New Linear Precursors of cIDPR Derivatives as Stable Analogs of cADPR: A Potent Second Messenger with Ca2+-Modulating Activity Isolated from Sea Urchin Eggs



 ,
,  ,
,  and
and 
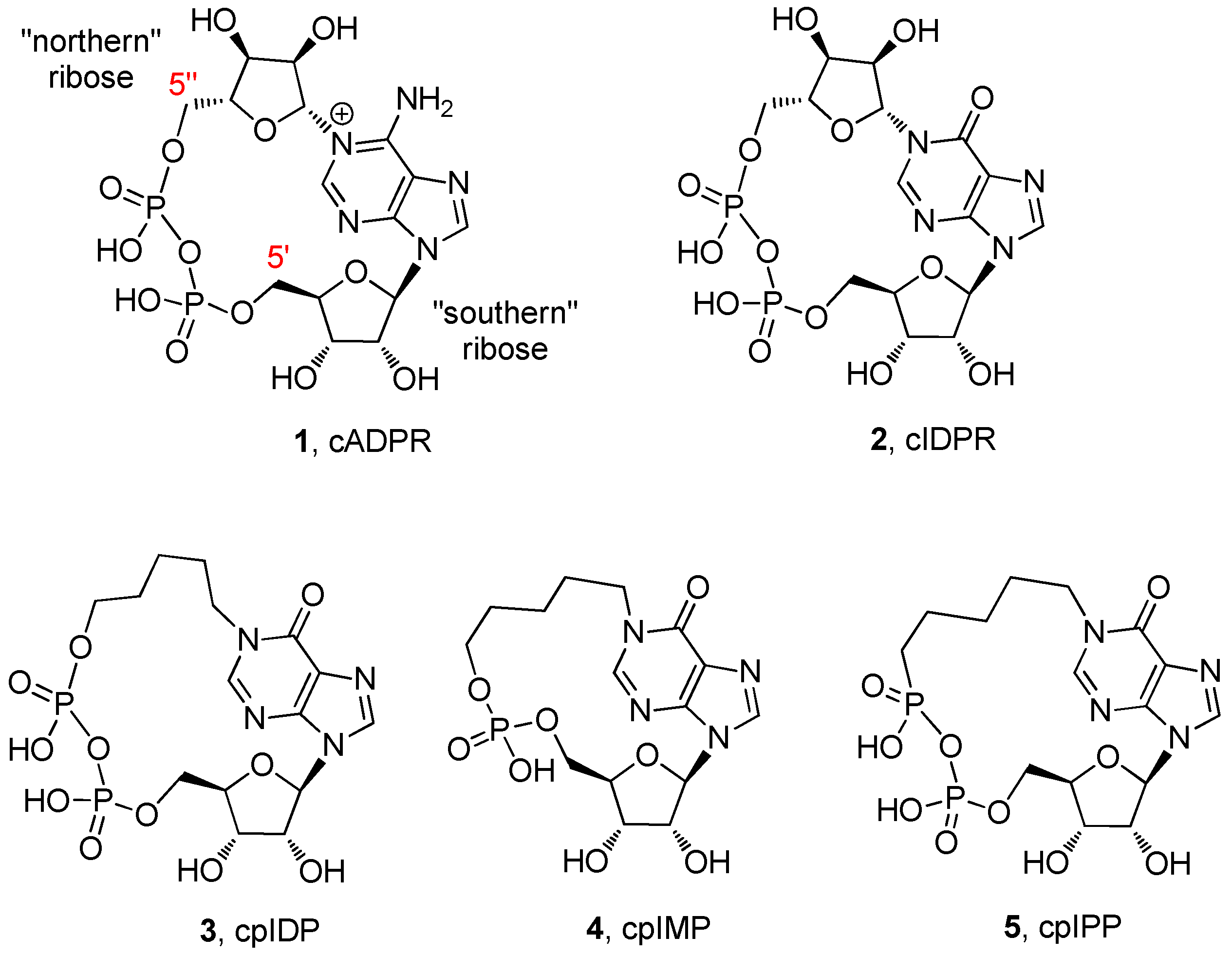{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
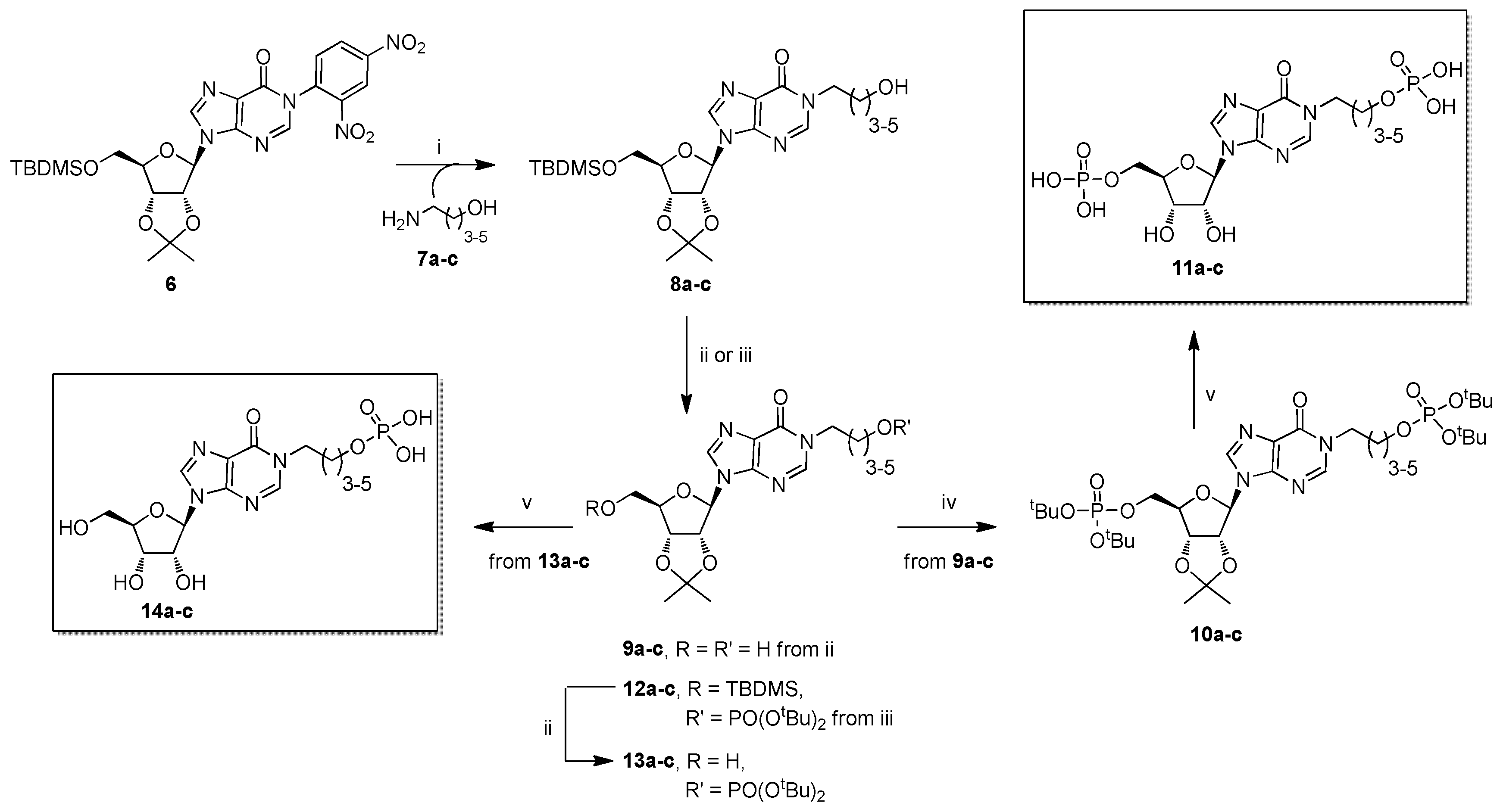
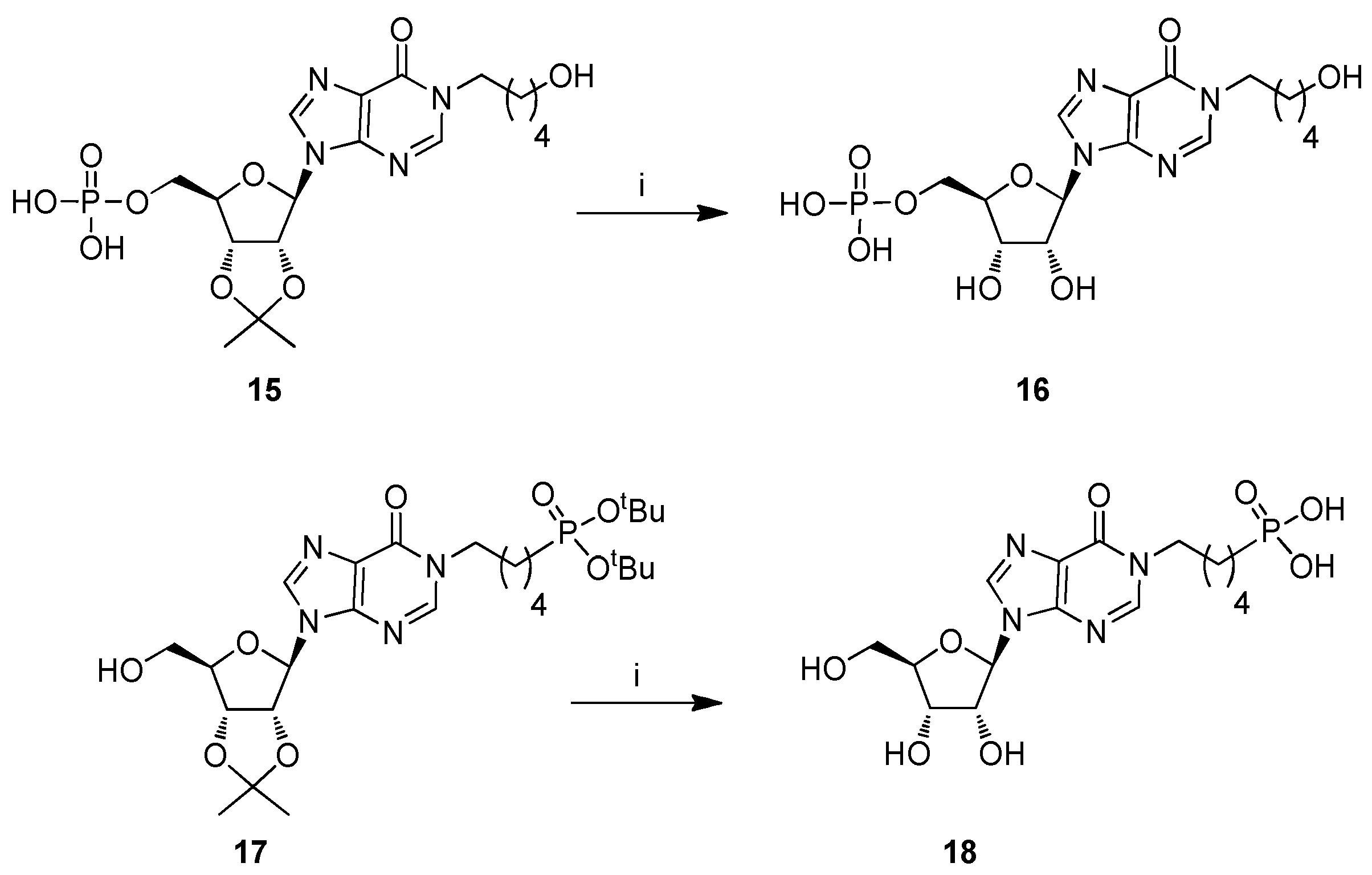
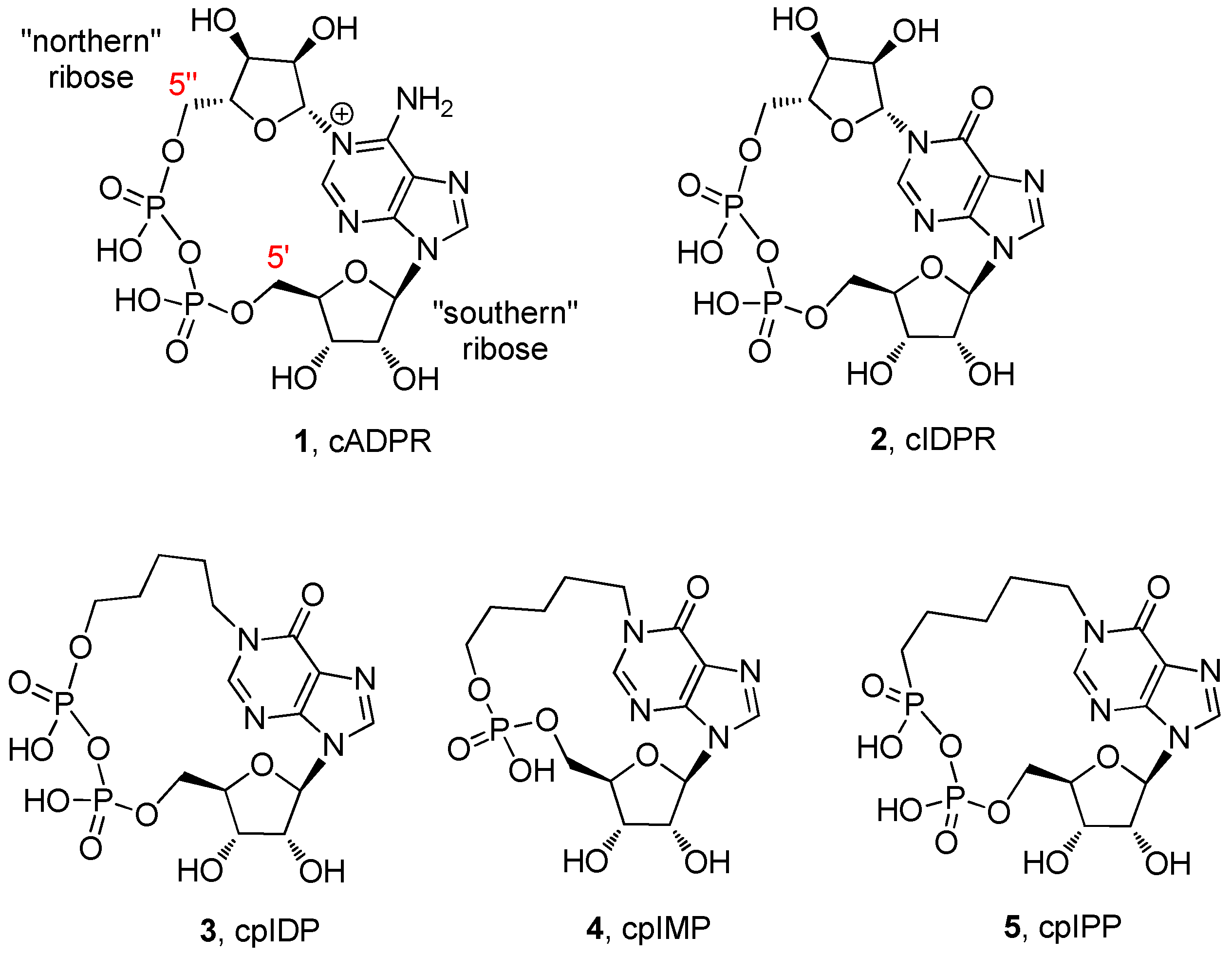
2. Results and Discussion
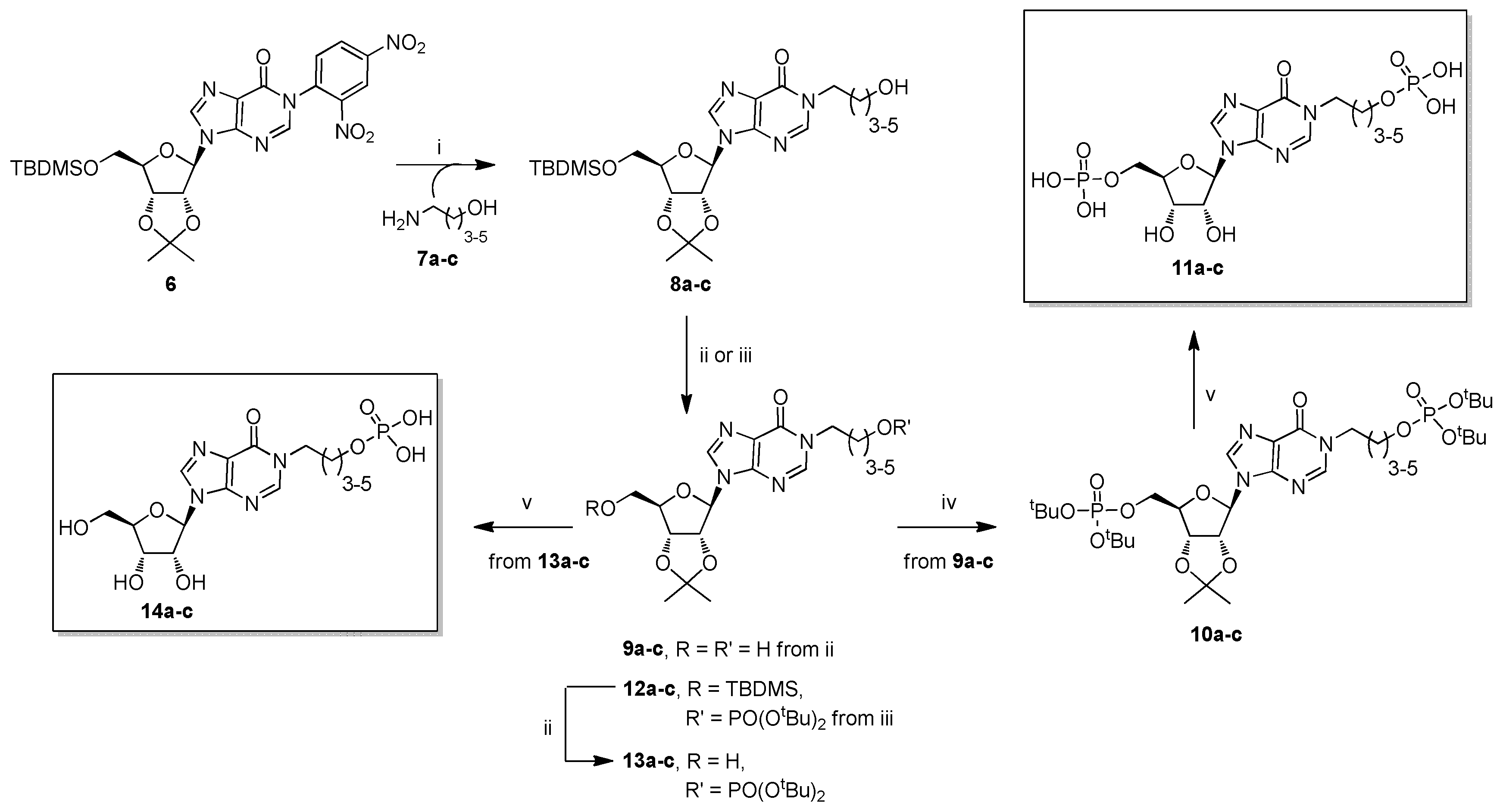
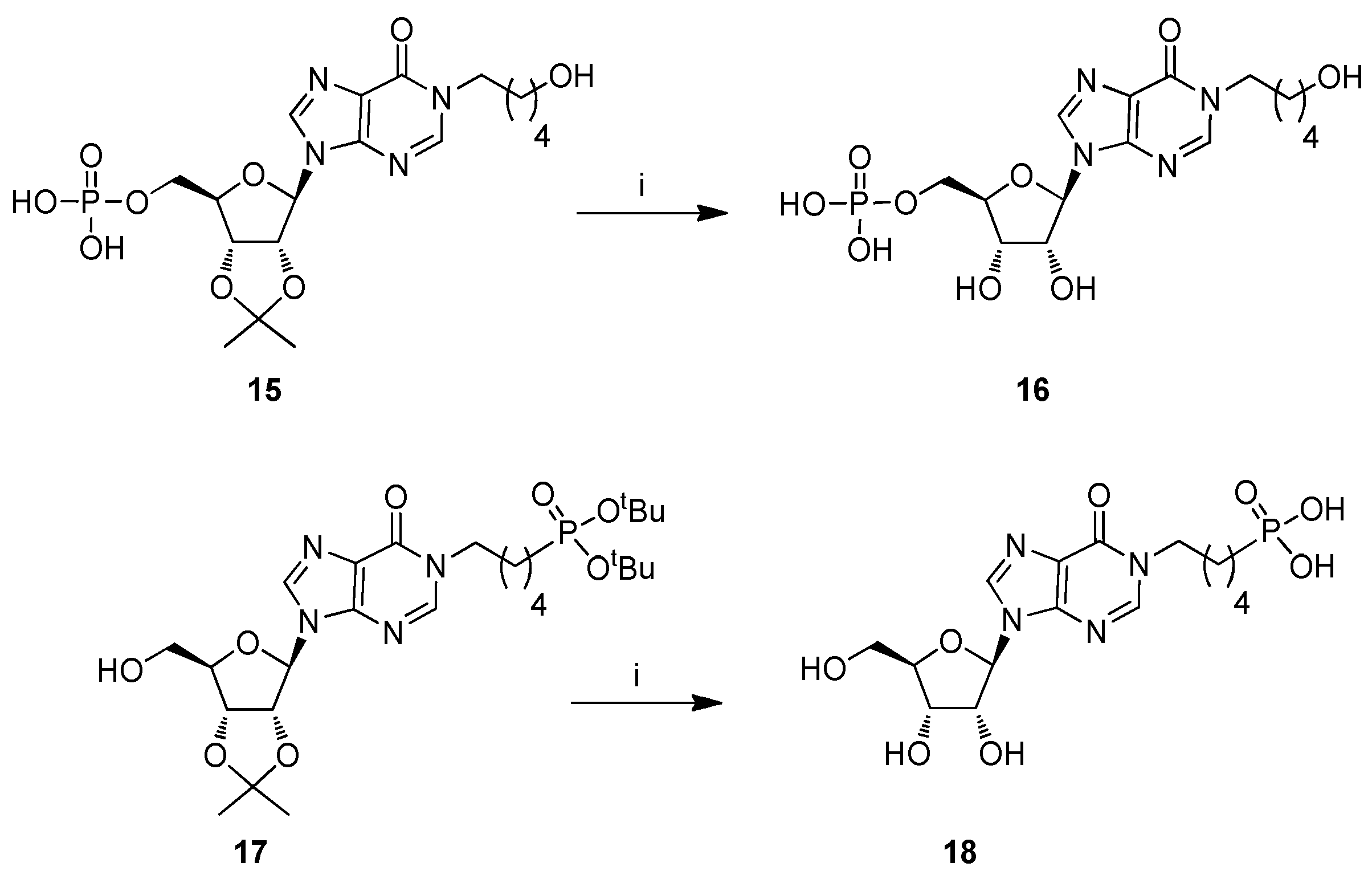
2.1. Chemistry
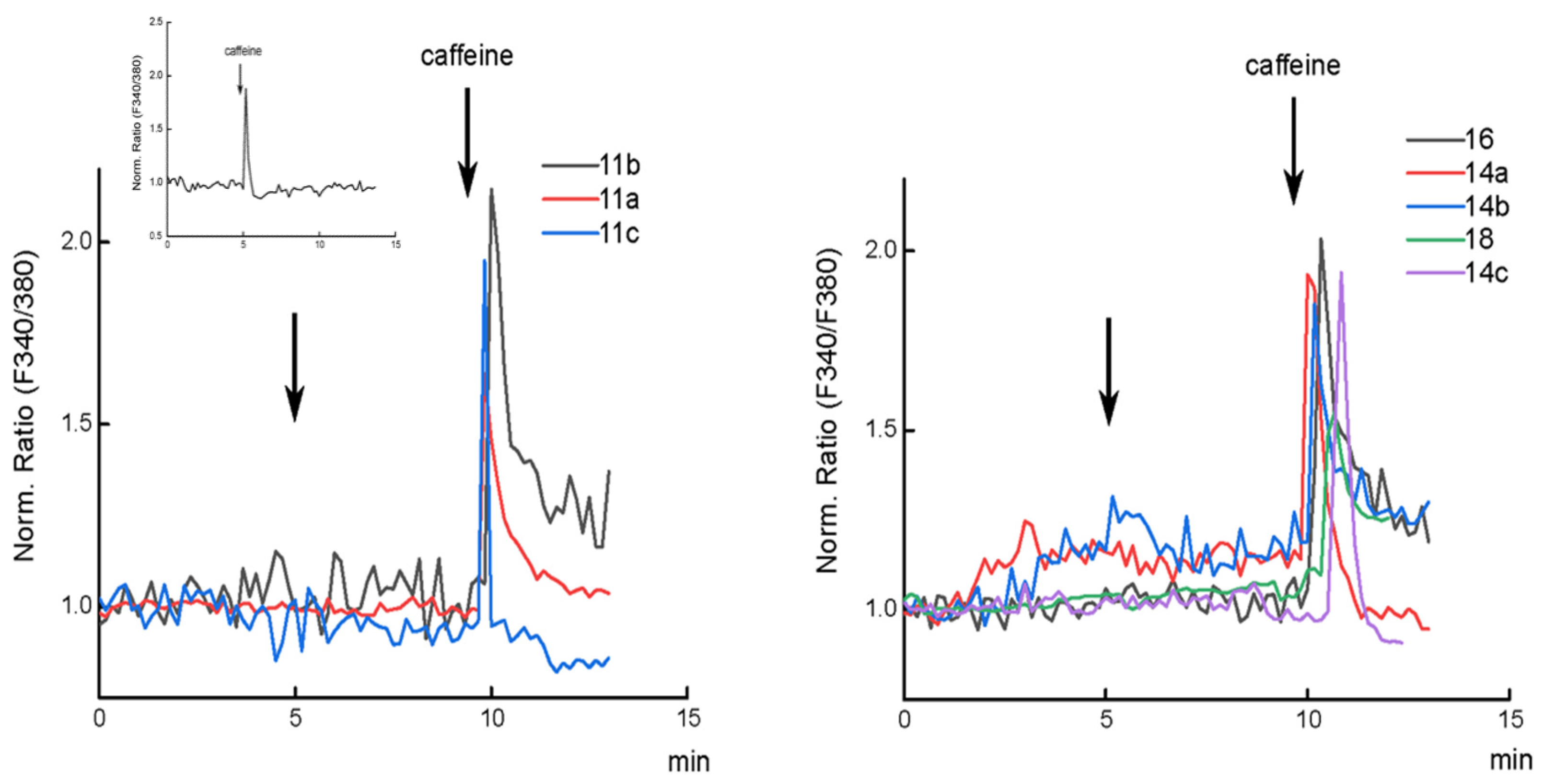
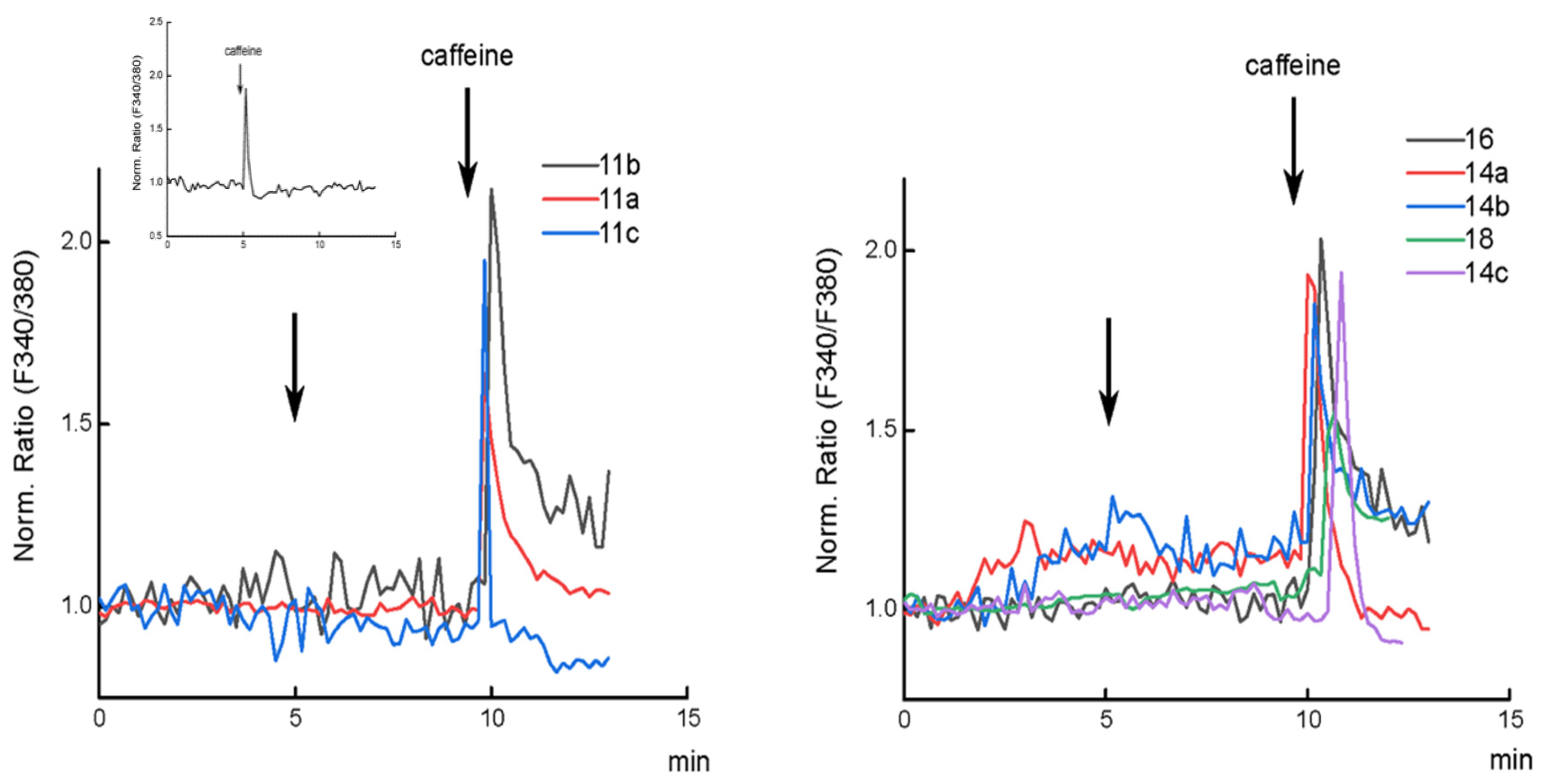
2.2. Biology
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemistry
3.2.1. General Procedure for the Preparation of Compounds 8a–c
3.2.2. General Procedure for the Preparation of Compounds 9a–c
3.2.3. General Procedure for the Preparation of Compounds 10a–c
3.2.4. General Procedure for the Preparation of Compounds 11a–c
