A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors

 ,
,  , , , and
, , , and 
Abstract

1. Introduction
2. Discussion
2.1. New Kinase Inhibitors Discovered from Marine Organisms
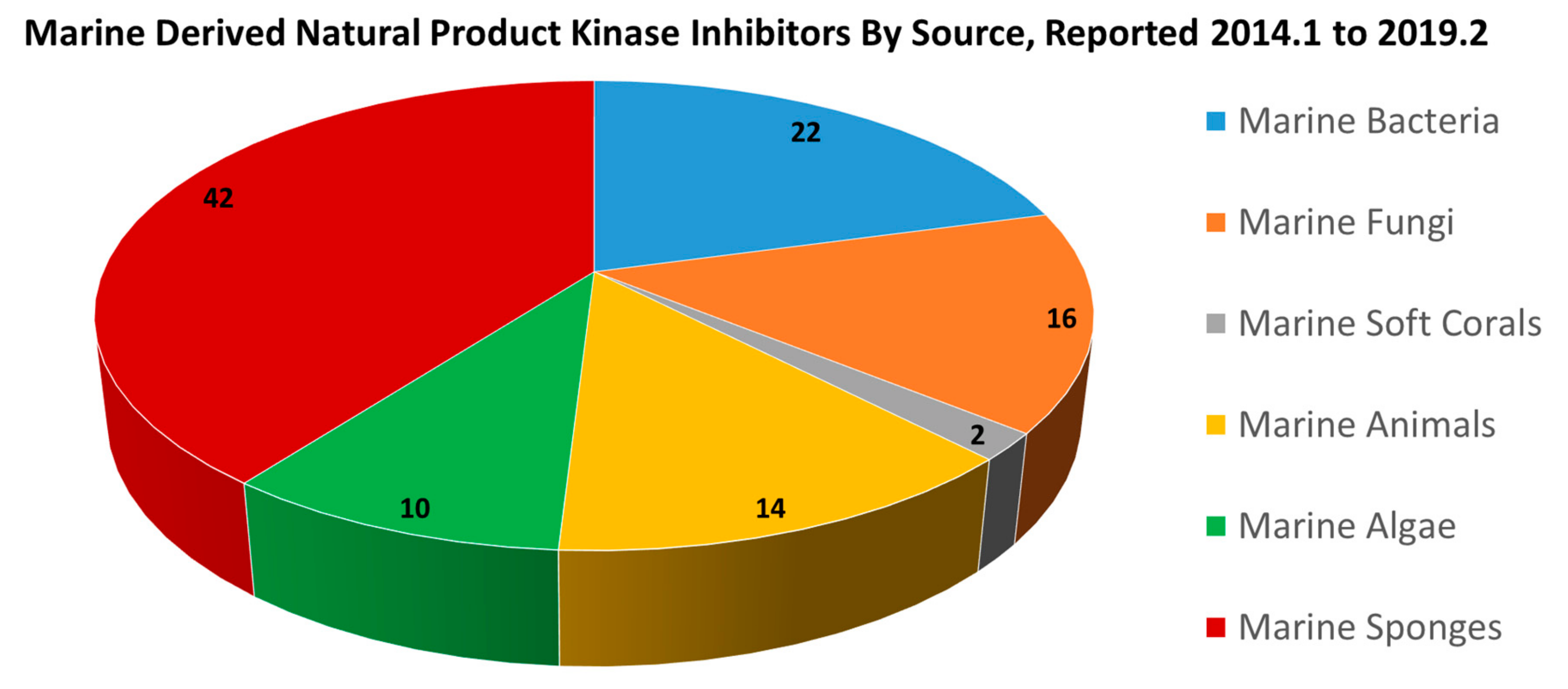
2.1.1. Kinase Inhibitors from Marine Bacteria
2.1.2. Kinase Inhibitors from Marine Fungi
2.1.3. Kinase Inhibitors from Marine Soft Coral
2.1.4. Kinase Inhibitors from Marine Animals
2.1.5. Kinase Inhibitors from Marine Algae
2.1.6. Kinase Inhibitors from Marine Sponges
2.2. Preclinical and Clinical Candidates
3. Conclusions
Funding
Conflicts of Interest
List of Abbreviations
| AKt | Serine/ threonine kinase |
| BTK | Brution tyrosine kinase |
| BrK | Breast tumor kinase |
| CLK1 | Cdc2-like kinase 1 |
| CLKs | Cdc2-like kinases |
| CDK1 | Cyclin-dependent kinase 1 |
| CDK2 | Cyclin-dependent kinase 2 |
| CDK4 | Cyclin-dependent kinase 4 |
| CDK5 | Cyclin-dependent kinase 5 |
| CDK6 | Cyclin-dependent kinase 6 |
| CDK9 | Cyclin-dependent kinase 9 |
| CK1 | Casein kinase 1 |
| CK5 | Casein kinase 5 |
| CAMK1a | Calcium/calmodulin-dependent protein kinase 1a |
| CDKN1C | Cyclin-dependent kinase inhibitor 1C |
| CDKN2B | Cyclin-dependent kinase inhibitor 2B |
| CHEK2 | Checkpoint kinase 2 |
| Class I PI3K | PI3-kinase class I |
| Chk | Csk homologous kinase |
| CaMKII | Calmodulin-dependent protein kinase II |
| DYRK1A | Dual-specificity tyrosine phosphorylation regulated kinase-1A |
| DYRKS | Dual-specificity tyrosine phosphorylation regulated kinases |
| ERK | Extracellular signal-regulated kinase |
| ERK1/2 | Extracellular signal-regulated kinase1/2 |
| EGFR | Epidermal growth factor receptor |
| FAK | Focal adhesion kinase |
| FGER | Fibroblast Growth Factor Receptor |
| GSK-3 | Glycogen synthase kinase 3 |
| GSK-3β | Glycogen synthase kinase 3 beta |
| IKK | IkappaB kinase |
| IGF1-R | Insulin-like growth factor 1 receptor |
| JNK | C-Jun NH-terminal kinase |
| LIMK | LIM kinase |
| MtSK | Shikimate kinase from mycobacterium tuberculosis |
| MRSA PK | MRSA pyruvate kinase |
| MAPK | Mitogen-activated protein kinases |
| MAP3K11 | Mitogen-activated protein kinase kinase kinase11 |
| MIC | Minimum inhibitory concentration |
| NF-κB | Nuclear factor – kappa B |
| PKC-α | Protein kinase C alpha |
| PKC-β | Protein kinase C beta |
| PKnG | Protein kinase G |
| PKB | Protein kinase B |
| p-AKt | Phosphorylated AKt |
| p-ERK1/2 | Phosphorylated extracellular signal-regulated kinase |
| PAK1 | p21-activated kinase 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PDGFR-α | Platelet-derived growth factor receptor alpha |
| PDGFR-β | Platelet-derived growth factor receptor beta |
| PDPK1 | Phosphatidylinositide-dependent protein kinase 1 |
| ROCK2 | Rho-associated protein kinase |
| RTKs | Receptor tyrosine kinase |
| RIPK2 | Receptor-interacting serine/threonine kinase 2 |
| STK | Serine/threonine kinase family |
| SIK2 | Salt-inducible kinase2 |
| SphKs | Sphingosine kinases |
| STAT3 | Signal transducer and activator of transcription 3 |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| JAK2 | Tyrosine-protein kinase JAK2 |
References
- Shang, J.; Hu, B.; Wang, J.; Zhu, F.; Kang, Y.; Li, D.; Sun, H.; Kong, D.X.; Hou, T. A cheminformatic insight into the differences between terrestrial and marine originated natural products. J. Chem. Inf. Model. 2018, 58, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C. Natural products as source of therapeutics against parasitic diseases. Angew. Chem. Int. Ed. 2015, 54, 14622–14624. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2014, 13, 123–137. [Google Scholar] [CrossRef]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Phytochem. Rev. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- El-Elimat, T.; Zhang, X.; Jarjoura, D.; Moy, F.J.; Orjala, J.; Kinghorn, A.D.; Pearce, C.J.; Oberlies, N.H. Chemical diversity of metabolites from fungi, cyanobacteria, and plants relative to FDA-approved anticancer agents. ACS Med. Chem. Lett. 2012, 3, 645–649. [Google Scholar] [CrossRef]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. Chem. Inf. Comput. 2003, 43, 218–227. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2008, 25, 35–94. [Google Scholar] [CrossRef]
- Rocha, L.A.; Pinheiro, H.T.; Shepherd, B.; Papastamatiou, Y.P.; Luiz, O.J.; Pyle, R.L.; Bongaerts, P. Mesophotic coral ecosystems are threatened and ecologically distinct from shallow water reefs. Science 2018, 361, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Airesdesousa, J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Kuttruff, C.A.; Eastgate, M.D.; Baran, P.S. Natural product synthesis in the age of scalability. Nat. Prod. Rep. 2014, 31, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase inhibitors of marine origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.K. Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Patterson, H.; Nibbs, R.; Mcinnes, I.; Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef]
- Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem.Sci. 2002, 27, 514–520. [Google Scholar]
- Ubersax, J.; Ferrell, J. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Official Website of U.S. Food and Drug Administration. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on 10 April 2019).
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353. [Google Scholar] [CrossRef]
- Rowinsky, E.K. The erbB family: Targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Annu. Rev. Med. 2004, 55, 433. [Google Scholar] [CrossRef]
- Yao, H.P.; Zhou, Y.Q.; Ma, Q.; Guin, S.; Padhye, S.S.; Zhang, R.W.; Wang, M.H. The monoclonal antibody Zt/f2 targeting RON receptor tyrosine kinase as potential therapeutics against tumor growth-mediated by colon cancer cells. Mol. Cancer 2011, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Q.; Sharma, S.V.; Markus, W. Allosteric inhibition of BCR-ABL. Cell Cycle 2010, 9, 3734–3738. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Wells, J.A. Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol. 2004, 14, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr. Pharm. Des. 2012, 18, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-competitive inhibitors of mTOR: An update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, P.; Mucs, D.; Bryce, R.A. Targeting the inactive conformation of protein kinases: Computational screening based on ligand conformation. MedChemComm 2012, 3, 434–440. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef]
- Simard, J.R.; Pawar, V.; Aust, B.; Wolf, A.; Rabiller, M.; Wulfert, S.; Robubi, A.; Klüter, S.; Ottmann, C.; Rauh, D. High-Throughput Screening To Identify Inhibitors Which Stabilize Inactive Kinase Conformations in p38α. J. Am. Chem. Soc. 2009, 51, 18478–18488. [Google Scholar] [CrossRef]
- Oleg, F.; Brian, M.; Vanda, P.; Peter, R.; Susanne, M.; Bullock, A.N.; Juerg, S.; Michael, S.M.; Stefan, K. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar]
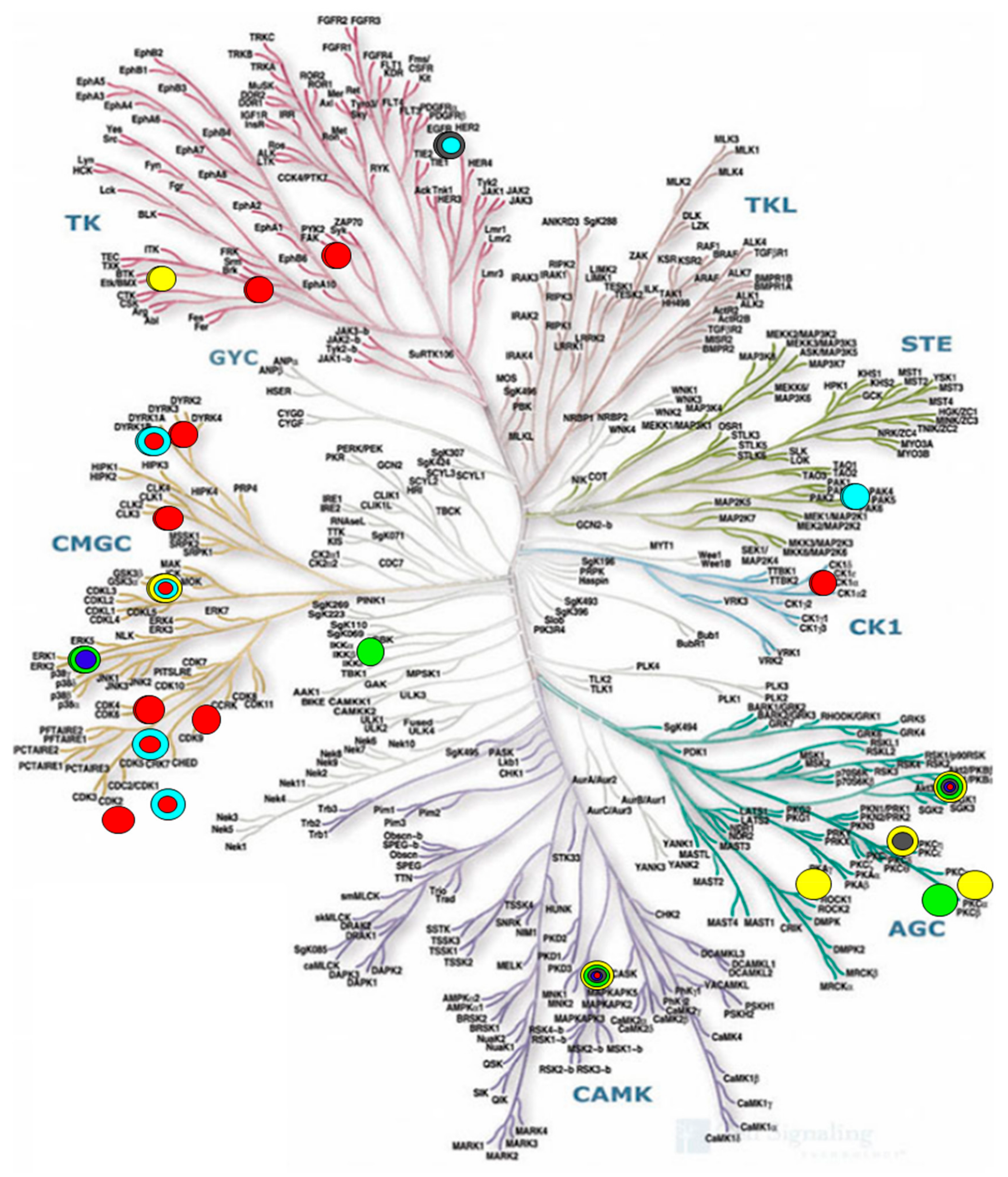
- Website of Cell Signaling Technology, Inc. Available online: https://media.cellsignal.com/www/images/science/kinases/kinome.jpg (accessed on 1 May 2019).
