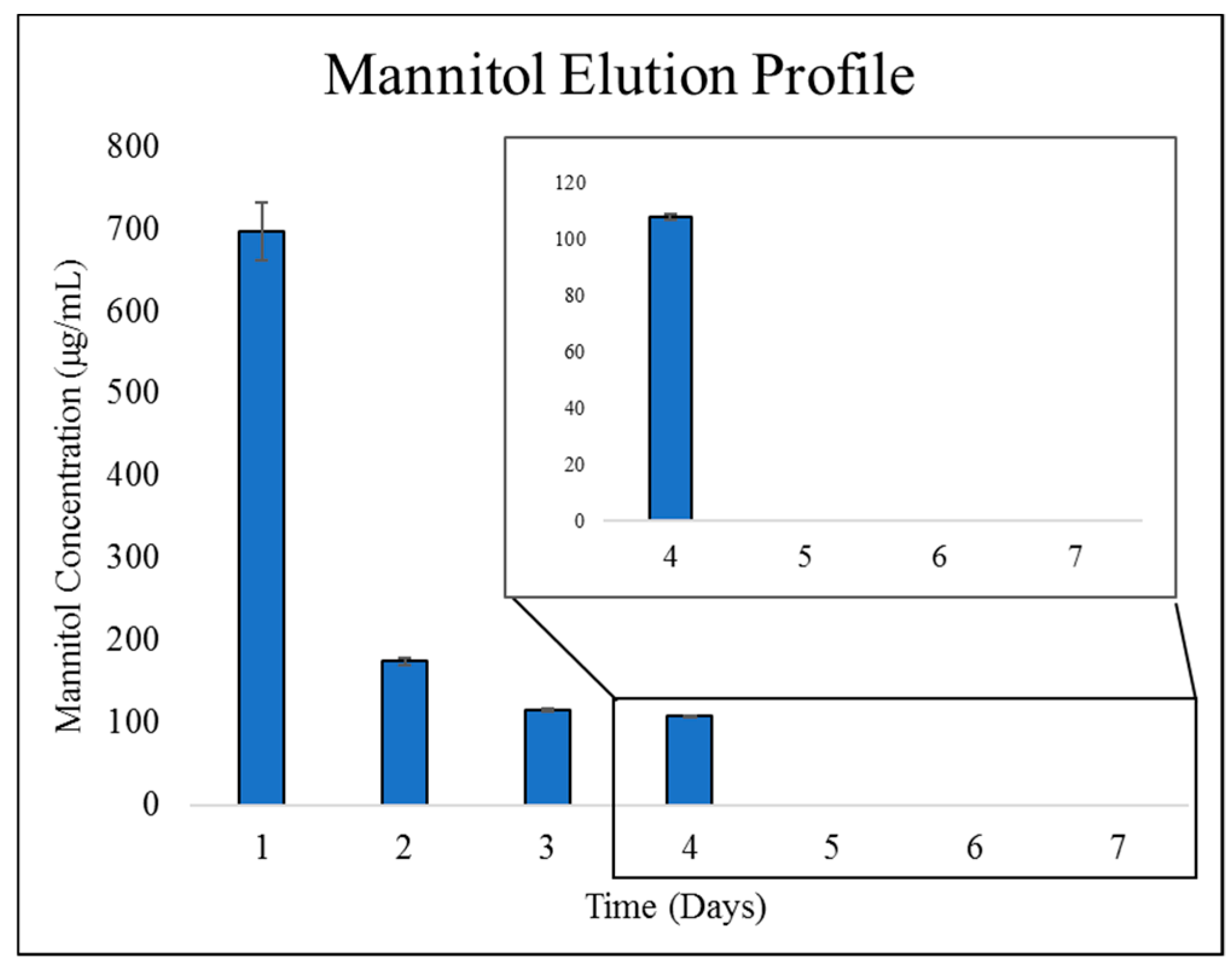
2.2.2. Mannitol Elution from ChMPEG Paste
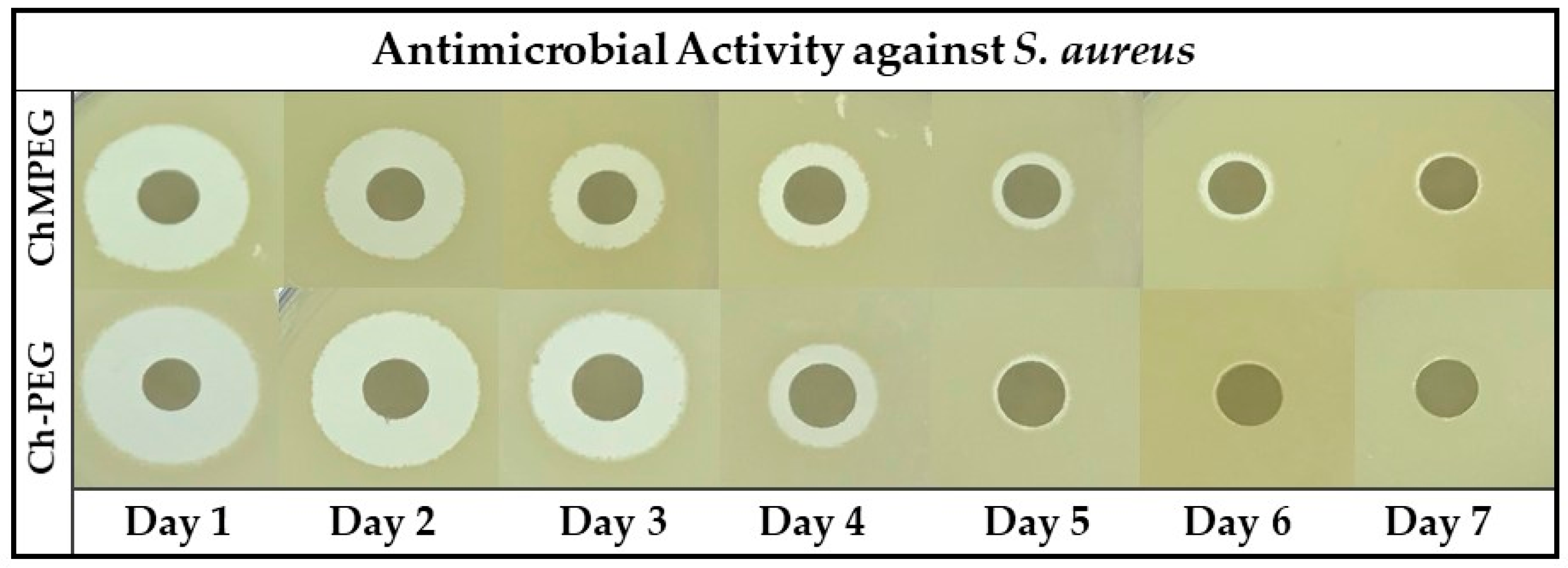
2.3. Antibiotic Activity Against Staphylococcus Aureus
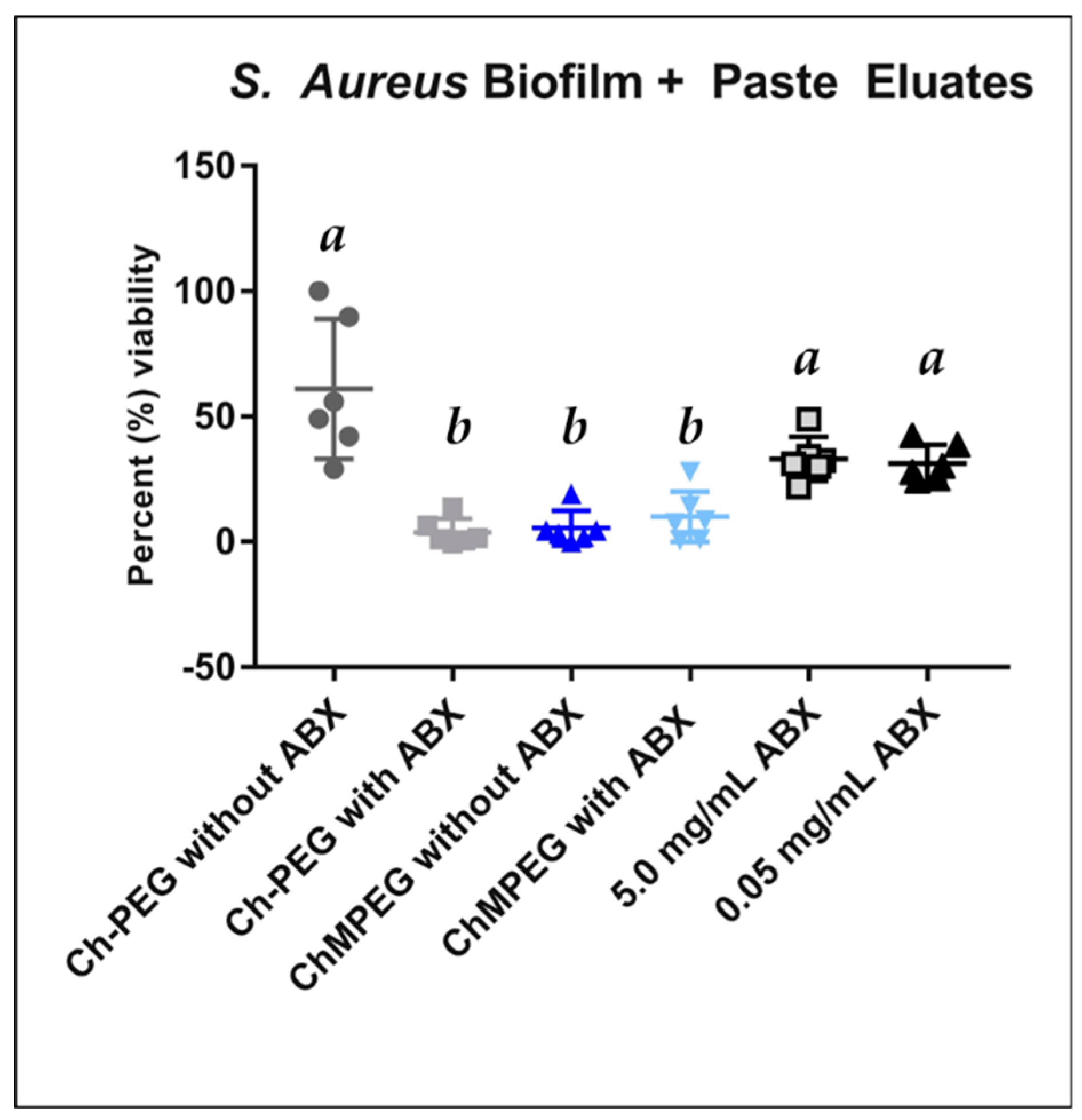
2.4. Biofilm Eradication
2.5. Cytocompatibility & Biocompatibility
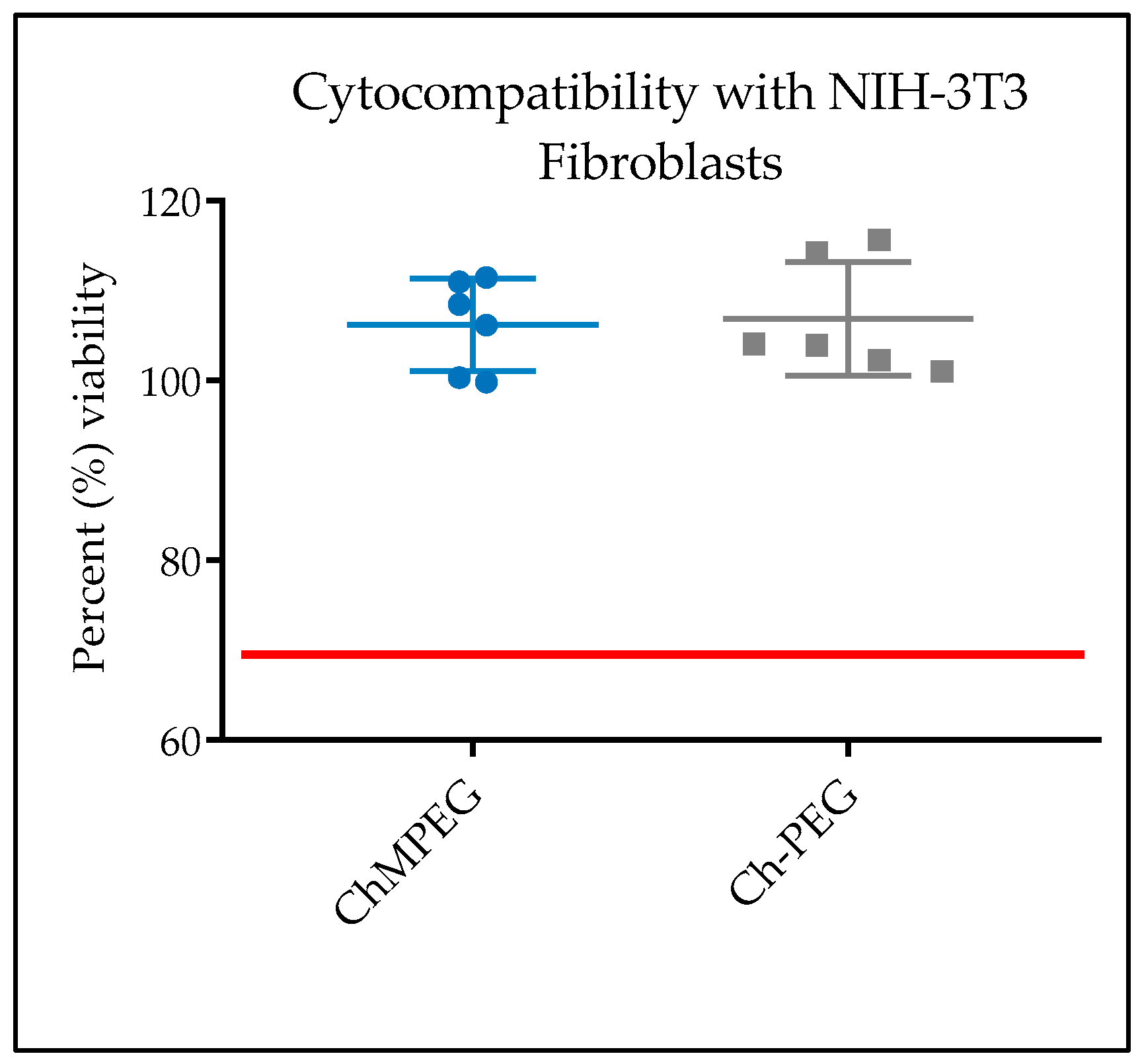
2.5.1. In Vitro Cytocompatibility with NIH-3T3 Cells
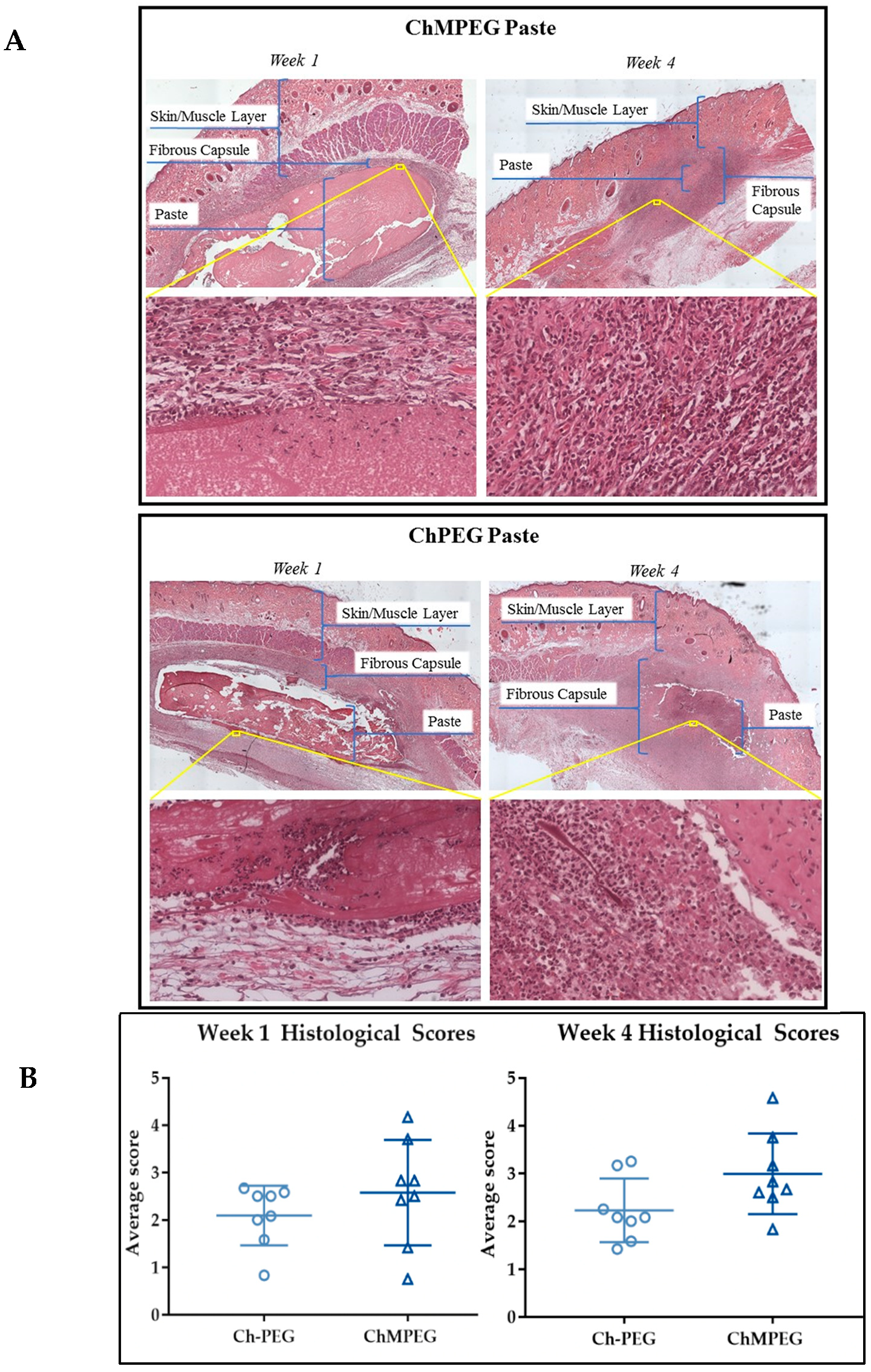
2.5.2. Biocompatibility Model with Sprague Dawley Rats
3. Discussion
4. Materials and Methods
4.1. Fabrication
4.2. Material Characteristics
4.2.1. Assessment of Injectability through 1 mL Syringe
4.2.2. Enzymatic Degradation of Paste
4.3. Elution Profiles
4.4. Antibiotic Activity
4.5. Biofilm Eradication
4.6. Cytocompatibility
4.7. Biocompatibility
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moon, G.H.; Cho, J.W.; Kim, B.S.; Yeo, D.H.; Oh, J.K. Analysis of risk factors for infection in orthopedic trauma patients. J. Trauma Inj. 2019, 32, 40–46. [Google Scholar] [CrossRef]
- Shirwaiker, R.A.; Springer, B.D.; Spangehl, M.J.; Garrigues, G.E.; Lowenberg, D.W.; Garras, D.N.; Yoo, J.U.; Pottinger, P.S. A clinical perspective on musculoskeletal infection treatment strategies and challenges. Jaaos J. Am. Acad. Orthop. Surg. 2015, 23, S44–S54. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Silva, J.; Teixeira, P. Staphylococcus aureus, a food pathogen: Virulence factors and antibiotic resistance. In Foodborne Diseases; Elsevier: Amsterdam, The Netherlands, 2018; pp. 213–238. [Google Scholar]
- Saeed, K.; McLaren, A.C.; Schwarz, E.M.; Antoci, V.; Arnold, W.V.; Chen, A.F.; Clauss, M.; Esteban, J.; Gant, V.; Hendershot, E.; et al. The 2018 international consensus meeting on musculoskeletal infection: Summary from the biofilm workgroup and consensus on biofilm related musculoskeletal infections. J. Orthop. Res. 2019, 37, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Stone, P.W. Economic burden of healthcare-associated infections: An american perspective. Expert