3.2.5. General Procedure for the Preparation of Compounds 12a–c
3.2.6. General Procedure for the Reparation of Compounds 13a–c
3.2.7. General Procedure for the Preparation of Compounds 14a–c
3.2.8. Procedure for the Preparation of Compound 16
3.2.9. Procedure for the Preparation of Compound 18
3.3. Biology
Cytosolic Ca2+ Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. Calcium signaling. Cell 2007, 14, 1047–1068. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L. Intracellular Calcium. In Signal Transduction; Audet, J., Ed.; Elsevier: London, UK, 2016; pp. 381–439. [Google Scholar]
- Barbado, M.; Fablet, K.; Ronjat, M.; De Waard, M. Gene regulation by voltage-dependent calcium channels. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1096–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.C.X.; Kihara, A.H.; Goulart, V.A.M.; Tonelli, F.M.P.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Woo, J.S.; Perez, C.F.; Lee, E.H. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 2017, 49, e378. [Google Scholar] [CrossRef] [PubMed]
- Neher, E.; Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [PubMed]
- Takasawa, S.; Okamoto, H. Pancreatic β-cell death, regeneration and insulin secretion: Roles of poly(ADP-ribose) polymerase and cyclic ADP-ribose. Int. J. Exp. Diabetes Res. 2002, 3, 79–96. [Google Scholar] [CrossRef]
- Guse, A.H.; Da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.L.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef]
- Gul, R.; Park, D.R.; Shawl, A.I.; Im, S.Y.; Nam, T.S.; Lee, S.H.; Ko, J.K.; Jang, K.Y.; Kim, D.; Kim, U.H. Nicotinic acid adenine dinucleotide phosphate (NAADP) and cyclic ADP-Ribose (cADPR) mediate Ca2+ signaling in cardiac hypertrophy induced by β-Adrenergic Stimulation. PLoS ONE 2016, 11, e0149125. [Google Scholar] [CrossRef]