- Dey, G.; Bharti, R.; Dhanarajan, G.; Das, S.; Dey, K.K.; Kumar, B.N.P.; Sen, R.; Mandal, M. Marine lipopeptide Iturin A inhibits Akt mediated GSK3β and FoxO3a signaling and triggers apoptosis in breast cancer. Sci. Rep. 2015, 5, 10316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qin, L.L.; Ding, W.J.; Ma, Z.J. Cytotoxic indolocarbazoles alkaloids from the streptomyces sp. A65. Tetrahedron 2018, 74, 726–730. [Google Scholar] [CrossRef]
- Qin, L.L.; Zhou, B.; Ding, W.; Ma, Z. Bioactive metabolites from marine-derived Streptomyces sp. A68 and its Rifampicin resistant mutant strain R-M1. Phytochem. Lett. 2018, 23, 46–51. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Li, J.Q.; Zhang, H.J.; Ding, W.J.; Ma, Z.J. Cyclizidine-Type Alkaloids from Streptomyces sp. HNA39. J. Nat. Prod. 2018, 81, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Freer, A.A.; Gardner, D.; Greatbanks, D.; Poyser, J.P.; Sim, G.A. Structure of cyclizidine (antibiotic M146791): X-ray crystal structure of an indolizidinediol metabolite bearing a unique cyclopropyl side-chain. J. Chem. Soc. Chem. Commun. 1982, 20, 1160–1162. [Google Scholar] [CrossRef]
- Gomi, S.; Ikeda, D.; Nakamura, H.; Naganawa, H.; Yamashita, F.; Hotta, K.; Kondo, S.; Okami, Y.; Umezawa, H.; Iitaka, Y. Isolation and structure of a new antibiotic, indolizomycin, produced by a strain SK2-52 obtained by interspecies fusion treatment. J. Antibiot. 1984, 37, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Miho, I.; Takahiro, H.; Motoki, T.; Kazuo, S.Y. A new cyclizidine analog-JBIR-102-from Saccharopolyspora sp. RL78 isolated from mangrove soil. J. Antibiot. 2012, 65, 41. [Google Scholar]
- Wang, J.N.; Zhang, H.J.; Li, J.Q.; Ding, W.J.; Ma, Z.J. Bioactive Indolocarbazoles from the Marine-Derived Streptomyces sp. DT-A61. J. Nat. Prod. 2018, 81, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhou, B.; Liu, H.; Huo, C.; Ding, W. One new indolocarbazole alkaloid from the Streptomyces sp. A22. Nat. Prod. Res. 2018, 32, 2583–2588. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, Z.J.; Zhang, H.J.; Li, J.Q.; Ding, W.J.; Ma, Z.J. Bioactive staurosporine derivatives from the Streptomyces sp. NB-A13. Bioorg. Chem. 2018, 82, 33–40. [Google Scholar] [CrossRef]
- Jiang, Y.-J.; Zhang, D.-S.; Zhang, H.-J.; Li, J.-Q.; Ding, W.-J.; Xu, C.-D.; Ma, A.Z.-J. Medermycin-Type Naphthoquinones from the Marine-Derived Streptomyces sp. XMA39. J. Nat. Prod. 2018, 9, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ma, S.; He, L.; Yuan, P.; She, Z.; Lu, Y. Sclerotiorin inhibits protein kinase G from Mycobacterium tuberculosis and impairs mycobacterial growth in macrophages. Curr. Top. Microbiol. Immunol. 2017, 103, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Dong-Cheol, K.; Hee-Suk, L.; Wonmin, K.; Dong-Sung, L.; Jae Hak, S.; Joung Han, Y.; Youn-Chul, K.; Hyuncheol, O. Anti-inflammatory effect of methylpenicinoline from a marine isolate of Penicillium sp. (SF-5995): Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced RAW264.7 macrophages and BV2 microglia. Molecules 2014, 19, 18073–18089. [Google Scholar]
- Li, W.; Li, M.; Su, X.; Qin, L.; Miao, M.; Yu, C.; Shen, Y.; Luo, Q.; Chen, Q. Mycoepoxydiene induces apoptosis and inhibits TPA-induced invasion in human cholangiocarcinoma cells via blocking NF-κB pathway. Biochimie 2014, 101, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Li, Y.; Guo, L.; Wang, Y.; Liu, P.; Zhu, W. Indole diterpenoids and isocoumarin from the fungus, Aspergillus flavus, isolated from the prawn, Penaeus vannamei. Mar. Drugs 2014, 12, 3970–3981. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Turlova, E.; Sun, C.L.F.; Kim, J.S.; Huang, S.; Zhong, X.; Guan, Y.Y.; Wang, G.L.; Rutka, J.T.; Feng, Z.P. Xyloketal B Suppresses Glioblastoma Cell Proliferation and Migration in Vitro through Inhibiting TRPM7-Regulated PI3K/Akt and MEK/ERK Signaling Pathways. Mar. Drugs 2015, 13, 2505–2525. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.C.; Quang, T.H.; Ngan, N.T.; Yoon, C.S.; Sohn, J.H.; Yim, J.H.; Feng, Y.; Che, Y.; Kim, Y.C.; Oh, H. Dihydroisocoumarin Derivatives from Marine-Derived Fungal Isolates and Their Anti-inflammatory Effects in Lipopolysaccharide-Induced BV2 Microglia. J. Nat. Prod. 2015, 78, 2948–2955. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Ko, S.K.; Kim, H.M.; Kim, G.H.; Son, S.; Kim, G.S.; Hwang, G.J.; Jeon, E.S.; Shin, K.S.; Ryoo, I.J. Stachybotrysin, an Osteoclast Differentiation Inhibitor from the Marine-Derived Fungus Stachybotrys sp. KCB13F013. J. Nat. Prod. 2016, 79, 2703–2708. [Google Scholar] [CrossRef] [PubMed]
- Wiese, J.; Imhoff, J.F.; Gulder, T.A.; Labes, A.; Schmaljohann, R. Marine Fungi as Producers of Benzocoumarins, a New Class of Inhibitors of Glycogen-Synthase-Kinase 3β. Mar. Drugs 2016, 14, 200. [Google Scholar] [CrossRef]
- Ko, W.; Sohn, J.H.; Jang, J.H.; Ahn, J.S.; Kang, D.G.; Lee, H.S.; Kim, J.S.; Kim, Y.C.; Oh, H. Inhibitory effects of alternaramide on inflammatory mediator expression through TLR4-MyD88-mediated inhibition of NF-кB and MAPK pathway signaling in lipopolysaccharide-stimulated RAW264.7 and BV2 cells. Chem. Biol. Interact. 2016, 244, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Kim, D.C.; Yoon, C.S.; Ko, W.M.; Lee, S.J.; Sohn, J.H.; Jang, J.H.; Ahn, J.S.; Kim, Y.C.; Oh, H. Anti-neuroinflammatory effects of citreohybridonol involving TLR4-MyD88-mediated inhibition of NF-кB and MAPK signaling pathways in lipopolysaccharide-stimulated BV2 cells. Neurochem. Int. 2016, 95, 55–62. [Google Scholar] [CrossRef]
- García-Caballero, M.; Blacher, S.; Paupert, J.; Quesada, A.R.; Medina, M.A.; Noël, A. Novel application assigned to toluquinol: Inhibition of lymphangiogenesis by interfering with VEGF-C/VEGFR-3 signaling pathway. Br. J. Pharmacol. 2016, 173, 1966–1987. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wiese, J.; Schmaljohann, R.; Imhoff, J. Biscogniauxone, a New Isopyrrolonaphthoquinone Compound from the Fungus Biscogniauxia mediterranea Isolated from Deep-Sea Sediments. Mar. Drugs 2016, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Ngan, N.T.T.; Quang, T.H.; Kim, K.W.; Kim, H.J.; Sohn, J.H.; Kang, D.G.; Lee, H.S.; Kim, Y.C.; Oh, H. Anti-inflammatory effects of secondary metabolites isolated from the marine-derived fungal strain Penicillium sp. SF-5629. Arch. Pharm. Res. 2017, 40, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Skropeta, D.; Wei, L. Recent advances in deep-sea natural products. Nat. Prod. Rep. 2014, 31, 999–1025. [Google Scholar] [CrossRef] [PubMed]
- Navarri, M.; Jégou, C.; Bondon, A.; Pottier, S.; Bach, S.; Baratte, B.; Ruchaud, S.; Barbier, G.; Burgaud, G.; Fleury, Y. Bioactive Metabolites from the Deep Subseafloor Fungus Oidiodendron griseum UBOCC-A-114129. Mar. Drugs 2017, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Sheu, M.J.; Ma, W.F.; Pan, C.H.; Sheu, J.H.; Wu, C.H. Anti-Restenotic Roles of Dihydroaustrasulfone Alcohol Involved in Inhibiting PDGF-BB-Stimulated Proliferation and Migration of Vascular Smooth Muscle Cells. Mar. Drugs 2015, 13, 3046–3060. [Google Scholar] [CrossRef]
- Mohyeldin, M.M.; Akl, M.R.; Siddique, A.B.; Hassan, H.M.; El Sayed, K.A. The marine-derived pachycladin diterpenoids as novel inhibitors of wild-type and mutant EGFR. Biochem. Pharmacol. 2017, 126, 51–68. [Google Scholar] [CrossRef]
- Hassan, H.M.; Khanfar, M.A.; Elnagar, A.Y.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.; Hifnawy, M.S.; El Sayed, K.A. Pachycladins A-E, prostate cancer invasion and migration inhibitory Eunicellin-based diterpenoids from the red sea soft coral Cladiella pachyclados. J. Nat. Prod. 2010, 73, 848. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R.M.; Badr, J.M.; Youssef, D.T.A. Didemnaketals D and E, bioactive terpenoids from a Red Sea ascidian Didemnum species. Tetrahedron 2014, 45, 35–40. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Mohamed, G.A.; Shaala, L.A.; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R.M. New purine alkaloids from the Red Sea marine tunicate Symplegma rubra. Phytochem. Lett. 2015, 13, 212–217. [Google Scholar] [CrossRef]
- Adrian, T.E.; Collin, P. The Anti-Cancer Effects of Frondoside A. Mar. Drugs 2018, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Bcq, N.; Yoshimura, K.; Kumazawa, S.; Tawata, S.; Maruta, H. Frondoside A from sea cucumber and nymphaeols from Okinawa propolis: Natural anti-cancer agents that selectively inhibit PAK1 in vitro. Drug Discov. Ther. 2017, 11, 110–114. [Google Scholar]
- Wätjen, W.; Ebada, S.S.; Bergermann, A.; Chovolou, Y.; Totzke, F.; Kubbutat, M.H.G.; Lin, W.; Proksch, P. Cytotoxic effects of the anthraquinone derivatives 1′-deoxyrhodoptilometrin and (S)-(−)-rhodoptilometrin isolated from the marine echinoderm Comanthus sp. Arch. Toxicol. 2016, 91, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.; Nielson, J.L.; Tapiolas, D.M.; Motti, C.A.; Ovenden, S.P.B.; Kearns, P.S.; Liptrot, C.H. Detailed NMR, Including 1,1-ADEQUATE, and Anticancer Studies of Compounds from the Echinoderm Colobometra perspinosa. Mar. Drugs 2009, 7, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.J.; Jiang, B.; Wu, N.; Li, X.; Liu, S.; Luo, J.; Shi, D. Anti-Angiogenic Properties of BDDPM, a Bromophenol from Marine Red Alga Rhodomela confervoides, with Multi Receptor Tyrosine Kinase Inhibition Effects. Int. J. Mol. Sci. 2015, 16, 13548–13560. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Luo, J.; Jiang, B.; Wang, L.; Wang, S.; Wang, C.; Fu, C.; Li, J.; Shi, D. Marine bromophenol bis (2,3-dibromo-4,5-dihydroxy-phenyl)-methane inhibits the proliferation, migration, and invasion of hepatocellular carcinoma cells via modulating β1-Integrin/FAK Signaling integrin/FAK signaling. Mar. Drugs 2015, 13, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.H.; Lee, E.H.; Park, K.; Kwon, J.Y.; Kim, P.H.; Jung, W.K.; Kim, Y.M. Eckol from Eisenia bicyclis Inhibits Inflammation Through the Akt/NF-kB Signaling in Propionibacterium acnes-Induced Human Keratinocyte Hacat Cells. J. Food Biochem. 2016, 41, e12312. [Google Scholar] [CrossRef]
- Lin, J.; Yu, J.; Zhao, J.; Zhang, K.; Zheng, J.; Wang, J.; Huang, C.; Zhang, J.; Yan, X.; Gerwick, W.H. Fucoxanthin, a Marine Carotenoid, Attenuates β-Amyloid Oligomer-Induced Neurotoxicity Possibly via Regulating the PI3K/Akt and the ERK Pathways in SH-SY5Y Cells. Oxidat. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Satomi, Y. Antitumor and Cancer-preventative Function of Fucoxanthin: A Marine Carotenoid. Anticancer Res. 2017, 37, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Kwon, S.H.; Chun, Y.S.; Gu, M.Y.; Yang, H.O. Anti-Neuroinflammatory Effects of Fucoxanthin via Inhibition of Akt/NF-κB and MAPKs/AP-1 Pathways and Activation of PKA/CREB Pathway in Lipopolysaccharide-Activated BV-2 Microglial Cells. Neurochem. Res. 2017, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Ye, B.R.; Kim, E.A.; Kim, J.; Kim, M.S.; Lee, W.W.; Ahn, G.N.; Kang, N.; Jung, W.K.; Heo, S.J. Bis (3-bromo-4,5-dihydroxybenzyl) ether, a novel bromophenol from the marine red alga Polysiphonia morrowii that suppresses LPS-induced inflammatory response by inhibiting ROS-mediated ERK signaling pathway in RAW 264.7 macrophages. Biomed. Pharmacother. 2018, 103, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Paudel, U.; Lee, Y.H.; Kwon, T.H.; Park, N.H.; Yun, B.S.; Hwang, P.H.; Yi, H.K. Eckols reduce dental pulp inflammation through the ERK1/2 pathway independent of COX-2 inhibition. Oral Dis. 2015, 20, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Chen, Q.Y.; Dang, L.H.; Luesch, H. Apratoxin S10, a Dual Inhibitor of Angiogenesis and Cancer Cell Growth To Treat Highly Vascularized Tumors. ACS Med. Chem. Lett. 2017, 8, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Mascuch, S.J.; Villa, F.A.; Byrum, T.; Gerwick, W.H. Honaucins A-C, Potent Inhibitors of Inflammation and Bacterial Quorum Sensing: Synthetic Derivatives and Structure-Activity Relationships. Chem. Biol. 2012, 19, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, M.; Li, L.; Choi, H.; Gerwick, W.H.; Soh, Y. Bromo-honaucin A inhibits osteoclastogenic differentiation in RAW 264.7 cells via Akt and ERK signaling pathways. Eur. J. Pharmacol. 2015, 769, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Hoiamide A, a Sodium Channel Activator of Unusual Architecture from a Consortium of Two Papua New Guinea Cyanobacteria. Chem. Biol. 2009, 16, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Xichun, L.; Xiaohan, Z.; Michael, G.; William, G.; Thomas, M. Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons. Mar. Drugs 2015, 13, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barri, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a Potent Antiproliferative Cyclic Depsipeptide from the Panamanian Marine Cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.G.; Mcphail, K.L.; Ishmael, J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Investig. New Drugs 2016, 34, 24–40. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Fabien, P.; Pritesh, P.; Xue, X.; Piggott, A.M.; Xiao-Cong, H.; Zeinab, K.; Capon, R.J. Callyspongisines A-D: Bromopyrrole alkaloids from an Australian marine sponge, Callyspongia sp. Org. Biomol. Chem. 2014, 12, 1579–1584. [Google Scholar]
- Cimino, G.; Rosa, S.D.; Stefano, S.D.; Mazzarella, L.; Puliti, R.; Sodano, G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 1982, 23, 767–768. [Google Scholar] [CrossRef]
- Williams, D.H.; Faulkner, D.J. Isomers and Tautomers of Hymenialdisine and Debromohymenialdisine. Nat. Prod. Lett. 1996, 9, 57–64. [Google Scholar] [CrossRef]
- Yu, Z.G.; Bi, K.S.; Guo, Y.W. Hyrtiosins A–E, Five New Scalarane Sesterterpenes from the South China Sea Sponge Hyrtios erecta. Helv. Chim. Acta 2005, 88, 1004–1009. [Google Scholar] [CrossRef]
- He, W.F.; Xue, D.Q.; Yao, L.G.; Li, J.Y.; Li, J.; Guo, Y.W. Hainanerectamines A–C, alkaloids from the Hainan sponge Hyrtios erecta. Mar. Drugs 2014, 12, 3982–3993. [Google Scholar] [CrossRef]
- Akl, M.R.; Foudah, A.I.; Ebrahim, H.Y.; Meyer, S.A.; El Sayed, K.A. The marine-derived sipholenol A-4-O-3’,4’-dichlorobenzoate inhibits breast cancer growth and motility in vitro and in vivo through the suppression of Brk and FAK signaling. Mar. Drugs 2014, 12, 2282–2304. [Google Scholar] [CrossRef]
- Lidgren, G.; Bohlin, L.; Bergman, J. Studies of swedish marine organisms VII. A novel biologically active indole alkaloid from the sponge geodia baretti. Tetrahedron Lett. 1986, 27, 3283–3284. [Google Scholar] [CrossRef]
- Lind, K.F.; Østerud, B.; Hansen, E.; Jørgensen, T.Ø.; Andersen, J.H. The immunomodulatory effects of barettin and involvement of the kinases CAMK1α and RIPK2. Immunopharmacol. Immunotoxicol. 2015, 5, 458–464. [Google Scholar] [CrossRef]
- Lind, K.F.; Espen, H.; Bjarne, S.; Karl-Erik, E.; Annette, B.; Magnus, E.; Kinga, L.; Andersen, J.H. Antioxidant and anti-inflammatory activities of barettin. Mar. Drugs 2013, 11, 2655–2666. [Google Scholar] [CrossRef]
- Rebecca, H.; Boris, P.; Eugen, J.; Joachim, S.; Dorian, S.; Daniel, C.; Frank, T.; Christoph, S.C.; Christian, P. Optimization of potent DFG-in inhibitors of platelet derived growth factor receptorβ (PDGF-Rβ) guided by water thermodynamics. J. Med. Chem. 2014, 58, 170–182. [Google Scholar]
- Segraves, N.L.; Lopez, S.; Johnson, T.A.; Said, S.A.; Fu, X.; Schmitz, F.J.; Crews, P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla-Fascaplysinopsis sponges and Didemnum tunicates. Tetrahedron Lett. 2003, 44, 3471–3475. [Google Scholar] [CrossRef]
- Emmanuel, A.; Thoma s, S.T.; Isabelle, M.; Hermann, E.; Menger, M.D.; Laschke, M.W. The Marine-Derived Kinase Inhibitor Fascaplysin Exerts Anti-Thrombotic Activity. Mar. Drugs 2015, 13, 6774–6791. [Google Scholar]
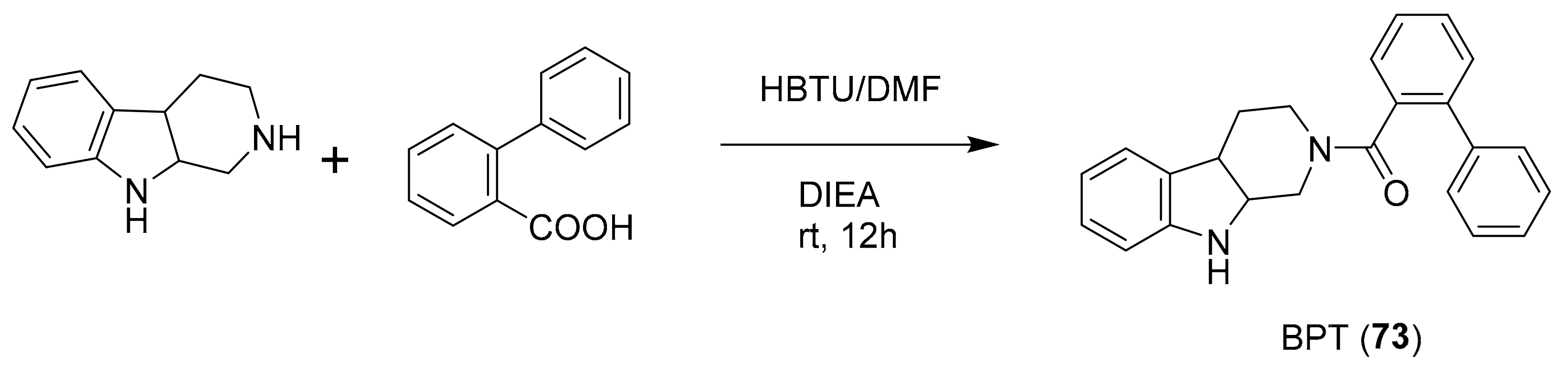
- Mahale, S.; Bharate, S.B.; Manda, S.; Joshi, P.; Jenkins, P.R.; Vishwakarma, R.A.; Chaudhuri, B. Antitumour potential of BPT: A dual inhibitor of cdk4 and tubulin polymerization. Cell Death Dis. 2015, 6, 1743. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G. Cytotoxic effects of fascaplysin against small cell lung cancer cell lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Santosh Kumar, G.; Anup Singh, P.; Sudhakar, M.; Ajay, K.; Bharate, S.B.; Vishwakarma, R.A.; Fayaz, M.; Shashi, B. Fascaplysin induces caspase mediated crosstalk between apoptosis and autophagy through the inhibition of PI3K/AKT/mTOR signaling cascade in human leukemia HL-60 cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Biol. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef]
- Veale, C.G.L.; Roya, Z.; Young, R.M.; Morrison, J.P.; Manoja, P.; Lobb, K.A.; Reiner, N.E.; Andersen, R.J.; Davies-Coleman, M.T. Synthetic analogues of the marine bisindole deoxytopsentin: Potent selective inhibitors of MRSA pyruvate kinase. J. Nat. Prod. 2015, 78, 355–362. [Google Scholar] [CrossRef]
- Akl, M.R.; Ayoub, N.M.; Ebrahim, H.Y.; Mohyeldin, M.M.; Orabi, K.Y.; Foudah, A.I.; El Sayed, K.A. Araguspongine C induces autophagic death in breast cancer cells through suppression of c-Met and HER2 receptor tyrosine kinase signaling. Mar. Drugs 2015, 13, 288–311. [Google Scholar] [CrossRef]
- Ebada, S.S.; Hoang, L.M.; Arlette, L.; De Voogd, N.J.; Emilie, D.; Laurent, M.; Marie-Lise, B.K.; Singab, A.N.B.; Müller, W.E.G.; Peter, P. Dispacamide E and other bioactive bromopyrrole alkaloids from two Indonesian marine sponges of the genus Stylissa. Nat. Prod. Res. 2015, 29, 231–238. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Gunasekera, S.P.; Lakshmi, V.; Tillekeratne, L.M. Marine natural products: Pyrrololactams from several sponges. J. Nat. Prod. 1985, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Elkhayat, E.S.; Edrada, R.A.; Ebel, R.; Proksch, P. New Bromopyrrole Alkaloids From The Marine Sponges Axinella Damicornis And Stylissa Flabelliformis. Nat. Prod. Commun. 2007, 2, 1149–1154. [Google Scholar] [CrossRef]
- Li, C.J.; Schmitz, F.J.; Kelly-Borges, M. A new lysine derivative and new 3-bromopyrrole carboxylic acid derivative from two marine sponges. J. Nat. Prod. 1998, 61, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Uemoto, H.; Tsuda, M.; Kobayashi, J.I. Mukanadins A−C, New Bromopyrrole Alkaloids from Marine Sponge Agelas nakamurai. J. Nat. Prod. 1999, 62, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Cafieri, F.; Fattorusso, E.; Mangoni, A.; Taglialatela-Scafati, O. Longamide and 3,7-dimethylisoguanine, two novel alkaloids from the marine sponge Agelas longissima. Tetrahedron Lett. 2010, 36, 7893–7896. [Google Scholar] [CrossRef]
- Fei, H.; Mai, L.H.; Longeon, A.; Copp, B.R.; Loaëc, N.; Bescond, A.; Meijer, L.; Bourguet-Kondracki, M.L. Novel Adociaquinone Derivatives from the Indonesian Sponge Xestospongia sp. Mar. Drugs 2015, 13, 2617–2628. [Google Scholar]
- Guzmán, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin A is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs 2015, 33, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 5, 3132–3153. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Bourguet-Kondracki, M.-L.; Mai, L.H.; Longeon, A.; Teta, R.; Meijer, L.; Soest, R.V.; Mangoni, A.; Costantino, V. Chloromethylhalicyclamine B, a Marine-Derived Protein Kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016, 79, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/mTOR Pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [PubMed]
- Roel, M.; Rubiolo, J.A.; Guerravarela, J.; Silva, S.B.L.; Thomas, O.P.; Cabezassainz, P.; Sánchez, L.; López, R.; Botana, L.M. Marine guanidine alkaloids crambescidins inhibit tumor growth and activate intrinsic apoptotic signaling inducing tumor regression in a colorectal carcinoma zebrafish xenograft model. Oncotarget 2016, 7, 83071. [Google Scholar] [CrossRef] [PubMed]
- Loaã, C.N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.L. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar]
- Li-Chun, L.; Tzu-Ting, K.; Hsin-Yi, C.; Wen-Shan, L.; Shih-Min, H.; Tsui-Chin, H. Manzamine A Exerts Anticancer Activity against Human Colorectal Cancer Cells. Mar. Drugs 2018, 16, 252. [Google Scholar]
- Simithy, J.; Fuanta, N.R.; Alturki, M.; Hobrath, J.V.; Calderón, A.I. Slow-Binding Inhibition of Mycobacterium tuberculosis Shikimate Kinase by Manzamine Alkaloids. Biochemistry 2018, 57, 4923–4933. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.S.; Dullaghan, E.M.; B Brett, F.; Huansheng, G.; Reiner, N.E.; Jon Paul Selvam, J.; Thorson, L.M.; Sara, C.; Nicholas, V.; Richardson, A.R. Discovery and optimization of a new class of pyruvate kinase inhibitors as potential therapeutics for the treatment of methicillin-resistant Staphylococcus aureus infections. Bioorg. Med. Chem. 2014, 22, 1708–1725. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, I.; Musman, M.; Ohtani, I.I.; Ichiba, T.; Tanaka, J. Pachastrissamine, a cytotoxic anhydrophytosphingosine from a marine sponge, Pachastrissa sp. J. Nat. Prod. 2002, 65, 1505–1506. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Song, J.; Bae, H.; Kim, W.J.; Lee, J.Y.; Han, G.H.; Lee, S.K.; Kim, S. Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine. Mar. Drugs 2015, 13, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Frederic, M.; Williams, D.E.; Patrick, B.O.; Irwin, H.; Robert, M.; Kim, S.C.; Roll, D.M.; Larry, F.; Rob, V.S.; Andersen, R.J. Liphagal, a Selective inhibitor of PI3 kinase alpha isolated from the sponge Aka coralliphaga: Structure elucidation and biomimetic synthesis. Cheminform 2006, 37, 321–324. [Google Scholar]
- Markwell-Heys, A.W.; Kuan, K.K.W.; George, J.H. Total Synthesis and Structure Revision of (-)-Siphonodictyal B and Its Biomimetic Conversion into (+)-Liphagal. Org. Lett. 2015, 17, 4228–4231. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, W.; Xu, T. Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B. Mar. Drugs 2018, 16, 115. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, L.; Xi, Y.; White, M.; Greenhaw, J.; Harris, T.; Wu, Q.; Bryant, M.; Papoian, T.; Mattes, W. Cytoto xicity of 34 FDA approved small—Molecule kinase inhibitor s in primary rat and human hepatocytes. Toxicol. Lett. 2018, 291, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Medical Data Website of China. Available online: https://data.pharmacodia.com (accessed on 5 May 2019).