Rev. Pharm. Outcomes Res. 2009, 9, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Berríos-Torres, S.I.; Umscheid, C.A.; Bratzler, D.W.; Leas, B.; Stone, E.C.; Kelz, R.R.; Reinke, C.E.; Morgan, S.; Solomkin, J.S.; Mazuski, J.E. Centers for disease control and prevention guideline for the prevention of surgical site infection, 2017. JAMA Surg. 2017, 152, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.M.; Lau, E.; Watson, H.; Schmier, J.K.; Parvizi, J. Economic burden of periprosthetic joint infection in the united states. J. Arthroplast. 2012, 27, 61–65. [Google Scholar] [CrossRef]
- Metsemakers, W.; Kuehl, R.; Moriarty, T.; Richards, R.; Verhofstad, M.; Borens, O.; Kates, S.; Morgenstern, M. Infection after fracture fixation: Current surgical and microbiological concepts. Injury 2018, 49, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, B.H.; Berg, R.A.; Daley, J.A.; Fritz, J.; Bhave, A.; Mont, M.A. Periprosthetic joint infection. Lancet 2016, 387, 386–394. [Google Scholar] [CrossRef]
- Renbarger, T.L.; Baker, J.M.; Sattley, W.M. Slow and steady wins the race: An examination of bacterial persistence. Aims Microbiol. 2017, 3, 171–185. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. Fems Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.L.; Hill, K.E.; Malic, S.; Thomas, D.W.; Williams, D.W. Antimicrobial tolerance and the significance of persister cells in recalcitrant chronic wound biofilms. Wound Repair Regen. 2011, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Montanaro, L. Implant infections: Adhesion, biofilm formation and immune evasion. Nat. Rev. Microbiol. 2018, 16, 397. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Davies, D.; Sauer, K. Control of biofilms with the fatty acid signaling molecule cis-2-decenoic acid. Pharmaceuticals 2015, 8, 816–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ji, H.; Liu, C.; Bing, W.; Wang, Z.; Qu, X. A multinuclear metal complex based dnase-mimetic artificial enzyme: Matrix cleavage for combating bacterial biofilms. Angew. Chem. Int. Ed. 2016, 55, 10732–10736. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.C.; Patel, S.K.; Kang, Y.C.; Lee, J.K. Quorum sensing inhibitors as antipathogens: Biotechnological applications. Biotechnol. Adv. 2018, 37, 68–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, S.; Chen, H.; Chen, T.; Yang, R.; Li, M.; Fu, Y.; Jia, A.Q. Anti-biofilm and antivirulence activities of metabolites from plectosphaerella cucumerina against pseudomonas aeruginosa. Front. Microbiol. 2017, 8, 769. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011, 473, 216. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Buson, A.; Jarolimek, W.; Rice, S.A. Mannitol enhances antibiotic sensitivity of persister bacteria in pseudomonas aeruginosa biofilms. PLoS ONE 2013, 8, e84220. [Google Scholar] [CrossRef]
- Cabral, D.; Wurster, J.; Belenky, P. Antibiotic persistence as a metabolic adaptation: Stress, metabolism, the host, and new directions. Pharmaceuticals 2018, 11, 14. [Google Scholar] [CrossRef]
- Percival, S. Importance of biofilm formation in surgical infection. Br. J. Surg. 2017, 104, e85–e94. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Rowe, S.E.; O’Gara, J.P.; Conlon, B.P. Convergence of staphylococcus aureus persister and biofilm research: Can biofilms be defined as communities of adherent persister cells? Plos Pathog. 2016, 12, e1006012. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry (Mosc.) 2005, 70, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.M.; Althausen, P.L.; Kellam, J.; Bosse, M.J.; Castillo, R.; Lower Extremity Assessment Project Study, G. Complications following limb-threatening lower extremity trauma. J. Orthop. Trauma 2009, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hospenthal, D.R.; Murray, C.K.; Andersen, R.C.; Bell, R.B.; Calhoun, J.H.; Cancio, L.C.; Cho, J.M.; Chung, K.K.; Clasper, J.C.; Colyer, M.H.; et al. Guidelines for the prevention of infections associated with combat-related injuries: 2011 update. J. Trauma: Inj. Infect. Crit. Care 2011, 71, S210–S234. [Google Scholar] [CrossRef] [PubMed]