- Higashida, H.; Hashii, M.; Yokoyama, S.; Hoshi, N.; Asai, K.; Kato, T. Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J. Neurochem. 2001, 76, 321–331. [Google Scholar] [CrossRef]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef]
- Fliegert, R.; Gasser, A.; Guse, A.H. Regulation of calcium signalling by adenine-based second messengers. Biochem. Soc. Trans. 2007, 35(PT 1), 109–114. [Google Scholar] [CrossRef] [Green Version]
- Galione, A.; Cui, Y.; Empson, R.; Iino, S.; Wilson, H.; Terrar, D. Cyclic ADP-Ribose and the Regulation of Calcium-Induced Calcium Release in Eggs and Cardiac Myocytes. Cell Biochem. Biophys. 1998, 28, 19–30. [Google Scholar] [CrossRef]
- Wei, W.; Graeff, R.; Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef]
- Okamoto, H.; Takasawa, S.; Sugawara, A. The CD38-Cyclic ADP-Ribose System in Mammals: Historical Background, Pathophysiology and Perspective. Messenger 2014, 3, 27–34. [Google Scholar] [CrossRef]
- Potter, B.V.L.; Walseth, T.F. Medicinal chemistry and pharmacology of cyclic ADP-ribose. Curr. Mol. Med. 2004, 4, 303–311. [Google Scholar] [CrossRef]
- Rosen, D.; Bloor-Young, D.; Squires, J.; Parkesh, R.; Waters, G.; Vasudevan, S.R.; Lewis, A.M.; Churchill, G.C. Synthesis and use of cell-permeant cyclic ADP-ribose. Biochem. Biophys. Res. Commun. 2012, 418, 353–358. [Google Scholar] [CrossRef]
- Guse, A.H.; Cakir-Kiefer, C.; Fukuoka, M.; Shuto, S.; Weber, K.; Bailey, V.C.; Matsuda, A.; Mayr, G.W.; Oppenheimer, N.; Schuber, F.; et al. Novel hydrolysis-resistant analogues of cyclic ADP-ribose: Modification of the “northern” ribose and calcium release activity. Biochemistry 2002, 41, 6744–6751. [Google Scholar] [CrossRef]
- Swarbrick, J.M.; Graeff, R.; Garnham, C.; Thomas, M.P.; Galione, A.; Potter, B.V.L. ‘Click cyclic ADP-ribose’: A neutral second messenger mimic. Chem. Commun. 2014, 50, 2458–2461. [Google Scholar] [CrossRef]