- Muñoz-Alonso, M.; Enrique, Á.; María José, G.N.; Marina, P.; Pablo, A.; Galmarini, C.M.; Alberto, M.O. c-Jun N-terminal kinase phosphorylation is a biomarker of plitidepsin activity. Mar. Drugs 2013, 11, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Maurizio, D.I.; Paola, A. Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Mar. Drugs 2014, 12, 719–733. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine, US. Database of Clinical Studies. Available online: http://www.clinicaltrial.gov (accessed on 15 May 2019).
- He, H.; Tran, P.; Gu, H.; Tedesco, V.; Zhang, J.; Lin, W.; Gatlik, E.; Klein, K.; Heimbach, T. Midostaurin, a Novel Protein Kinase Inhibitor for the Treatment of Acute Myelogenous Leukemia: Insights from Human Absorption, Metabolism, and Excretion Studies of a BDDCS II Drug. Drug Metab. Dispos. 2017, 45, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Knapper, S.; Russell, N.; Gilkes, A.; Hills, R.K.; Gale, R.E.; Cavenagh, J.D.; Jones, G.; Kjeldsen, L.; Grunwald, M.R.; Thomas, I. A randomised assessment of adding the kinase inhibitor lestaurtinib to 1st-line chemotherapy for FLT3-mutated AML. Blood 2016, 129, 1143–1154. [Google Scholar] [CrossRef]
- Bourhill, T.; Narendran, A.; Johnston, R.N. Enzastaurin: A lesson in drug development. Crit. Rev. Oncol. Hematol. 2017, 112, 72–79. [Google Scholar] [CrossRef]
- Bernardo, M.L.; Belén, V.; José Esteban, P.R.; Arturo, S.M.; Juan José, P.R. Population pharmacokinetics of kahalalide F in advanced cancer patients. Cancer Chemother. Pharmacol. 2015, 76, 365–374. [Google Scholar]
- Lefranc, F.; Koutsaviti, A.; Ioannou, E.; Kornienko, A.; Newman, D. Algae metabolites: From in vitro growth inhibitory effects to promising anticancer activity. Nat. Prod. Rep. 2019, 36, 810–841. [Google Scholar] [CrossRef]
- Lien, W.C.; Chen, T.Y.; Sheu, S.Y.; Lin, T.C.; Kang, F.C.; Yu, C.H.; Kuan, T.S.; Huang, B.M.; Wang, C.Y. 7-hydroxy-staurosporine, UCN-01, induces DNA damage response and autophagy in human osteosarcoma U2-OS cells. J. Cell. Biochem. 2018, 119, 4729–4741. [Google Scholar] [CrossRef]