- McConoughey, S.J.; Howlin, R.P.; Wiseman, J.; Stoodley, P.; Calhoun, J.H. Comparing pmma and calcium sulfate as carriers for the local delivery of antibiotics to infected surgical sites. J. Biomed. Mater. Res. B Appl. Biomater. 2014, 103, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Hafeman, A.E.; Zienkiewicz, K.J.; Carney, E.; Litzner, B.; Stratton, C.; Wenke, J.C.; Guelcher, S.A. Local delivery of tobramycin from injectable biodegradable polyurethane scaffolds. J. Biomater. Sci. Polym. Ed. 2010, 21, 95–112. [Google Scholar] [CrossRef]
- Gogia, J.S.; Meehan, J.P.; Di Cesare, P.E.; Jamali, A.A. Local antibiotic therapy in osteomyelitis. Semin. Plast. Surg. 2009, 23, 100–107. [Google Scholar] [CrossRef]
- Jackson, S.R.; Richelsoph, K.C.; Courtney, H.S.; Wenke, J.C.; Branstetter, J.G.; Bumgardner, J.D.; Haggard, W.O. Preliminary in vitro evaluation of an adjunctive therapy for extremity wound infection reduction: Rapidly resorbing local antibiotic delivery. J. Orthop. Res. 2009, 27, 903–908. [Google Scholar] [CrossRef]
- Overstreet, D.; McLaren, A.; Calara, F.; Vernon, B.; McLemore, R. Local gentamicin delivery from resorbable viscous hydrogels is therapeutically effective. Clin. Orthop. Relat. Res. 2015, 473, 337–347. [Google Scholar] [CrossRef]
- Harris, M.; Alexander, C.; Wells, C.; Bumgardner, J.; Carpenter, D.; Jennings, J. Chitosan for the delivery of antibiotics. In Chitosan Based Biomaterials Volume 2; Elsevier: Amsterdam, The Netherlands, 2017; pp. 147–173. [Google Scholar]
- Cheung, R.C.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An update on potential biomedical and pharmaceutical applications. Mar Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Berretta, J.M.; Jennings, J.A.; Courtney, H.S.; Beenken, K.E.; Smeltzer, M.S.; Haggard, W.O. Blended chitosan paste for infection prevention: Preliminary and preclinical