- Qi, N.; Jung, K.; Wang, M.; Na, L.X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. A novel membrane-permeant cADPR antagonist modified in the pyrophosphate bridge. Chem. Commun. 2011, 47, 9462–9464. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Z.; Dammermann, W.; Zhang, L.; Guse, A.H.; Zhang, L.H. Synthesis and agonist activity of cyclic ADP-ribose analogues with substitution of the northern ribose by ether or alkane chains. J. Med. Chem. 2006, 49, 5501–5512. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Zhang, K.; Hu, J.; Liu, Z.; Jin, H.; Zhang, L.; Zhang, L. Calcium mobilizing behaviors of neutral cyclic ADP-ribose mimics which integrate the modifications of nucleobase, northern ribose and pyrophosphate. ChemBioChem 2018, 19, 1444–1451. [Google Scholar] [CrossRef]
- Guse, A. Biochemistry, Biology, and Pharmacology of Cyclic Adenosine Diphosphoribose (cADPR). Curr. Med. Chem. 2004, 11, 847–855. [Google Scholar] [CrossRef]
- Guse, A.H. Structure-activity relationship of cyclic ADP-ribose, an update. J. Chin. Pharm. Sci. 2013, 22, 127–136. [Google Scholar] [CrossRef]
- Wagner, G.K.; Guse, A.H.; Potter, B.V.L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5’-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [Google Scholar] [CrossRef]
- Zhang, L.; Yue, J.; Zhang, L.H. Cyclic adenosine 5’-diphosphoribose (cADPR) mimics used as molecular probes in cell signaling. Chem. Rec. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Shuto, S.; Matsuda, A. Chemistry of Cyclic ADP-Ribose and Its Analogs. Curr. Med. Chem. 2004, 11, 827–845. [Google Scholar] [CrossRef]
- Guse, A.H. The Ca2+-Mobilizing Second Messenger Cyclic ADP-Ribose. In Calcium: The Molecular Basis of Calcium Action in Biology and Medicine; Pochet, R., Donato, R., Haiech, J., Heizmann, C.W., Gerke, V., Eds.; Springer: The Netherlands, 2000; pp. 109–128. Available online: https://www.springer.com/gp/book/9780792364214 (accessed on 15 August 2019).