- Pujari, R.; Jose, J.; Bhavnani, V.; Kumar, N.; Shastry, P.; Pal, J.K. Tamoxifen-induced cytotoxicity in breast cancer cells is mediated by glucose-regulated protein 78 (GRP78) via AKT (Thr308) regulation. Int. J. Biochem. Cell Biol. 2016, 77, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Andel, L.V.; Rosing, H.; Schellens, J.; Beijnen, J. Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment. Mar. Drugs 2018, 16, 246. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Lazarus, H.M.; Cooper, B.W.; Schluchter, M.D.; Panneerselvam, A.; Jacobberger, J.W.; Hsu, J.W.; Janakiraman, N.; Simic, A.; Dowlati, A. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009, 84, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Burns, J.M.; Gorelick, K.J.; Crockford, D.R.; Grenier, E.; Wilke, S.; Cooper, E.C.; Alkon, D.L. Bryostatin-1 improves cognition and daily living tasks in moderate to severe Alzheimer’s disease: Preliminary report of a phase 2 study. J. Alzheimer’s Assoc. 2017, 13, 1476. [Google Scholar] [CrossRef]
- Keck, G.E.; Poudel, Y.B.; Rudra, A.; Stephens, J.C.; Kedei, N.; Lewin, N.E.; Peach, M.L.; Blumberg, P.M. Molecular modeling, total synthesis, and biological evaluations of C9-deoxy bryostatin 1. Angew. Chem. Int. Ed. 2010, 49, 4580–4584. [Google Scholar] [CrossRef] [PubMed]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A. Integrating Molecular Networking and Biological Assays To Target the Isolation of a Cytotoxic Cyclic Octapeptide, Samoamide A, from an American Samoan Marine Cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
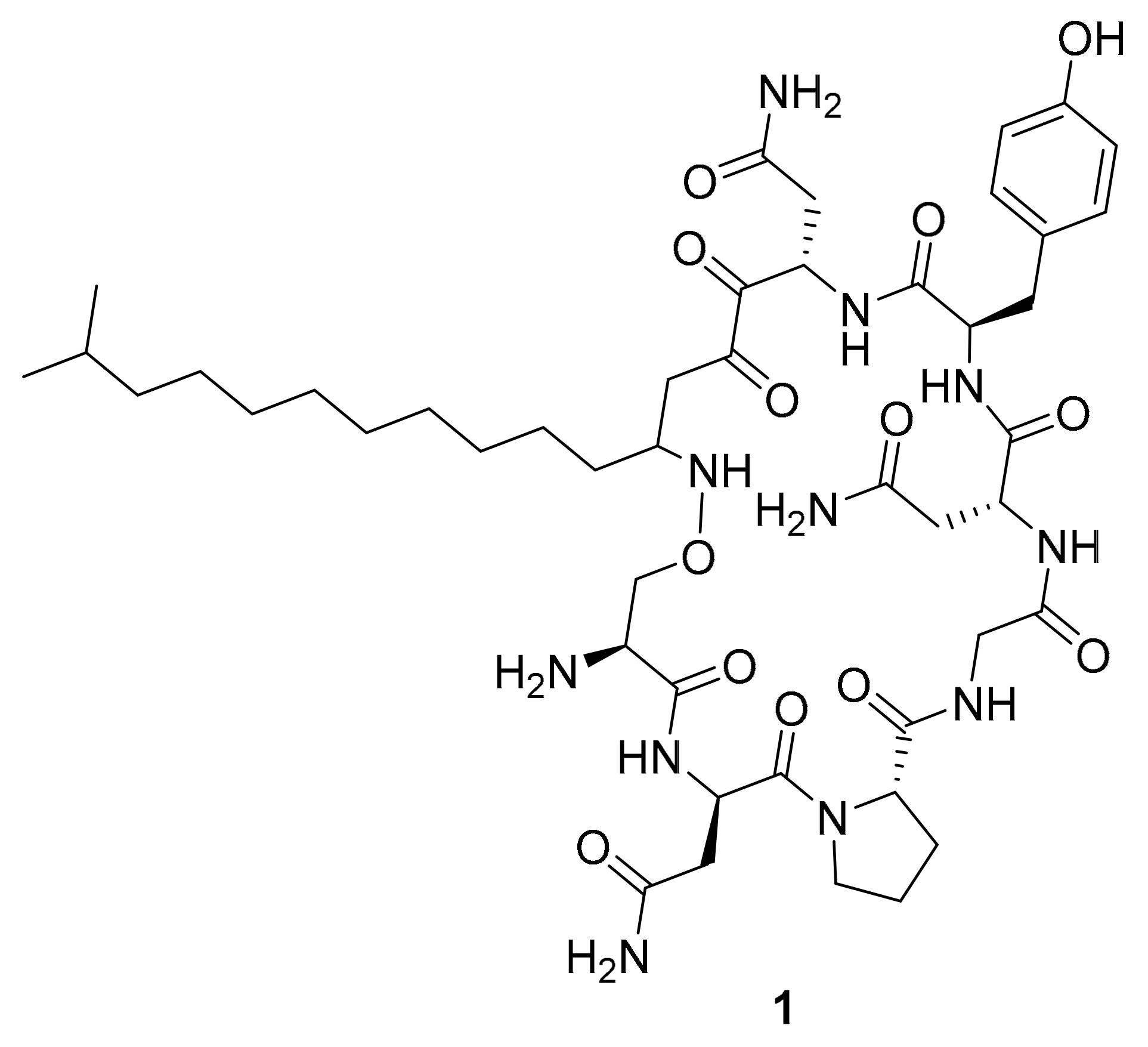
| 1 | lipopeptide | Bacillus megaterium | p-Akt, p-MAPK, p-GSK-3β | [33] |
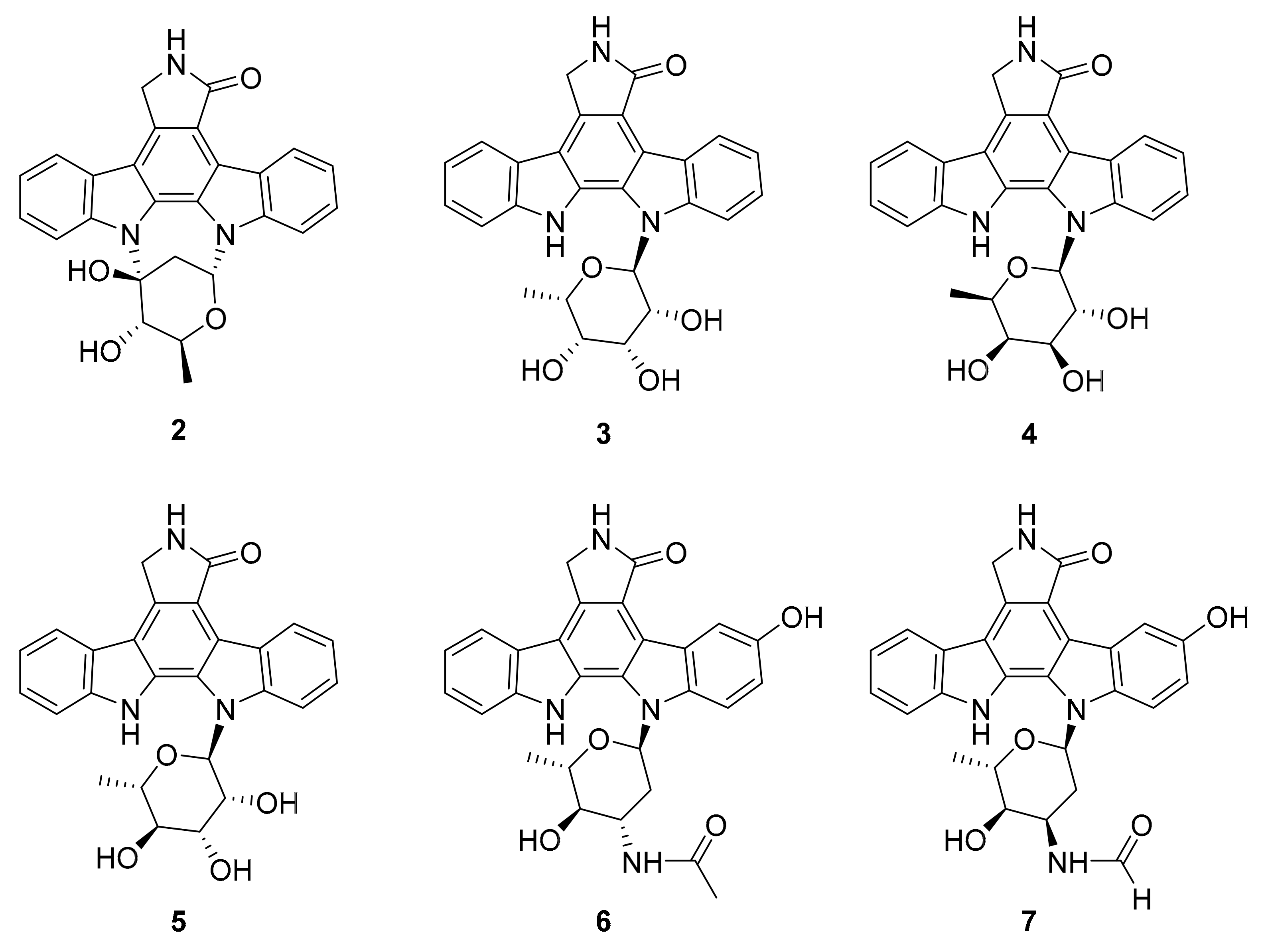
| 2 3–5 | indolocarbazole alkaloids | Streptomyces sp. A65 | inactive analogue PKC, BTK (0.25–1.91 μM) | [34] |
| 6–8 | indolocabazole derivatives | Streptomyces sp. A68 | PKC-α, BTK, ROCK2 (0.91–1.84 μM) | [35] |
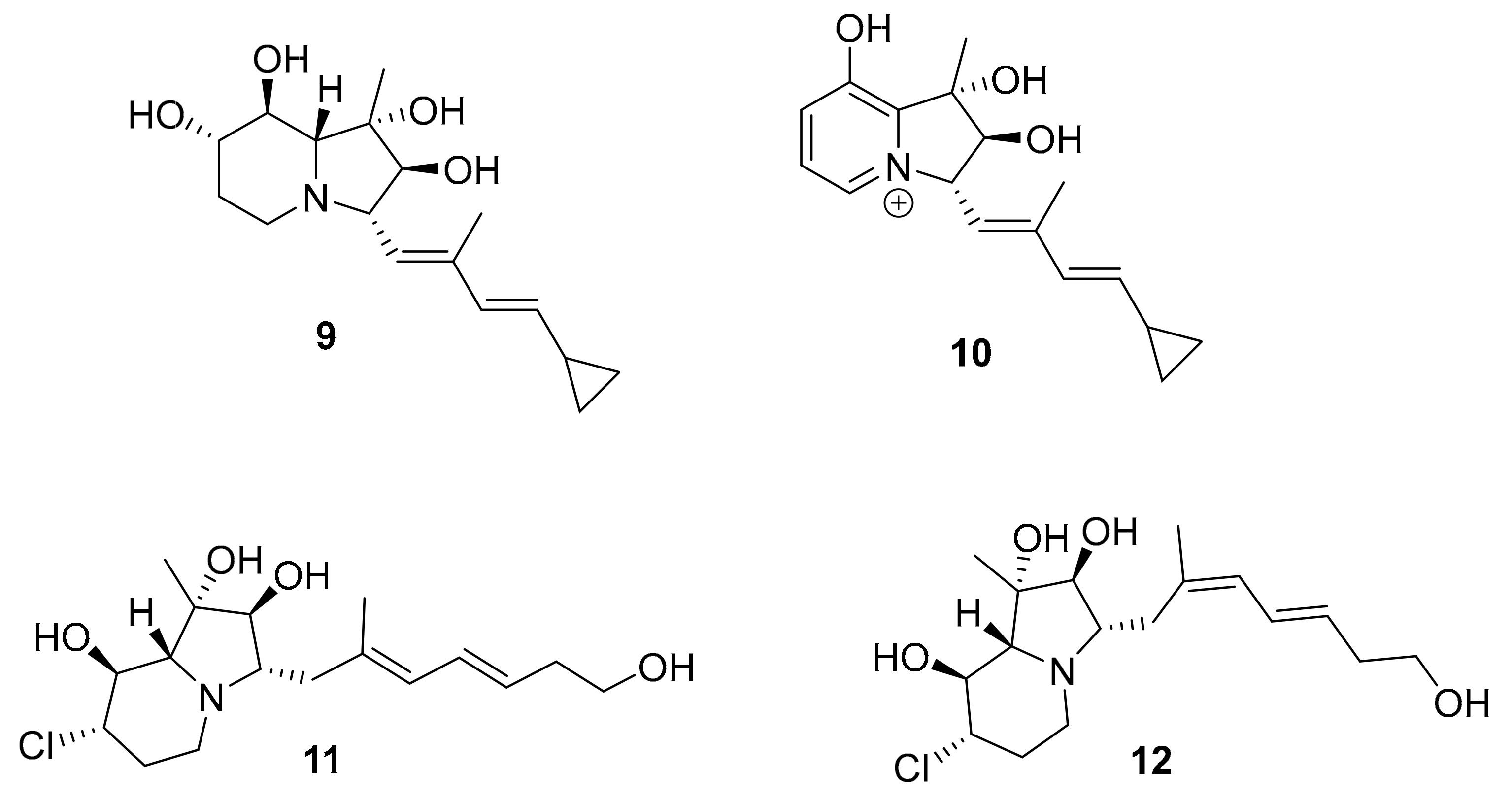
| 9–12 | cyclizidine alkaloid | Streptomyces sp. HNA39 | ROCK2 (7.0–42 μM) | [36,37,38,39] |
| 13 | Indolocarbazole alkaloid indolocarbazole alkaloid indolocarbazole alkaloid | Streptomyces sp. DT-A61 | ROCK2 (5.7 nM) | [40] |
| 14 | Streptomyces sp. DT-A61 | PKC-α (92 nM) | [40] | |
| 15 | Streptomyces sp. A22 | PKC, BTK, ROCK2 (0.91–1.84 μM) | [41] | |