evaluations. Clin. Orthop. Relat. Res. 2017, 475, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.S.; Alexander, C.M.; Berretta, J.M.; Courtney, H.S.; Beenken, K.E.; Smeltzer, M.S.; Bumgardner, J.D.; Haggard, W.O.; Jennings, J.A. Evaluation of a chitosan-polyethylene glycol paste as a local antibiotic delivery device. World J. Orthop 2017, 8, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Boles, L.; Alexander, C.; Pace, L.; Haggard, W.; Bumgardner, J.; Jennings, J. Development and evaluation of an injectable chitosan/beta-glycerophosphate paste as a local antibiotic delivery system for trauma care. J. Funct Biomater 2018, 9. [Google Scholar] [CrossRef]
- Boles, L.R.; Bumgardner, J.D.; Fujiwara, T.; Haggard, W.O.; Guerra, F.D.; Jennings, J.A. Characterization of trimethyl chitosan/polyethylene glycol derivatized chitosan blend as an injectable and degradable antimicrobial delivery system. Int. J. Biol. Macromol. 2019, 133, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.P.; Courtney, H.S.; Bumgardner, J.D.; Haggard, W.O. Chitosan sponges to locally deliver amikacin and vancomycin: A pilot in vitro evaluation. Clin. Orthop. Relat. Res. 2010, 468, 2074–2080. [Google Scholar] [CrossRef]
- Smith, J.K.; Moshref, A.R.; Jennings, J.A.; Courtney, H.S.; Haggard, W.O. Chitosan sponges for local synergistic infection therapy: A pilot study. Clin. Orthop. Relat. Res. 2013, 471, 3158–3164. [Google Scholar] [CrossRef] [PubMed]
- Boles, L.R.; Awais, R.; Beenken, K.E.; Smeltzer, M.S.; Haggard, W.O.; Jennings, J.A. Local delivery of amikacin and vancomycin from chitosan sponges prevent polymicrobial implant-associated biofilm. Mil. Med. 2018, 183, 459–465. [Google Scholar] [CrossRef]
- Jennings, J.A.; Beenken, K.E.; Parker, A.C.; Smith, J.K.; Courtney, H.S.; Smeltzer, M.S.; Haggard, W.O. Polymicrobial biofilm inhibition effects of acetate-buffered chitosan sponge delivery device. Macromol. Biosci. 2016, 16, 591–598. [Google Scholar] [CrossRef]