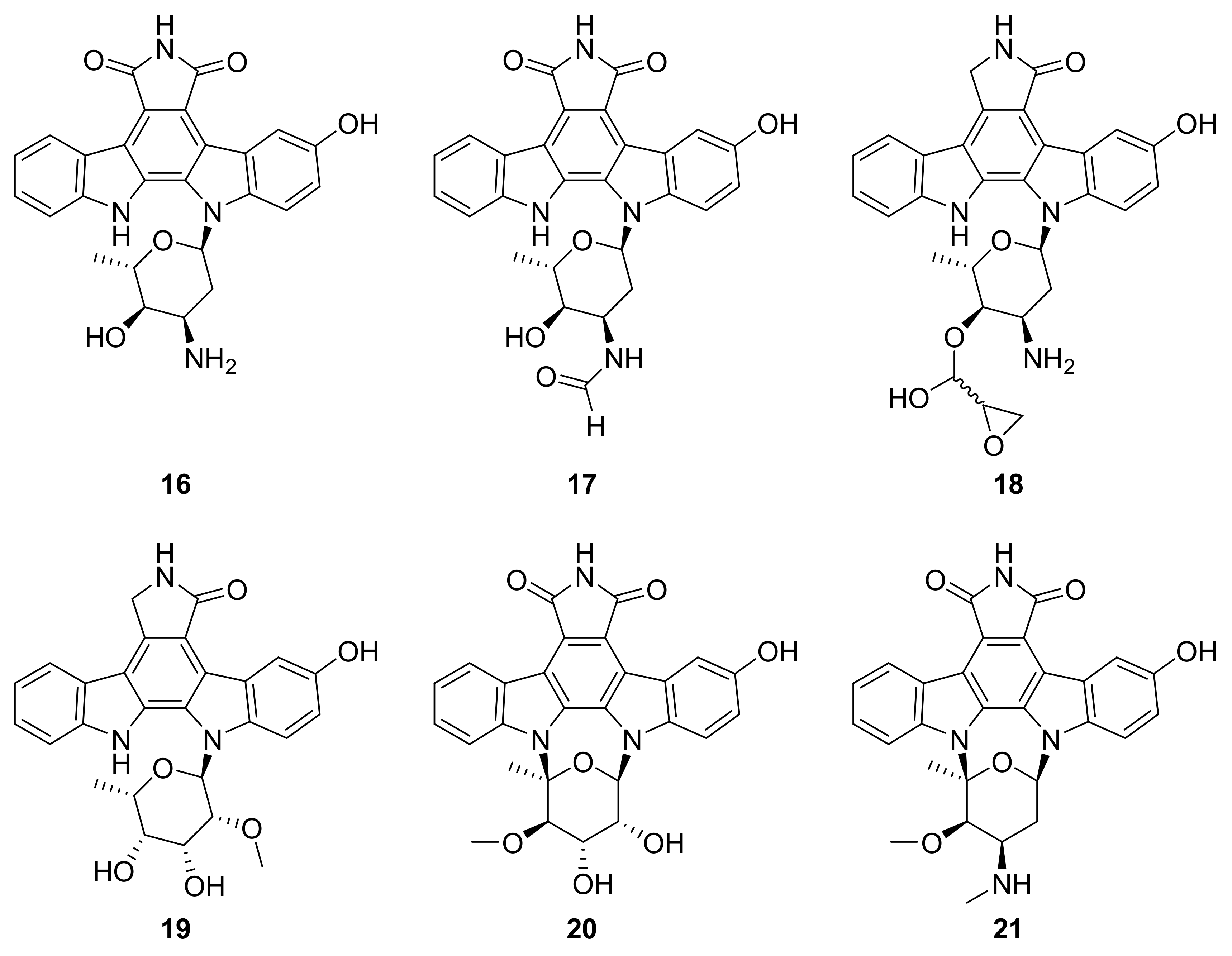
| 16 17–21 | staurosporine derivatives | Streptomyces sp. NB-A13 | inactive analogue PKC-θ (0.06–9.43 μM) | [42] |
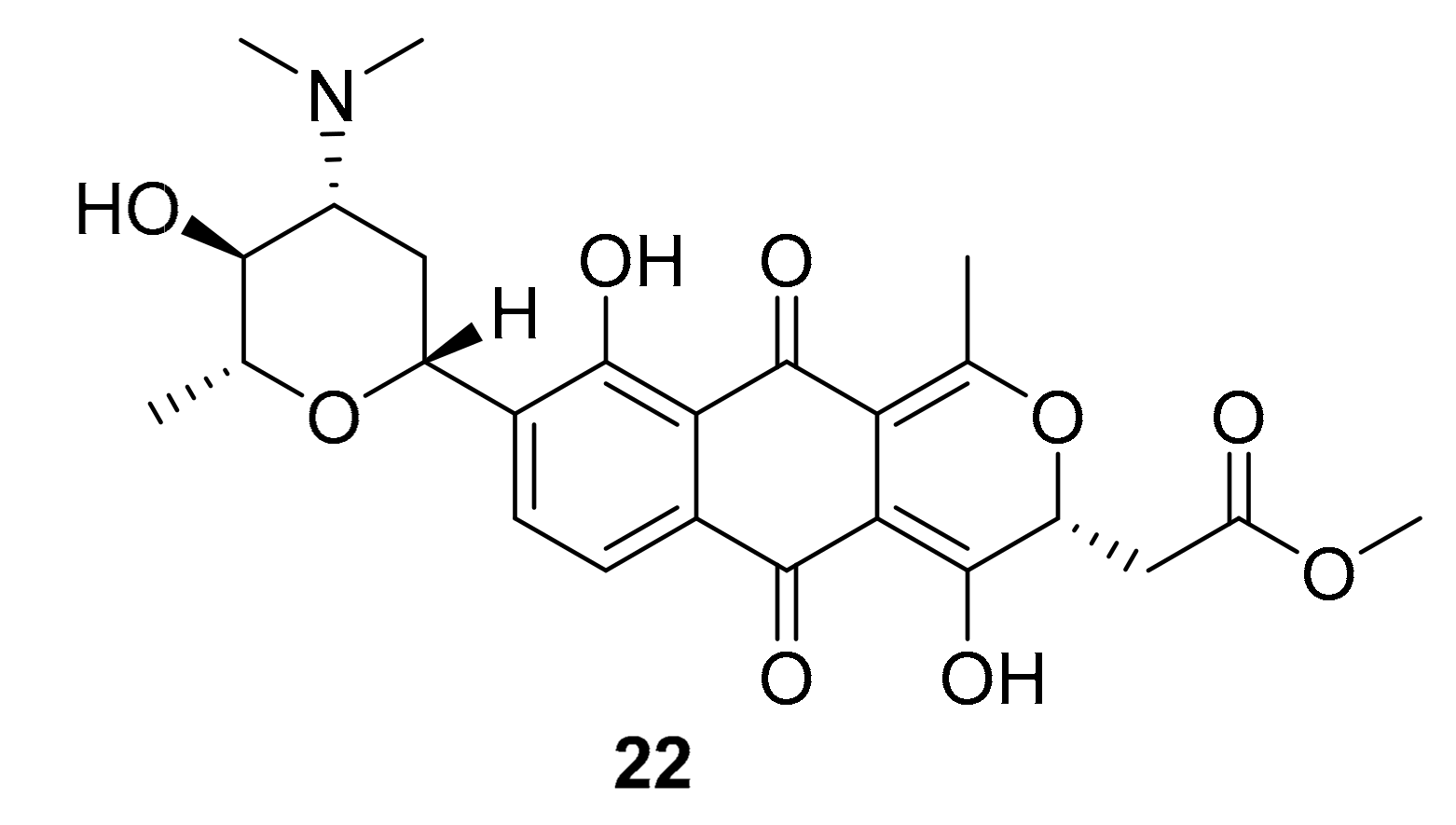
| 22 | naphthoquinone | Streptomyces sp. XMA39 | ROCK2 (>20 μM) | [43] |
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
| 23 | isochromen-6-one | Penicillium sp. ZJ27 | PKnG (76.5 μM) | [41] |
| 24 | alkaloid | Penicillium sp. | p-p38 MAPK | [45] |
| 25 | polyketide | Diaporthe sp. HLY-1 | IKK | [46] |
| 26 | indole diterpenoid | Aspergillus flavus OUCMDZ-2205. | PKC-β (15.6 μM) | [47] |
| 27 | furan derivative | Xylaria sp. (No. 2508) | p-Akt, p-ERK1/2 | [48] |
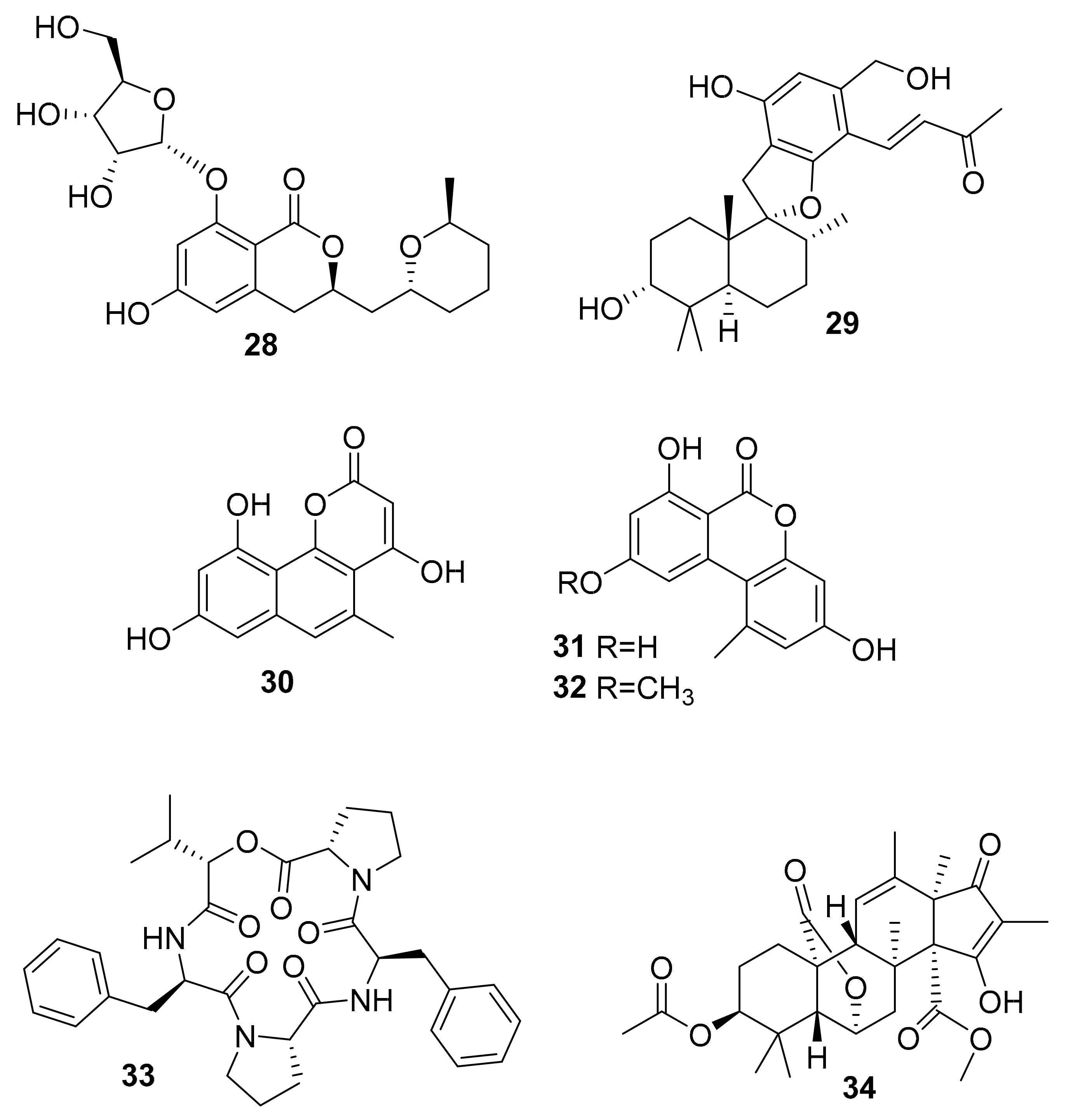
| 28 | dihydro-isocoumarin | Aspergillus sp. SF-5976 | p-p38 MAPK | [49] |
| 29 | phenylspiro-drimane derivative | Stachybotrys sp. KCB13F013 | p-ERK, p-JNK, p-p38 MAPK | [50] |
| 30 | benzocoumarin | Aspergillus sp. LF660 | GSK-3β (0.35 ± 0.04 μM) | [51] |
| 31 | GSK-3β (0.13 ± 0.04 μM) | |||
| 32 | GSK-3β (0.20 ± 0.04 μM) | |||
| 33 | lipophilic depsipeptide | Alternaria sp. SF-5016 | p-JNK, p-p38 MAPK | [52] |
| 34 | citreohybridonol | Toxicocladosporium sp. SF-5699 | p-p38 MAPK | [53] |
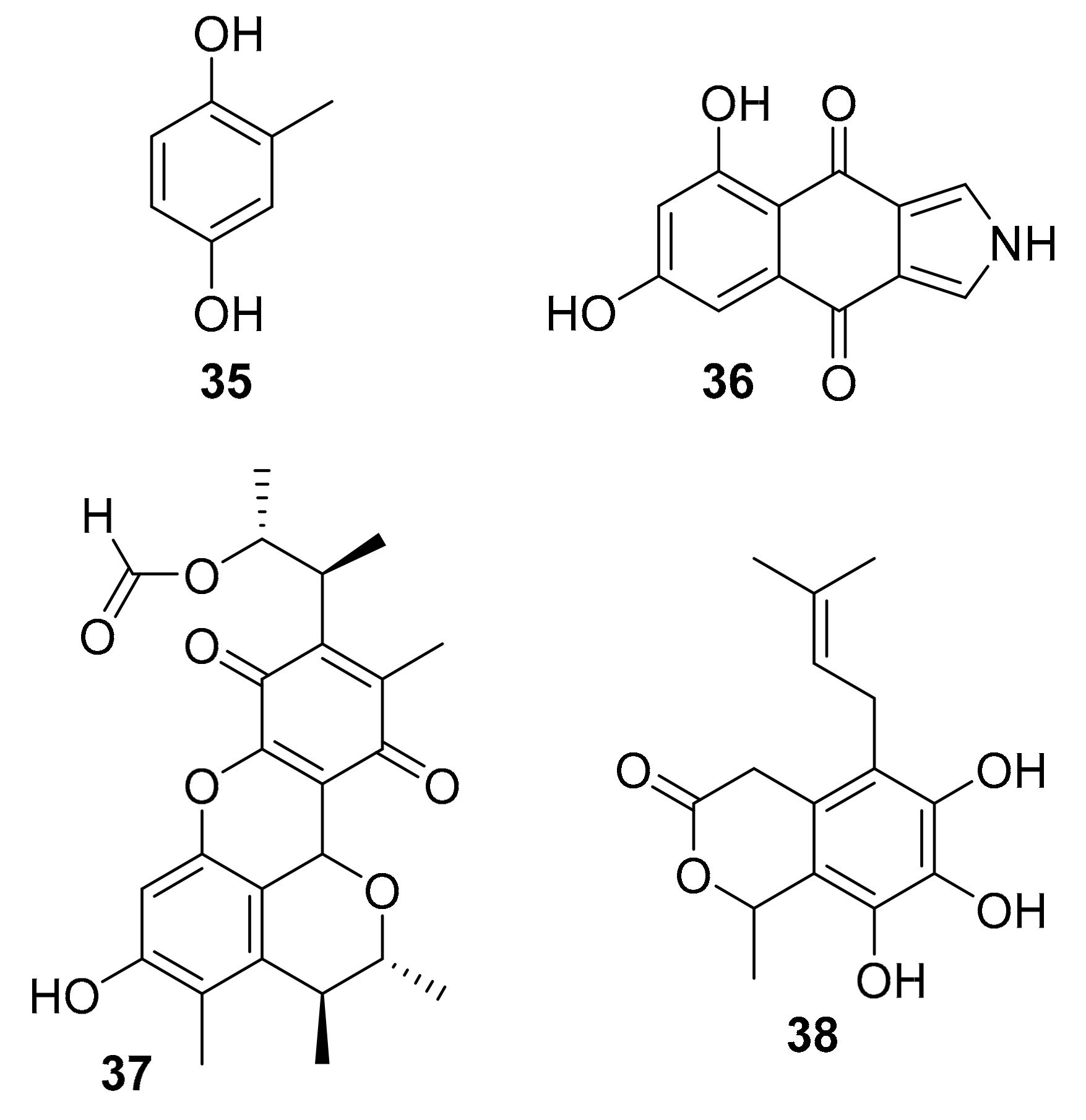
| 35 | 2-methyl-hydroquinone | Penicillium sp. HL-85-ALS5-R004 | p-Akt, p-ERK1/2 | [54] |
| 36 | isopyrrolo-naphthoquinone | Biscogniauxia mediterranea | GSK-3β (8.04 μM) | [55] |
| 37 | polyketides | Penicillium sp. SF-5629 | p-p38 MAPK | [56] |
| 38 | Oidiodendron griseum UBOCC-A-114129 | CLK1 (15.6 g/mL) | [58] |
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
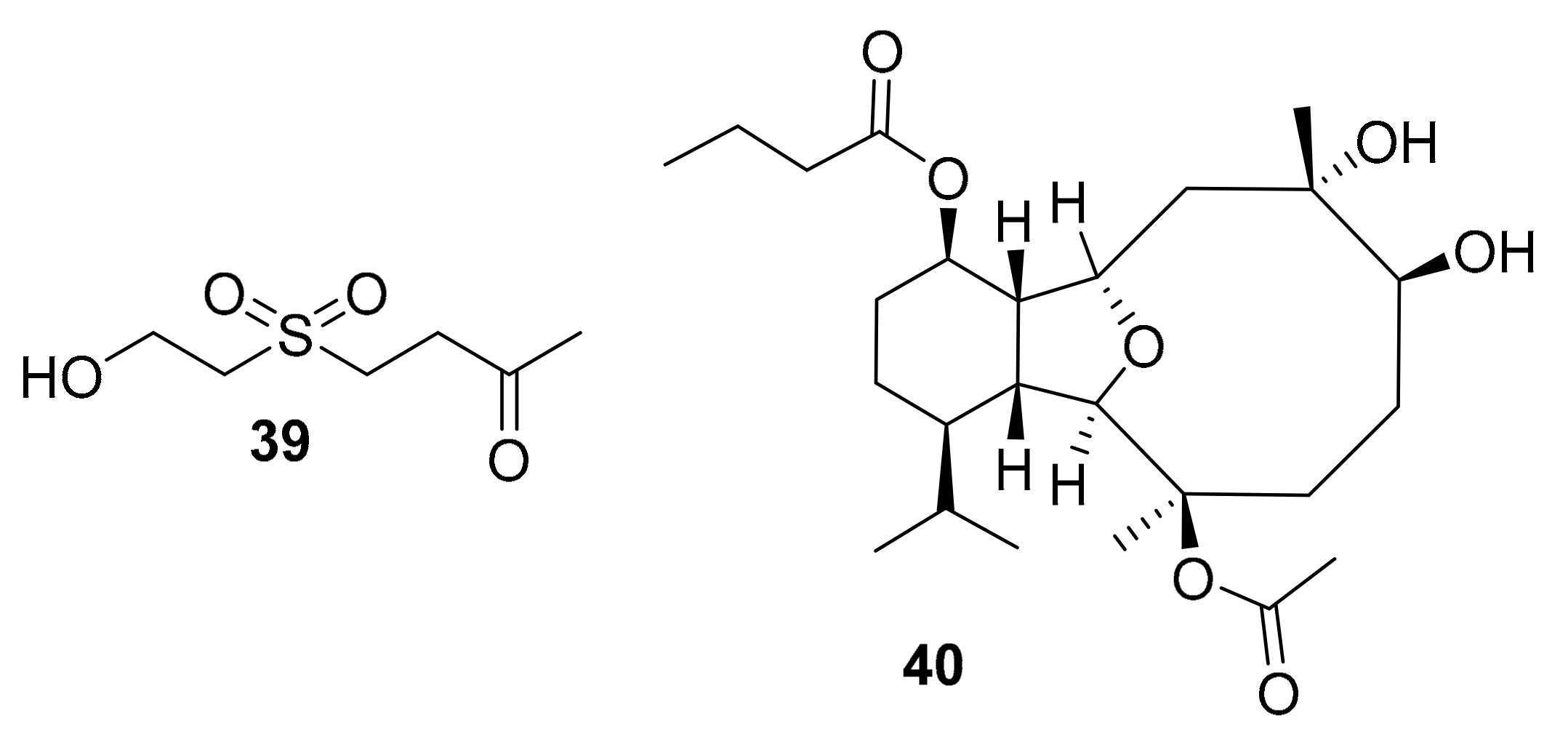
| 39 | simple sulfone alcohol | Cladiella australis | MAPK, AKt | [59] |
| 40 | eunicellin diterpenoid | Cladiella pachyclados | EGFR | [60] |
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
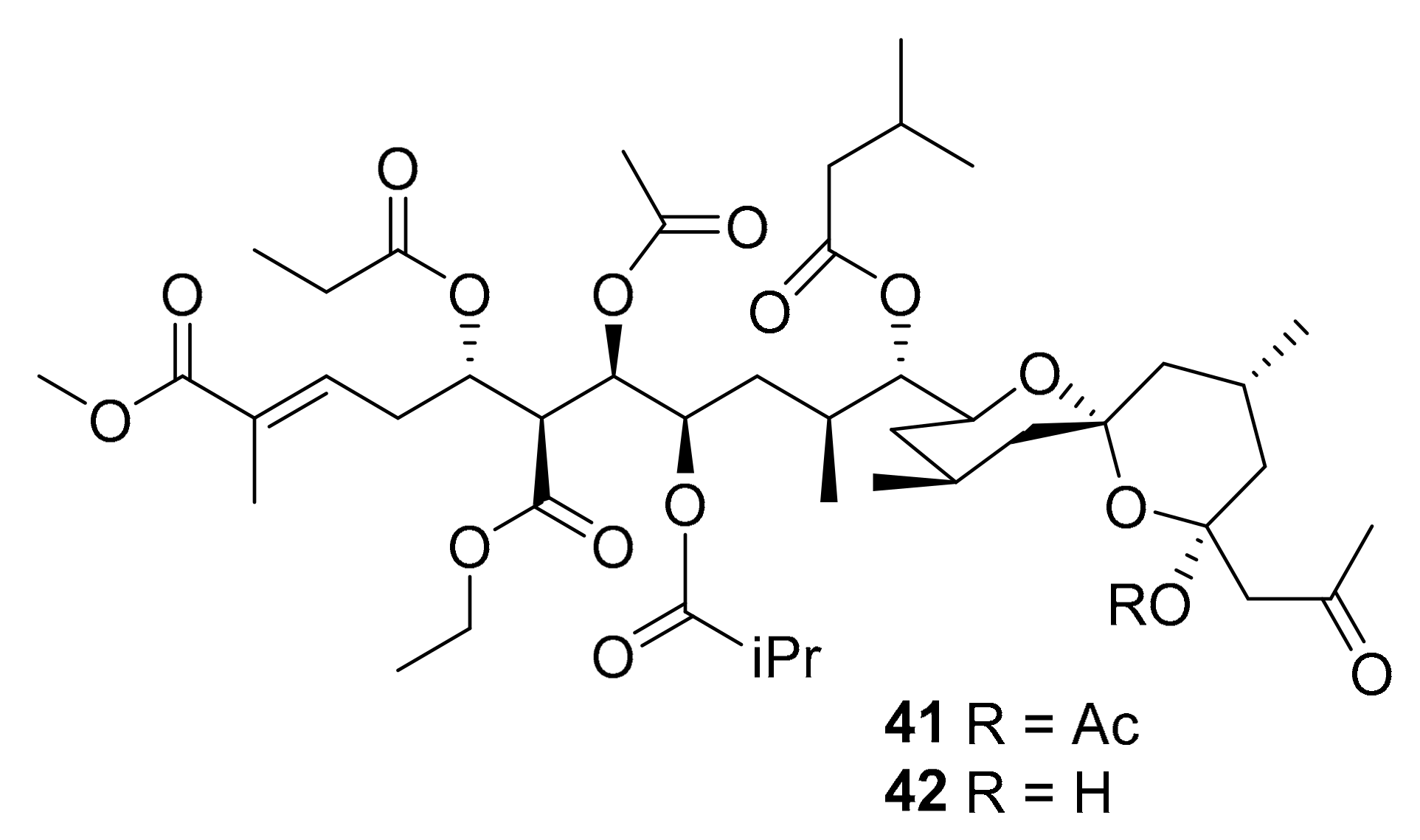
| 41–42 | spiroketals | marine ascidian genus Didemnum | CDK5, CK1, DyrK1A, GSK-3 (10 μg/mL) | [62] |
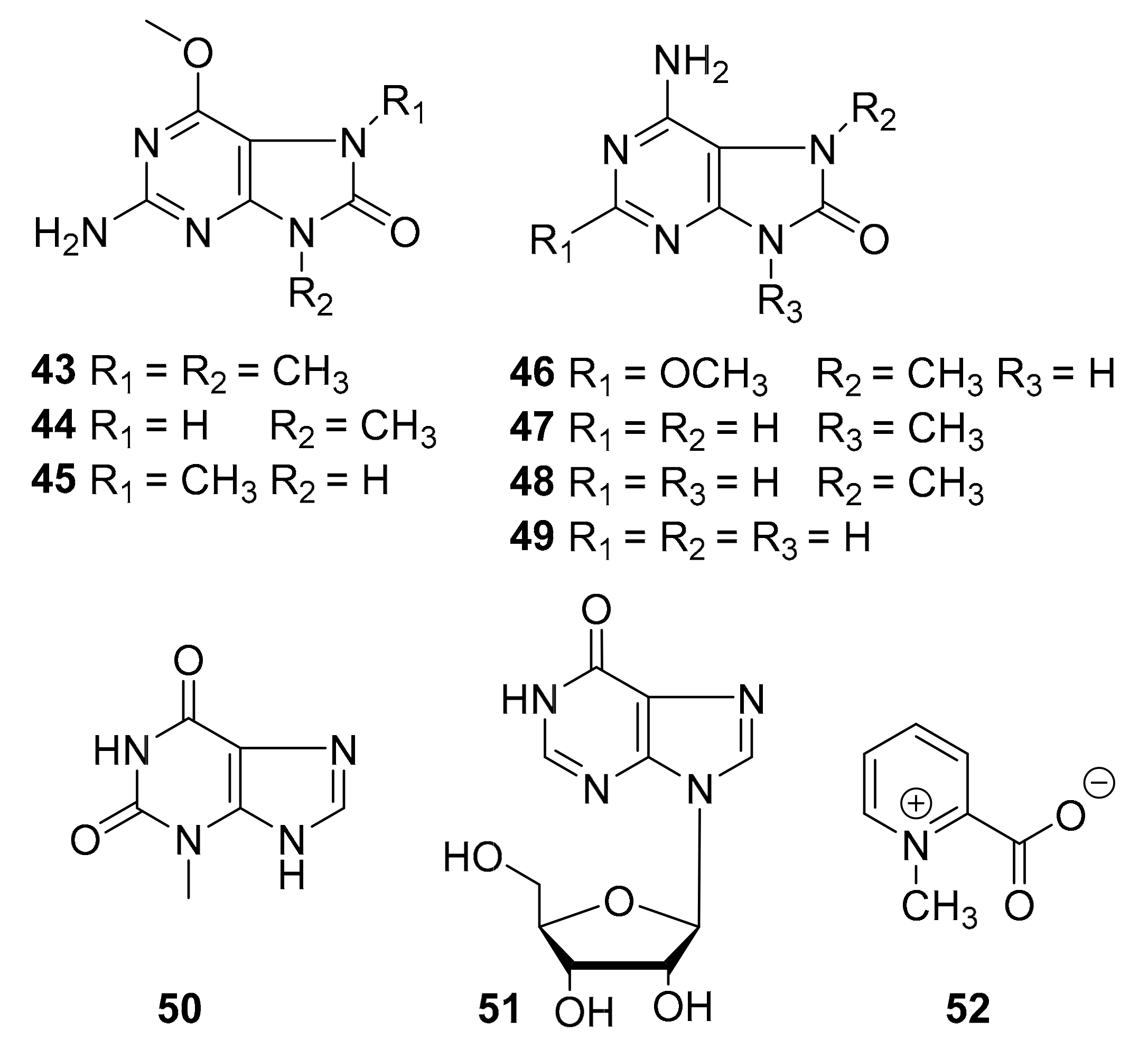
| 43–52 | purine alkaloids | marine tunicate Symplegma rubra | CDK5, CK1, DyrK1A, GSK-3 (10 μg/mL) | [63] |
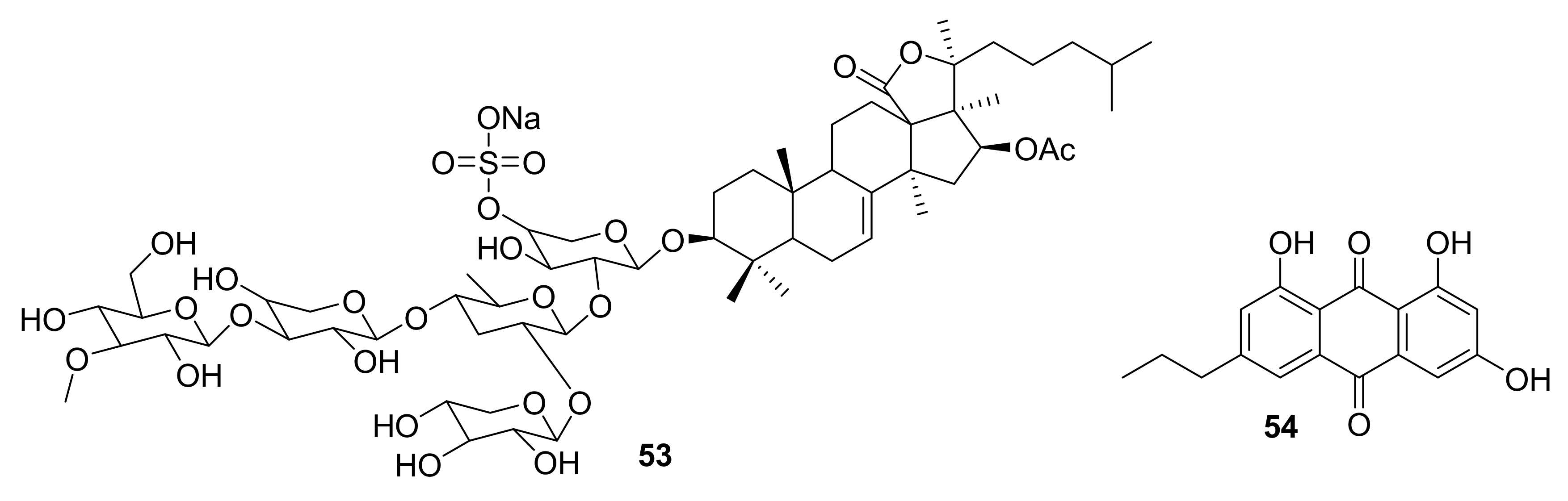