- Hoemann, C.; Sun, J.; Legare, A.; McKee, M.; Buschmann, M. Tissue engineering of cartilage using an injectable and adhesive chitosan-based cell-delivery vehicle. Osteoarthr. Cartil. 2005, 13, 318–329. [Google Scholar] [CrossRef]
- Chenite, A.; Chaput, C.; Wang, D.; Combes, C.; Buschmann, M.; Hoemann, C.; Leroux, J.; Atkinson, B.; Binette, F.; Selmani, A. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials 2000, 21, 2155–2161. [Google Scholar] [CrossRef]
- Dang, Q.; Liu, K.; Zhang, Z.; Liu, C.; Liu, X.; Xin, Y.; Cheng, X.; Xu, T.; Cha, D.; Fan, B. Fabrication and evaluation of thermosensitive chitosan/collagen/α, β-glycerophosphate hydrogels for tissue regeneration. Carbohydr. Polym. 2017, 167, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.H.; Yang, S.H.; Liu, C.C.; Gefen, A.; Lin, F.H. Thermosensitive hydrogel made of ferulic acid-gelatin and chitosan glycerophosphate. Carbohydr. Polym. 2013, 92, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Zhang, H.; Neau, S.H. In vitro degradation of chitosan by a commercial enzyme preparation: Effect of molecular weight and degree of deacetylation. Biomaterials 2001, 22, 1653–1658. [Google Scholar] [CrossRef]
- Yuan, Y.; Chesnutt, B.M.; Wright, L.; Haggard, W.O.; Bumgardner, J.D. Mechanical property, degradation rate, and bone cell growth of chitosan coated titanium influenced by degree of deacetylation of chitosan. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 86, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, T.; Okamura, Y.; Shigemasa, Y.; Minami, S.; Okamoto, Y. Effects of molecular weight and deacetylation degree of chitin/chitosan on wound healing. Carbohydr. Polym. 2007, 67, 640–644. [Google Scholar] [CrossRef]
- Kong, M.; Chen, X.G.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef]
- Sudarshan, N.; Hoover, D.; Knorr, D. Antibacterial action of chitosan. Food Biotechnol. 1992, 6, 257–272. [Google Scholar] [CrossRef]
- Zheng, L.Y.; Zhu, J.F. Study on antimicrobial activity of chitosan with different molecular weights. Carbohydr. Polym. 2003, 54, 527–530. [Google Scholar] [CrossRef]