| 53 | saponin sulfate | sea cucumber | PAK1 (1.2 μM) LIMK (60 μM) AKT (59 μM) | [64,65] |
| 54 | anthraquinone derivative | marine echinoderm Comanthus sp. | IGF1-R (5 μM) FAK (8.4 μM) EGFR (4 μM) | [66] |
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
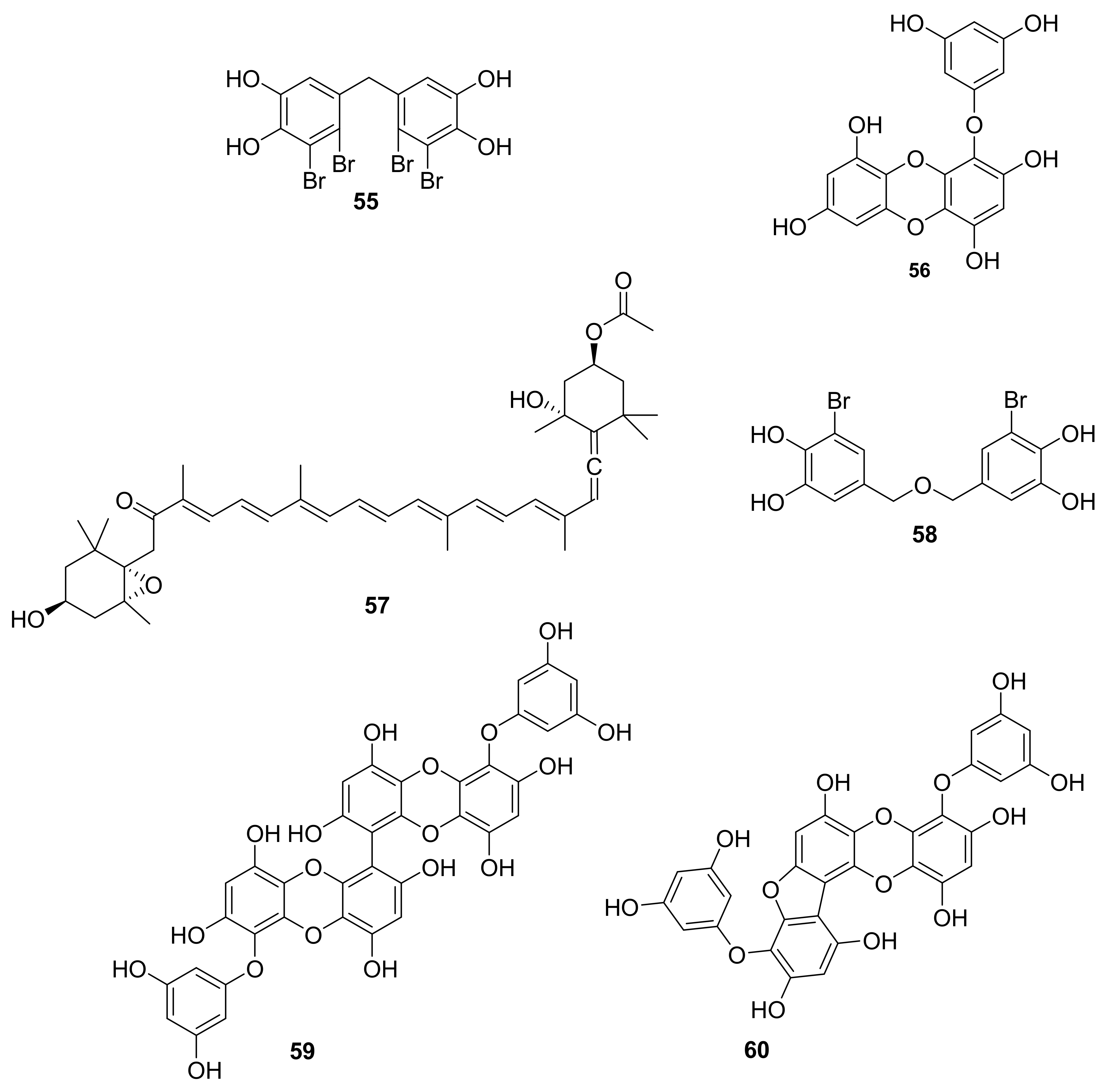
| 55 | bromophenol | red alga Rhodomelaceae confervoides. | RTKs, p-PKB/Akt | [68,69] |
| 56 | phlorotannin | brown seaweed Eisenia bicyclis | p-AKt | [70] |
| 57 | carotenoid | brown alga wakame seaweeds wakame (Undaria pinnatifida) and kombu (Laminaria japonica) | AKt, MAPK | [71,72,73] |
| 58 | bromophenol | red alga Polysiphonia morrowii | p-ERK | [74] |
| 59–60 | lipopoly-saccharide | brown seaweed marine alga (Eisenia bicyclis) | p-ERK1/2, p-JNK | [75] |
| 61a | cyclic depsipeptide | marine cyanobacterium Moorea producens (formerly Lyngbya majuscula) | RTKs | [76] |
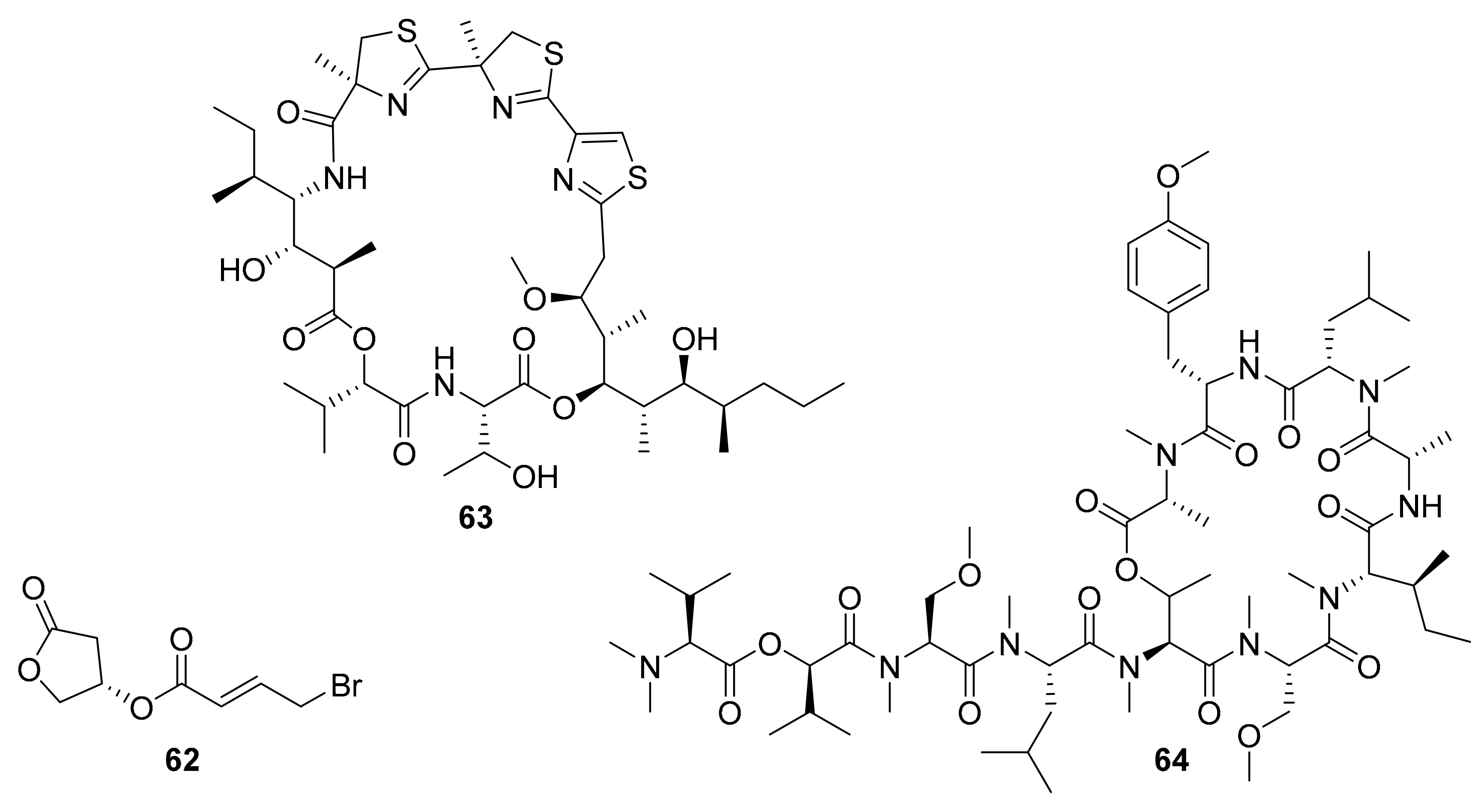
| 62 a | bromo-honaucin | marine cyanobacterium Leptolyngbya crossbyana | AKt, ERK | [77,78] |
| 63 b | cyclic depsipeptide | marine cyanobacterium Moorea producens | JNK | [79,80] |
| 64 | cyclic depsipeptide | marine cyanobacterium Leptolyngbya sp. | VEGFA/VEGFR2 | [81,82] |
| Compound | Chemical Class | Marine Source | Drug Targets/Inhibitory Activity | References |
|---|---|---|---|---|
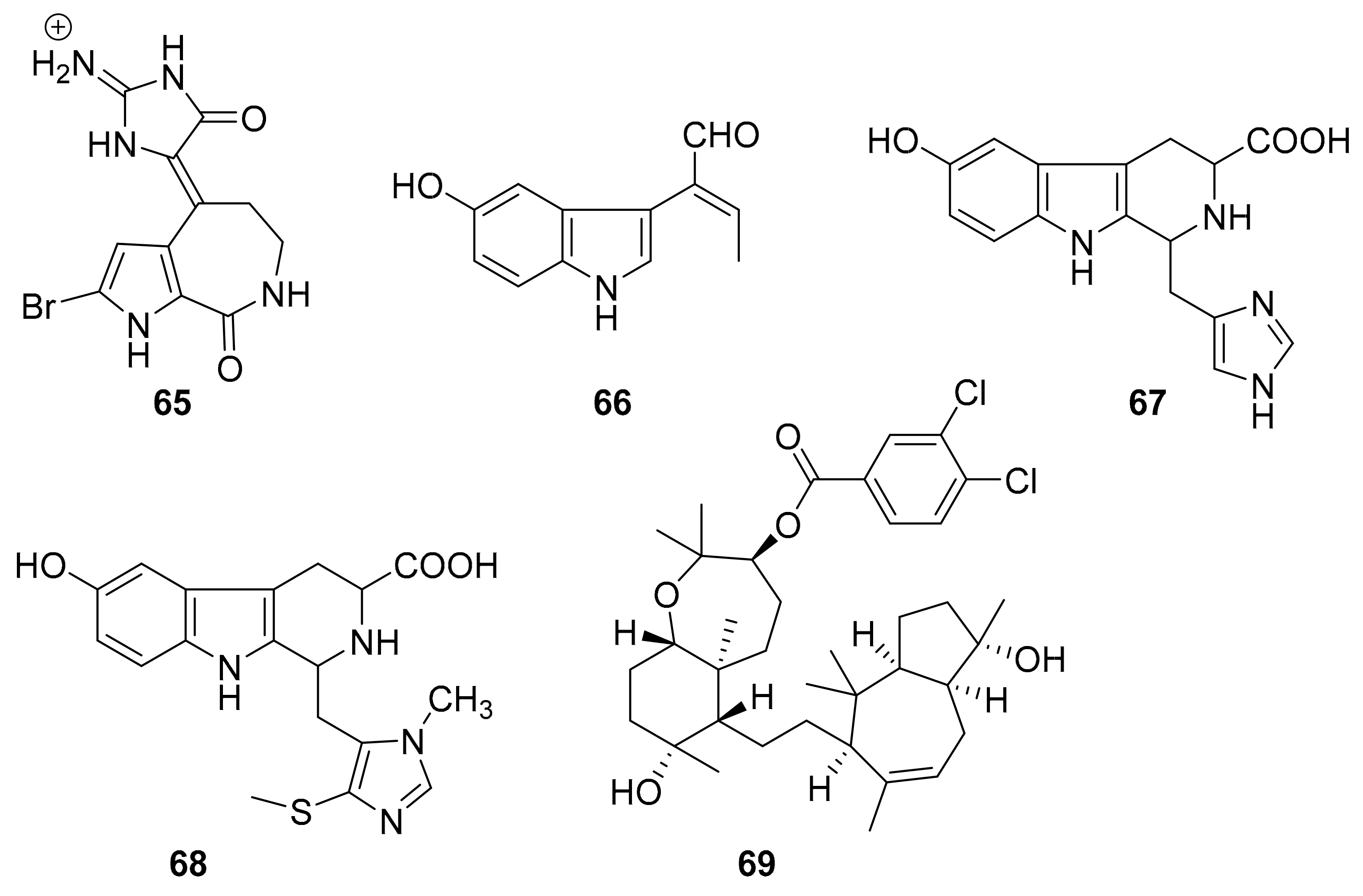
| 65 | bromopyrrole alkaloid | Callyspongia sp. (CMB-01152). | CK1, CK5, GSK3β | [84] |
| 66 | indole alkaloids | Hyrtios erecta | STK (24.5 μg/mL) | [88] |
| 67 | STK (13.6 μg/mL) | |||
| 68 | STK (18.6 μg/mL) | |||
| 69 | sipholane triterpenoid | Callyspongia siphonella. | p-Brk, p-FAK | [89] |
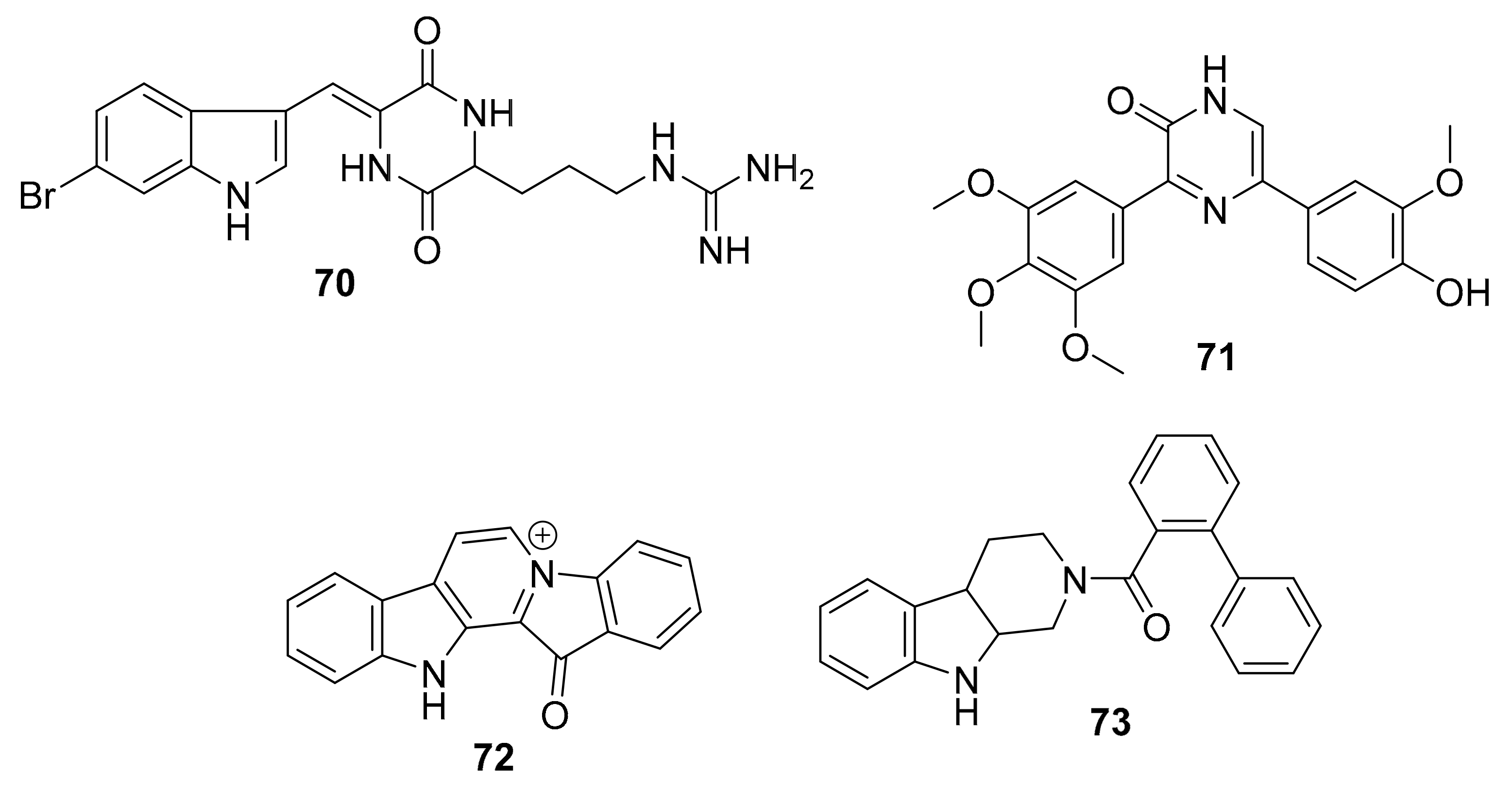
| 70 | indole alkaloid | Geodia barretti | RIPK2 (8.0 μM) CAMK1a (5.7 μM) SIK2 (6.1 μM) | [90,92,93] |
| 71 | diarylpyrazine | Hamacanthins | PDGF-Rβ (0.02 μM) | [93] |
| 72 | tryptoline analogues | Fascaplysinopsis sp. | CDK4/D1 (0.35 μM) | [94,95,96,97,98] |
| 73 a | CDK4 (10 μM) | |||
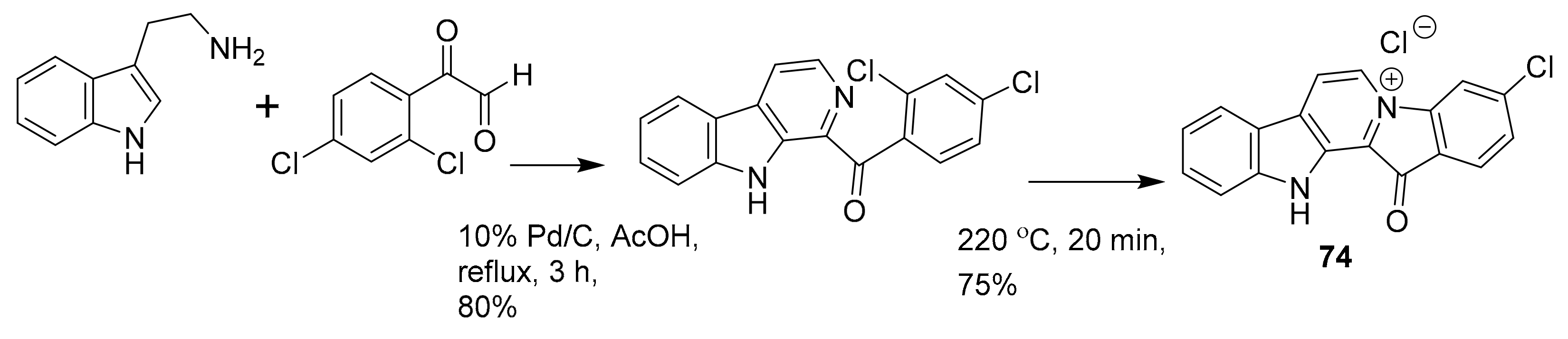
| 74 a | alkaloid derivative | Fascaplysinopsis sp. | PI3K/Akt | [99] |
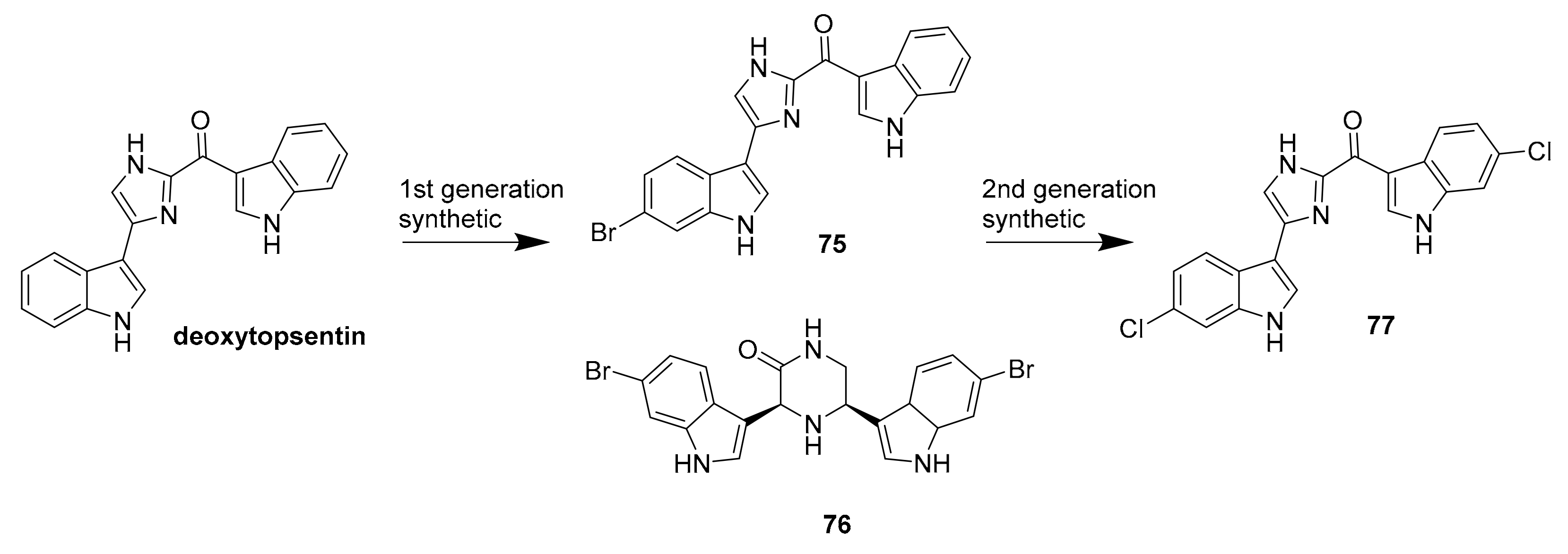
| 75 | bisindole alkaloids | Topsentia pachastrelloides | MRSA PK (60 nM) | [100] |