- Muzzarelli, R.; Tarsi, R.; Filippini, O.; Giovanetti, E.; Biagini, G.; Varaldo, P. Antimicrobial properties of n-carboxybutyl chitosan. Antimicrob. Agents Chemother. 1990, 34, 2019–2023. [Google Scholar] [CrossRef] [PubMed]
- Shawkat, H.; Westwood, M.M.; Mortimer, A. Mannitol: A review of its clinical uses. Contin. Educ. Anaesth. Crit. Care Pain 2012, 12, 82–85. [Google Scholar] [CrossRef]
- Zhang, W.; Neal, J.; Lin, L.; Dai, F.; Hersey, D.P.; McDonagh, D.L.; Su, F.; Meng, L. Mannitol in critical care and surgery over 50+ years: A systematic review of randomized controlled trials and complications with meta-analysis. J. Neurosurg. Anesthesiol. 2018, 31, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Nissenson, A.R.; Weston, R.E.; Kleeman, C.R. Mannitol. West J. Med. 1979, 131, 277–284. [Google Scholar] [PubMed]
- Nordtveit, R.J.; Varum, K.M.; Smidsrod, O. Degradation of partially n-acetylated chitosans with hen egg white and human lysozyme. Carbohydr. Polym. 1996, 29, 163–167. [Google Scholar] [CrossRef]
- Pangburn, S.H.; Trescony, P.V.; Heller, J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 1982, 3, 105–108. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro-and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wei, J.; Li, H.; Wang, Y.L.; Xie, D.X.; Yang, T.; Gao, S.G.; Li, Y.S.; Luo, W.; Lei, G.H. Effectiveness and safety of glucosamine, chondroitin, the two in combination, or celecoxib in the treatment of osteoarthritis of the knee. Sci. Rep. 2015, 5, 16827. [Google Scholar] [CrossRef]
- VandeVord, P.J.; Matthew, H.W.; DeSilva, S.P.; Mayton, L.; Wu, B.; Wooley, P.H. Evaluation of the biocompatibility of a chitosan scaffold in mice. J. Biomed Mater Res. 2002, 59, 585–590. [Google Scholar] [CrossRef]
- Javorska, L.; Krcmova, L.K.; Solichova, D.; Solich, P.; Kaska, M. Modern methods for vancomycin determination in biological fluids by methods based on high-performance liquid chromatography--a review. J. Sep. Sci. 2016, 39, 6–20. [Google Scholar] [CrossRef]