| 76 | MRSA PK (16 nM) | |||
| 77 | MRSA PK (1.4 nM) | |||
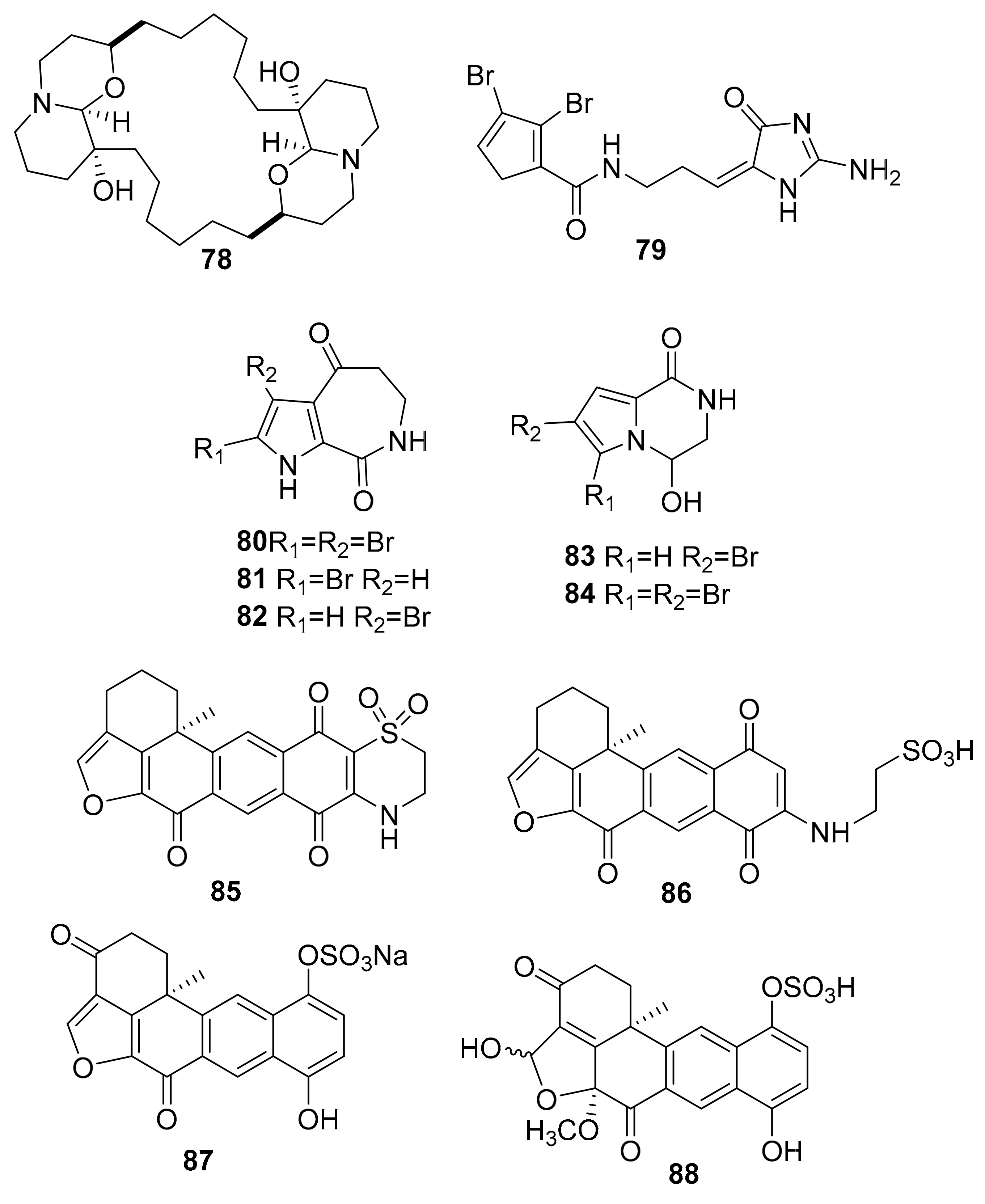
| 78 | macrocyclic oxaquinolizidine alkaloid | Xestospongia species. | PI3K, Akt | [101] |
| 79–84 | bromopyrrole alkaloids | Stylissa massa and Stylissa flabelliformis | GSK-3, DYRK1A, CK-1 (0.6–6.4 μM) | [102] |
| 85 | adociaquinone derivatives | Xestospongia sp. | inactive analogue | [108] |
| 86 | CDK9/cyclin T (3 μM) | |||
| 87–88 | CDK5/p25 (6 μM) | |||
| 89 | cyclic peptide | Amphibleptula | GSK-3β | [109] |
| 90 | polycyclic quinone | Petrosia | hexokinase II | [110] |
| 91 | halogenated alkaloid | Acanthostrongylophora ingens | CK1δ/ε (6 μM) | [111] |
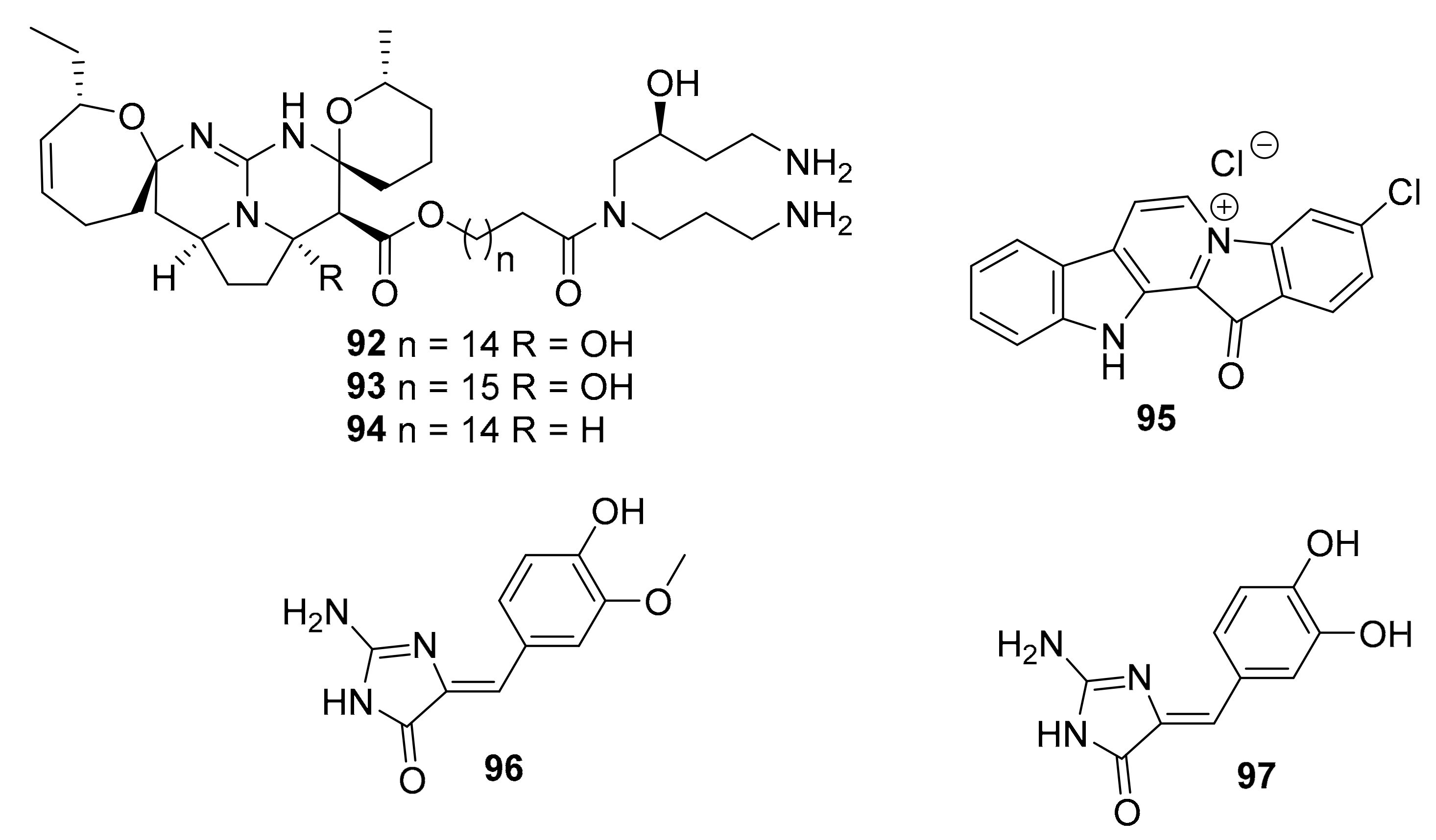
| 92 | isomalabaricane triterpenoid | Jaspis stellifera | PI3K, Akt | [112] |
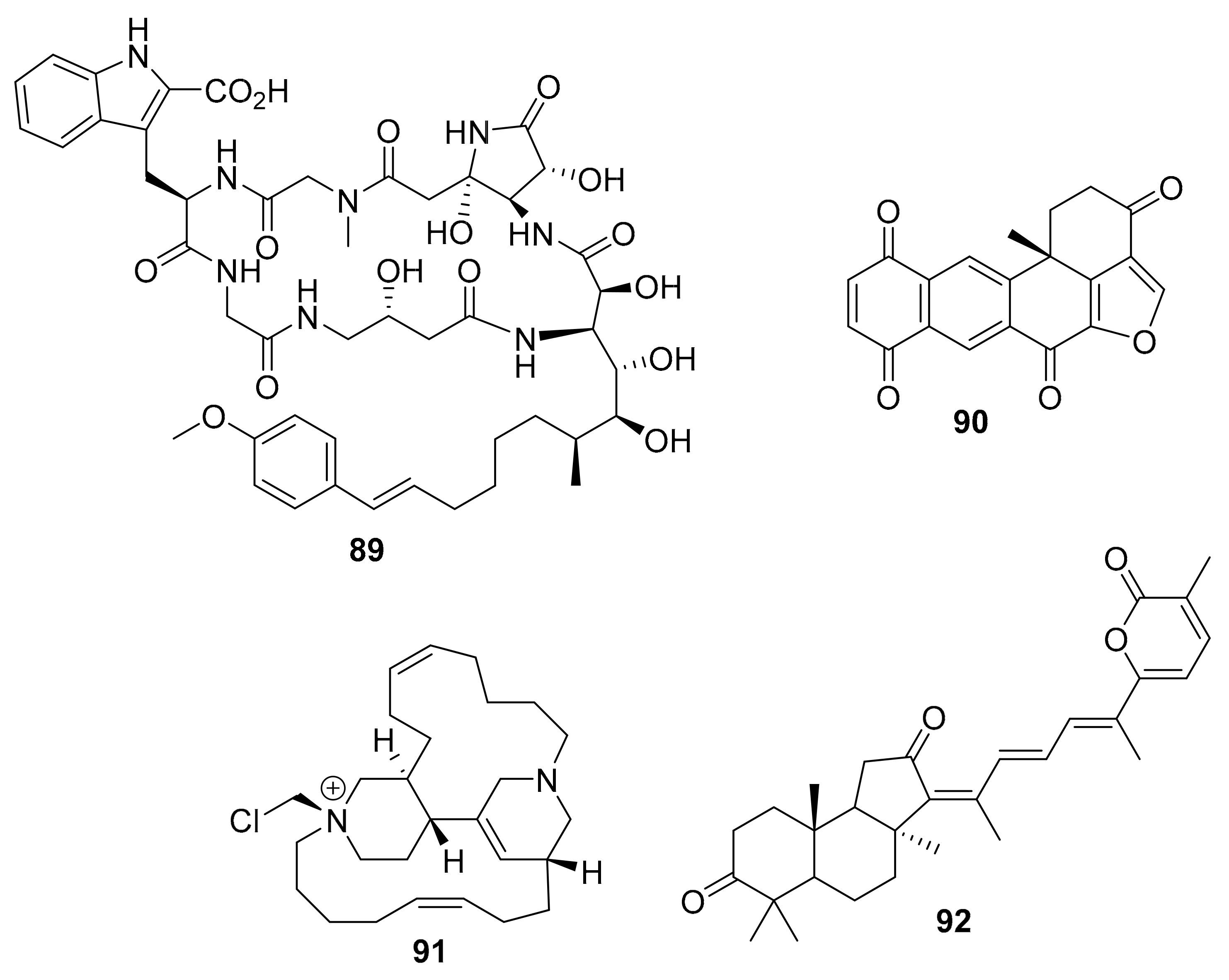
| 93–95 | guanidine alkaloids | Smenospongia sp. | CDK2/6 | [113] |
| 96–97 | 2-aminoimidazolone alkaloids | Leucetta and Clathrina | DYRKs (0.17 ~ 0.88 μM) CLKs (0.32 ~ 8.6 μM) | [114] |
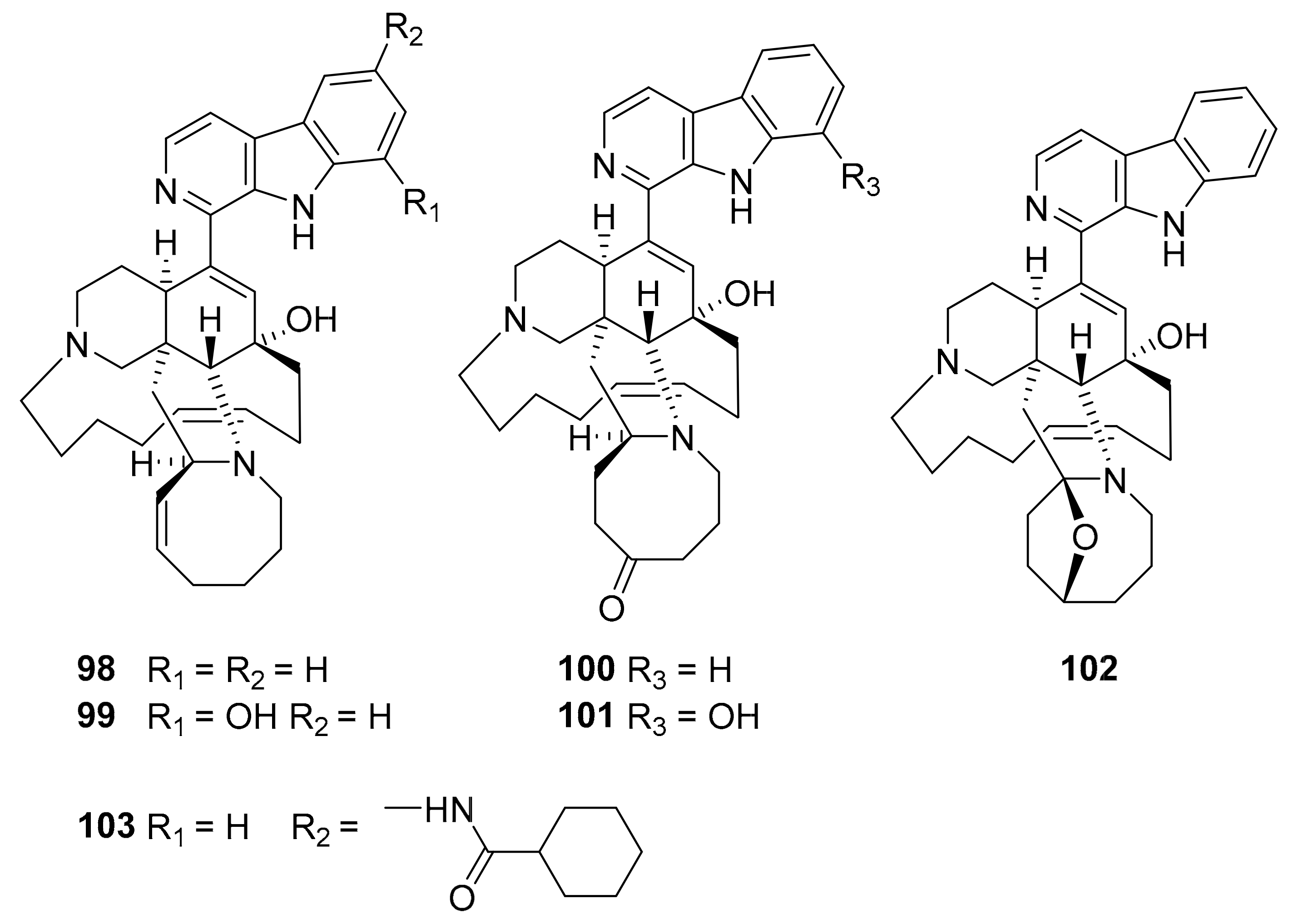
| 98–103 | manzamine alkaloids | Acanthostrongylophora sp. | MtSK | [115,116] |
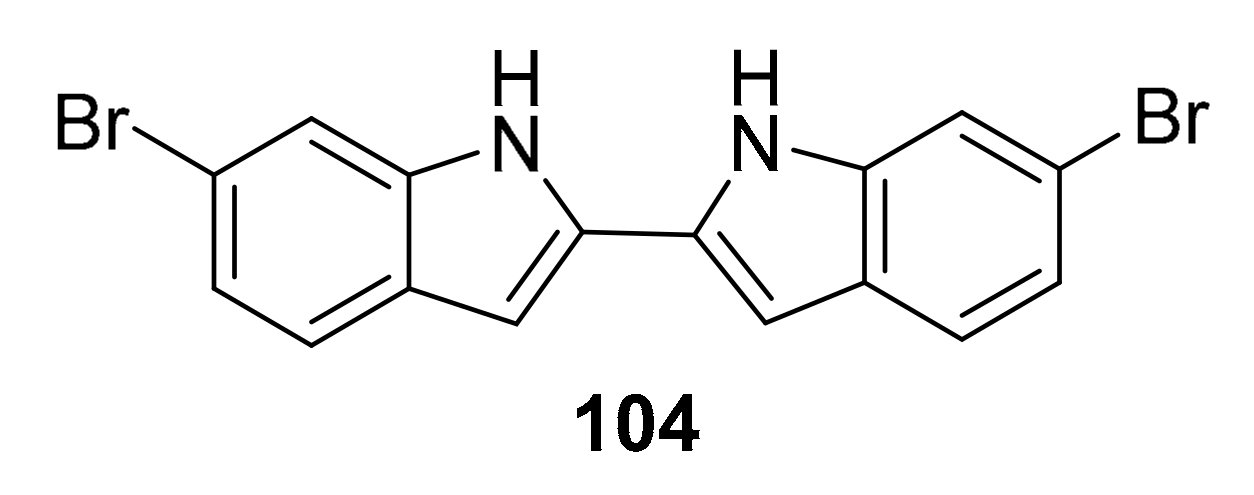
| 104 | bis-indole alkaloid | Topsentia pach astrelloides | MRSA PK | [117] |
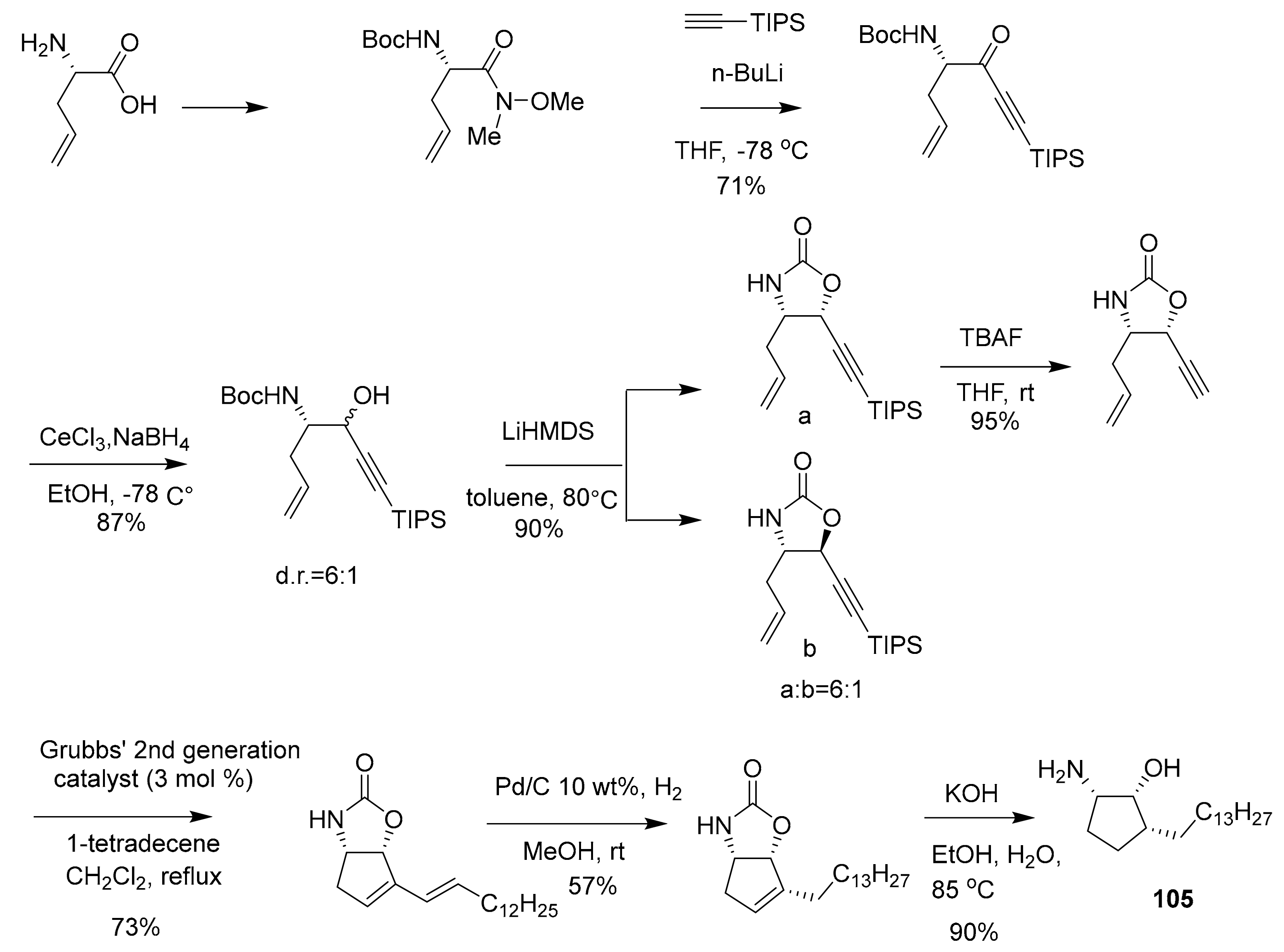
| 105 | sphingolipid | Pachastrissa sp. | SphK1 (7.5 μM) SphK1 (20.1 μM) | [118,119] |
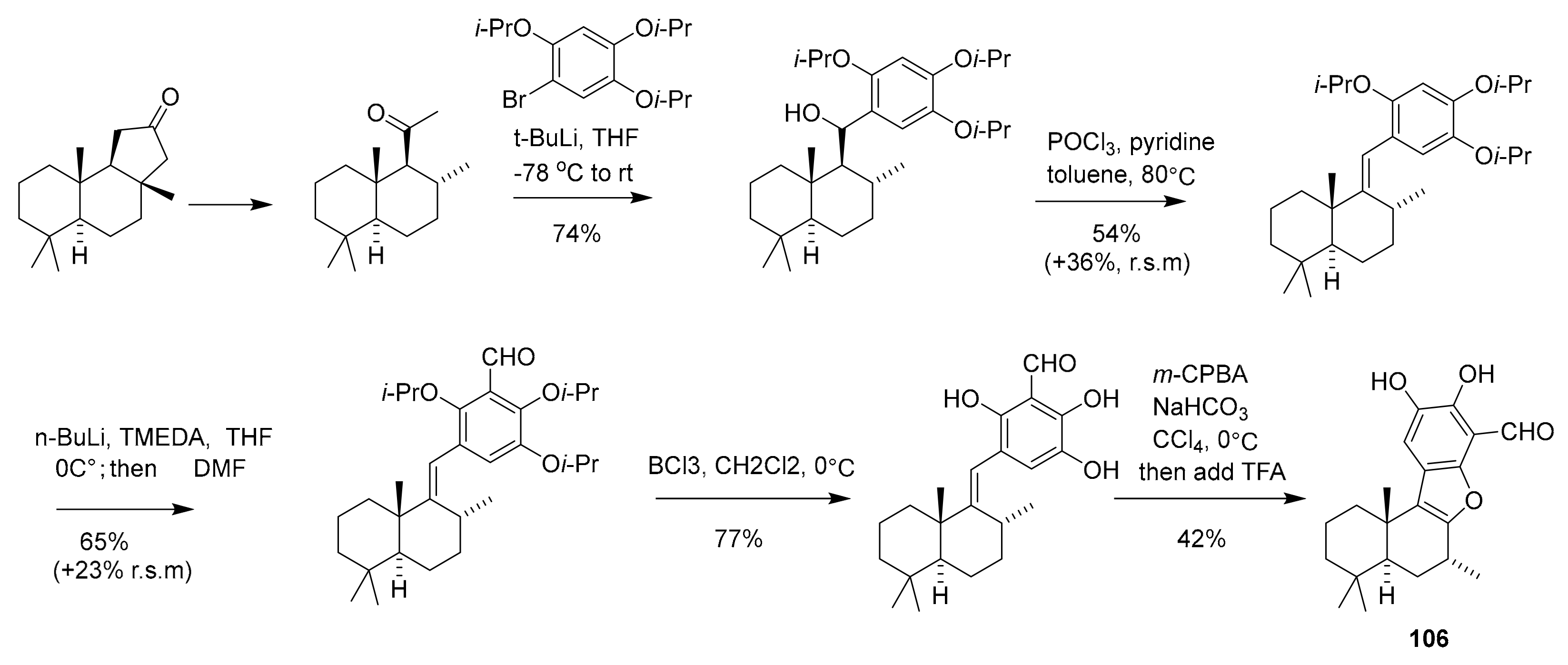
| 106 | polyfused-benzofuran | Aka coralliphaga | PIK-α (100 nM) | [122] |
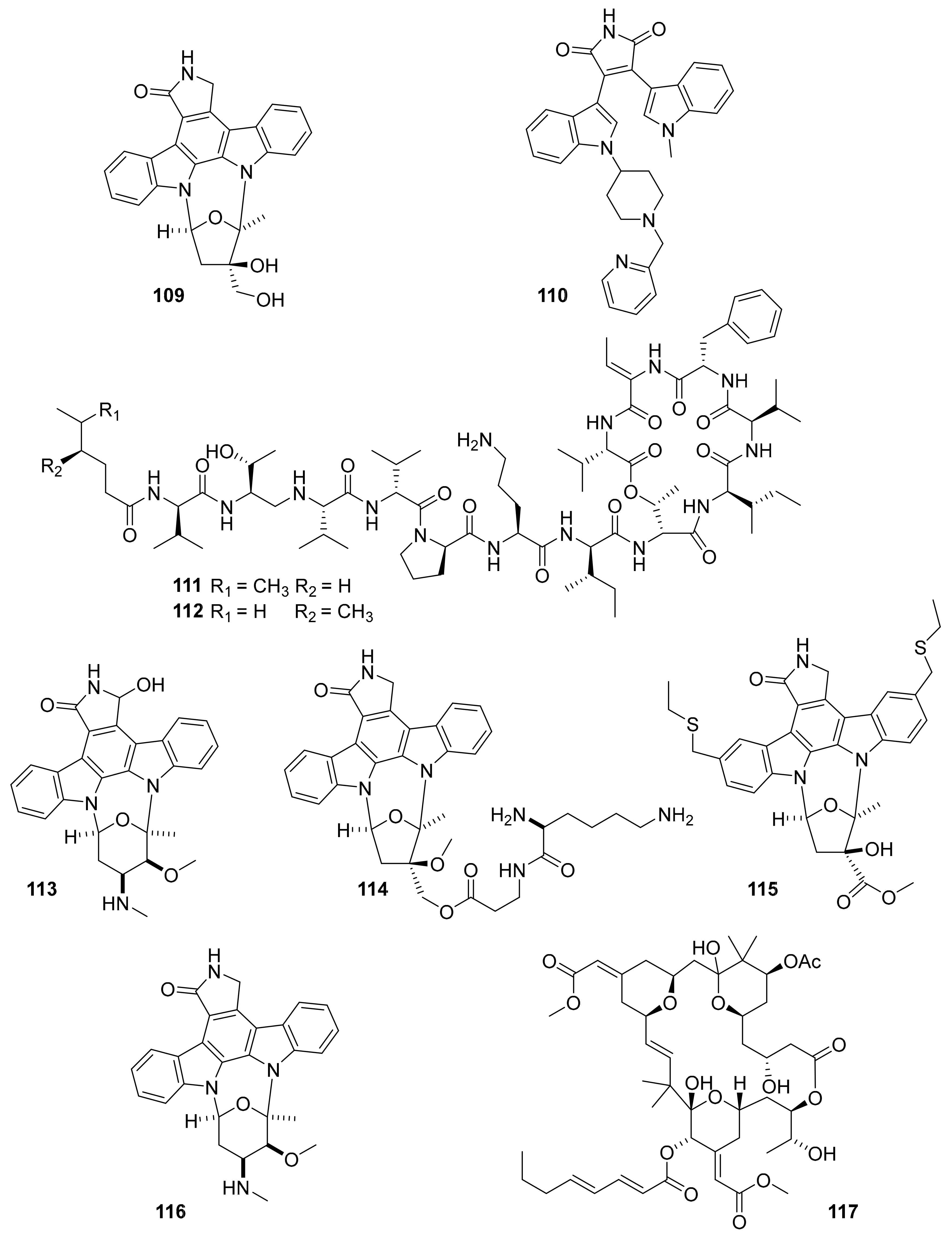
| Compound Name (Trademark) | Target | Marine Source | Chemical Class | Company or Institution | Therapeutic Area | Status | References |
|---|---|---|---|---|---|---|---|
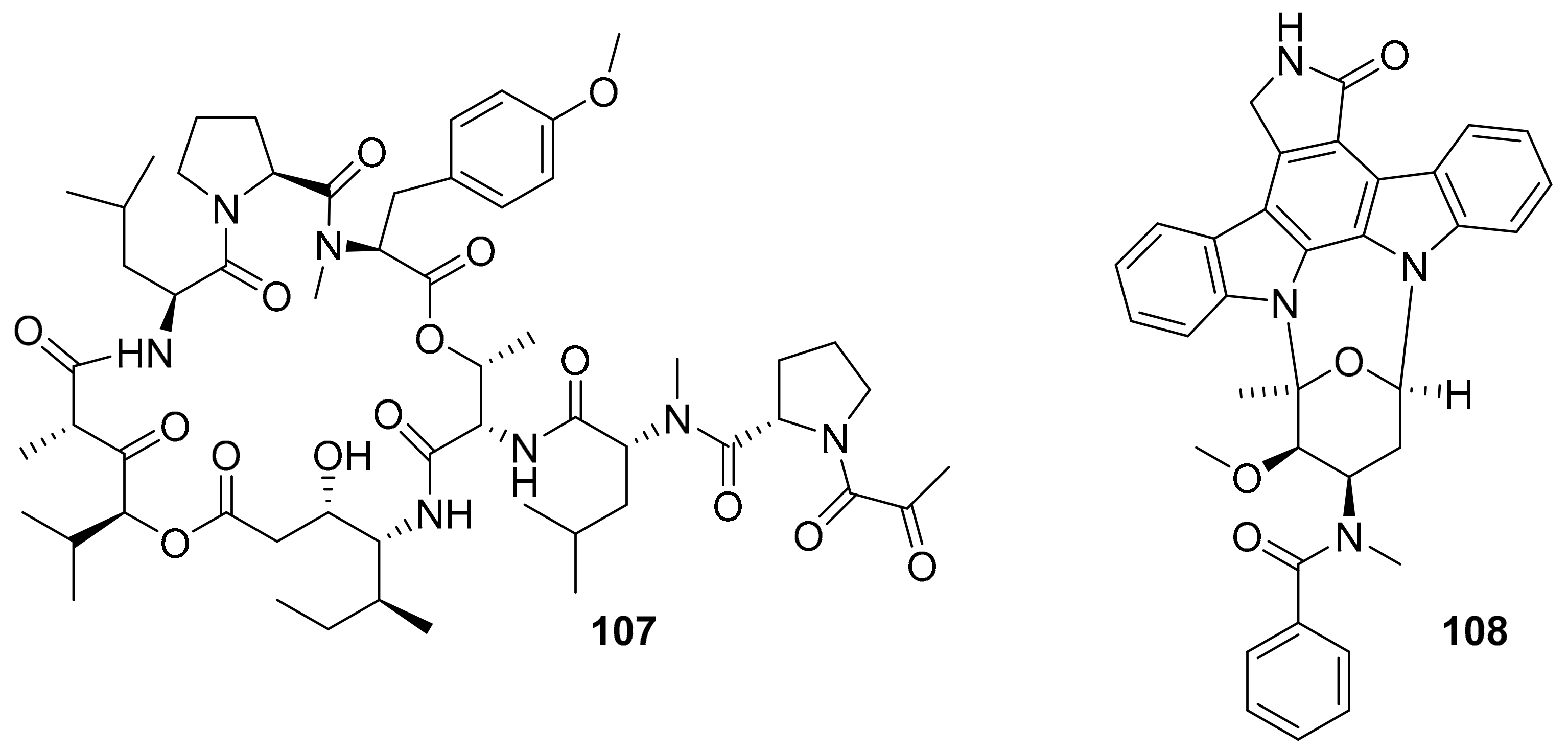
| plitidepsin (aplidin, 107) | VEGFR1 | ascidian | depsipeptide | PharmaMar | multiple myeloma | FDA-approved (2018) | [124,125,126,127] |
| midostaurin (108) | PKC-α, KIT, FLT3, VEGFR2, PDGFR-β | ascidian / bacteria | indolocarbazole | Novartis | acute myeloid leukemia, | FDA-approved (2017) | [124,127,128] |
| lestaurtinib (CEP-701, 109) | Flt-3, JAK-2 | ascidian / bacteria | indolocarbazole | Kyowa kirin | acute lymphoblast leukemia | Phase III | [127,130] |