- Lai, F.; Sheehan, T. Enhancement of detection sensitivity and cleanup selectivity for tobramycin through pre-column derivatization. J. Chromatogr. 1992, 609, 173–179. [Google Scholar] [CrossRef]
- Jansen, J.; De Ruijter, J.; Janssen, P.; Paquay, Y. Histological evaluation of a biodegradable polyactive®/hydroxyapatite membrane. Biomaterials 1995, 16, 819–827. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paste Group | Force (N) |
|---|---|
| ChMPEG | 1.20 N ± 0.20 N |
| Ch-PEG (control) | 1.82 N ± 0.38 N |
| ChMPEG ZOI | Ch-PEG ZOI | |
|---|---|---|
| Day 1 | 5.31 ± 0.19 mm | 6.18 ± 0.16 mm * |
| Day 2 | 4.37 ± 0.31 mm | 4.92 ± 0.29 mm * |
| Day 3 | 2.92 ± 0.19 mm | 1.99 ± 0.08 mm * |
| Day 4 | 2.40 ± 0.20 mm | 0.42 ± 0.19 mm * |
| Day 5 | 1.18 ± 0.17 mm | 0 mm * |
| Day 6 | 0.61 ± 0.32 mm | 0 mm * |
| Day 7 | 0.22 ± 0.04 mm | 0 mm * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pace, L.R.; Harrison, Z.L.; Brown, M.N.; Haggard, W.O.; Jennings, J.A. Characterization and Antibiofilm Activity of Mannitol–Chitosan-Blended Paste for Local Antibiotic Delivery System. Mar. Drugs 2019, 17, 517. https://doi.org/10.3390/md17090517
Pace LR, Harrison ZL, Brown MN, Haggard WO, Jennings JA. Characterization and Antibiofilm Activity of Mannitol–Chitosan-Blended Paste for Local Antibiotic Delivery System. Marine Drugs. 2019; 17(9):517. https://doi.org/10.3390/md17090517
Chicago/Turabian StylePace, Leslie R., Zoe L. Harrison, Madison N. Brown, Warren O. Haggard, and J. Amber Jennings. 2019. "Characterization and Antibiofilm Activity of Mannitol–Chitosan-Blended Paste for Local Antibiotic Delivery System" Marine Drugs 17, no. 9: 517. https://doi.org/10.3390/md17090517
APA StylePace, L. R., Harrison, Z. L., Brown, M. N., Haggard, W. O., & Jennings, J. A. (2019). Characterization and Antibiofilm Activity of Mannitol–Chitosan-Blended Paste for Local Antibiotic Delivery System. Marine Drugs, 17(9), 517. https://doi.org/10.3390/md17090517