| enzastaurin (LY317615, 110) | PKCβ, Class I PI3K | ascidian / bacteria | indolocarbazole | Eli Lilly | diffuse large B cell lymphoma | Phase III | [127,131] |
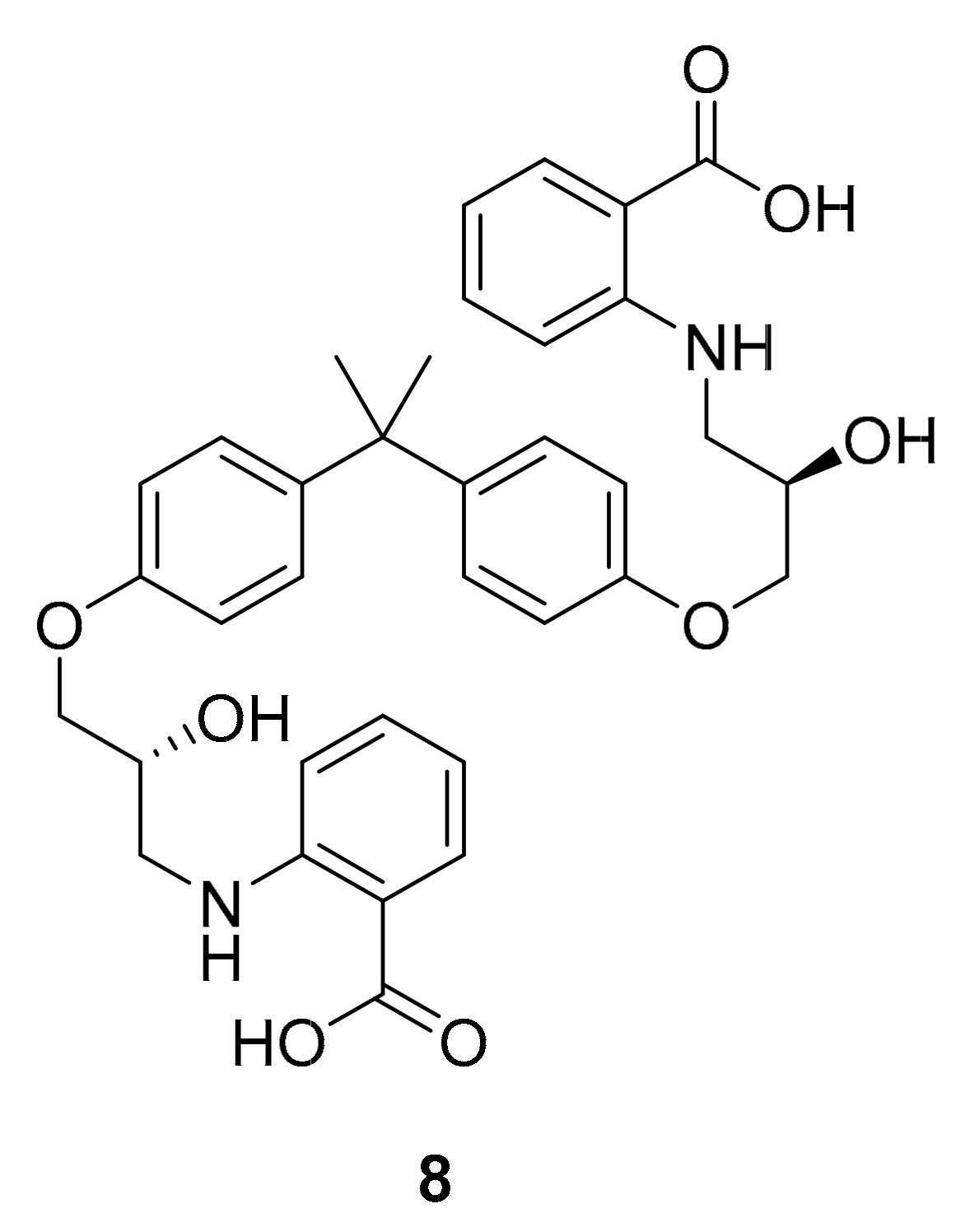
| kahalalide F (PM-92102, 111) | EGFR | mollusk and algae | tridecapeptide | PharmaMar Cephalon | cancar | Phase II (terminated) | [127,132,133] |
| isokahalalide F (PM02734, elisidepsin, 112) | ErbB2 | mollusk and algae | tridecapeptide | PharmaMar | cancer | Phase I (terminated) | [14,124,127] |
| 7-hydroxystaurosporine (UCN-01, 113) | Chk2, PDPK1, Chk1 PKC | ascidian / bacteria | indolocarbazole | Kyowa kirin | cancer | Phase II (terminated) | [127,134,135,] |
| CEP-2563 (114, prodrug of CEP-751) | Trk-A, Trk-B, Trk-C | ascidian / bacteria | indolocarbazole | Cephalon | cancer | Phase I (terminated) | [14,124,127] |
| CEP-1347 (KT7515; 115) | MAP3K11 | ascidian | indolocarbazole | Teva | Parkinson’s | Phase III (terminated) | [14,124,127] |
| staurosporine (AM-2282, 116) | PKC, JAK2, CamKIII | ascidian / bacteria | indolocarbazole | Kyowa Hakko Kirin (originator) | multiple myeloma | Phase II (terminated) | [127,136] |
| bryostatin 1 b (117) | PKC | bryozoan | macrocytic lactone | Neurotrope Bioscience | Alzheimer’s | Phase II (terminated) | [127,137,138,139] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Mar. Drugs 2019, 17, 493. https://doi.org/10.3390/md17090493
Li T, Wang N, Zhang T, Zhang B, Sajeevan TP, Joseph V, Armstrong L, He S, Yan X, Naman CB. A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Marine Drugs. 2019; 17(9):493. https://doi.org/10.3390/md17090493
Chicago/Turabian StyleLi, Te, Ning Wang, Ting Zhang, Bin Zhang, Thavarool P. Sajeevan, Valsamma Joseph, Lorene Armstrong, Shan He, Xiaojun Yan, and C. Benjamin Naman. 2019. "A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors" Marine Drugs 17, no. 9: 493. https://doi.org/10.3390/md17090493
APA StyleLi, T., Wang, N., Zhang, T., Zhang, B., Sajeevan, T. P., Joseph, V., Armstrong, L., He, S., Yan, X., & Naman, C. B. (2019). A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Marine Drugs, 17(9), 493. https://doi.org/10.3390/md17090493