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a New N1-Pentyl Analogue of Cyclic Inosine Diphosphate Ribose (cIDPR) as a Stable Potential Mimic of Cyclic ADP Ribose (cADPR). Eur. J. Org. Chem. 2002, 2002, 4234–4238. [Google Scholar] [CrossRef]
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a novel N-1 carbocyclic, N-9 butyl analogue of cyclic ADP ribose (cADPR). Tetrahedron 2002, 58, 363–368. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Varra, M.; Piccialli, G.; Mayol, L. Synthesis of a new N-9 ribityl analogue of cyclic inosine diphosphate ribose (cIDPR) as a mimic of cyclic ADP ribose (cADPR). Nucleos. Nucleot. Nucl. 2005, 24, 735–738. [Google Scholar] [CrossRef]
- Oliviero, G.; Borbone, N.; Amato, J.; D’Errico, S.; Piccialli, G.; Varra, M.; Mayol, L. Synthesis of A New Ribose Modified Analogue of Cyclic Inosine Diphosphate Ribose. Nucleos. Nucleot. Nucl. 2007, 26, 1321–1324. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Piccialli, G.; Mayol, L. A solid-phase approach to the synthesis of N-1-alkyl analogues of cyclic inosine-diphosphate-ribose (cIDPR). Tetrahedron 2010, 66, 1931–1936. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Solid-phase synthesis of a new diphosphate 5-aminoimidazole-4-carboxamide riboside (AICAR) derivative and studies toward cyclic AICAR diphosphate ribose. Molecules 2011, 16, 8110–8118. [Google Scholar] [CrossRef]
- Mahal, A.; D’Errico, S.; Borbone, N.; Pinto, B.; Secondo, A.; Costantino, V.; Tedeschi, V.; Oliviero, G.; Piccialli, V.; Piccialli, G. Synthesis of cyclic N 1 -pentylinosine phosphate, a new structurally reduced cADPR analogue with calcium-mobilizing activity on PC12 cells. Beilstein J. Org. Chem. 2015, 11, 2689–2695. [Google Scholar] [CrossRef]
- D’Errico, S.; Borbone, N.; Catalanotti, B.; Secondo, A.; Petrozziello, T.; Piccialli, I.; Pannaccione, A.; Costantino, V.; Mayol, L.; Piccialli, G.; et al. Synthesis and Biological Evaluation of a New Structural Simplified Analogue of cADPR, a Calcium-Mobilizing Secondary Messenger Firstly Isolated from Sea Urchin Eggs. Mar. Drugs 2018, 16, 89. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- Swarbrick, J.M.; Riley, A.M.; Mills, S.J.; Potter, B.V.L. Designer small molecules to target calcium signalling. Biochem. Soc. Trans. 2015, 43, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695. [Google Scholar] [CrossRef]
- Watt, J.M.; Rozewitz, M.D.; Guse, A.H.; Fliegert, R.; Potter, B.V.L. Synthesis of Terminal Ribose Analogues of Adenosine 5′-Diphosphate Ribose as Probes for the Transient Receptor Potential Cation Channel TRPM2. J. Org. Chem. 2019, 84, 6143–6157. [Google Scholar]
- Moreau, C.; Liu, Q.; Graeff, R.; Wagner, G.K.; Thomas, M.P.; Swarbrick, J.M.; Shuto, S.; Lee, H.C.; Hao, Q.; Potter, B.V.L. CD38 Structure-Based Inhibitor Design Using the N1-Cyclic Inosine 5′-Diphosphate Ribose Template. PLoS ONE 2013, 8, e66247. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D’Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. A facile synthesis of 5’-Fluoro-5’-deoxyacadesine (5’-F-AICAR): A novel non-phosphorylable AICAR Analogue. Molecules 2012, 17, 13036–13044. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Piccialli, V.; Piccialli, G. Synthesis of 5-Aminoimidazole-4-Carboxamide Riboside (AICAR) and Its Derivatives Using Inosine as Starting Material. Curr. Protoc. Nucleic Acid Chem. 2015, 63, 1.35.1–1.35.24. [Google Scholar]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Piccialli, G.; Luciano, M. Facile solid-phase synthesis of AICAR 5-monophosphate (ZMP) and its 4-N-Alkyl derivatives. Eur. J. Org. Chem. 2010, 2010, 1517–1524. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Synthesis of new acadesine (AICA-riboside) analogues having acyclic D-ribityl or 4-hydroxybutyl chains in place of the ribose. Molecules 2013, 18, 9420–9431. [Google Scholar] [CrossRef]
- Ribose, S.; Cyclase, I.A.; Cd, H.; Swarbrick, J.M.; Grae, R.; Zhang, H.; Thomas, M.P.; Hao, Q.; Potter, B.V.L. Cyclic Adenosine 5′-Diphosphate Ribose Analogs without a “Southern” Ribose Inhibit ADP-ribosyl Cyclase—Hydrolase CD38. J. Med. Chem. 2014, 57, 8517–8529. [Google Scholar]
- Swarbrick, J.M.; Potter, B.V.L. Total Synthesis of a Cyclic Adenosine 5′-Diphosphate Ribose Receptor Agonist. J. Org. Chem. 2012, 77, 4191–4197. [Google Scholar] [CrossRef]
- Mangoni, O.; Imperatore, C.; Tomas, C.R.; Costantino, V.; Saggiomo, V.; Mangoni, A. The new carotenoid pigment moraxanthin is associated with toxic microalgae. Mar. Drugs 2011, 9, 242–255. [Google Scholar] [CrossRef]
- Airey, J.A.; Baring, M.D.; Sutko, J.L. Ryanodine receptor protein is expressed during differentiation in the muscle cell lines BC3H1 and C2C12. Dev. Biol. 1991, 148, 365–374. [Google Scholar] [CrossRef]
- Ríos, E. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 2018, 150, 521–537. [Google Scholar] [CrossRef] [Green Version]
- Dutka, T.L.; Lamboley, C.R.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. Effects of carnosine on contractile apparatus Ca2+ sensitivity and sarcoplasmic reticulum Ca2+ release in human skeletal muscle fibers. J. Appl. Physiol. 2011, 112, 728–736. [Google Scholar] [CrossRef]
- Wissing, F.; Nerou, E.P.; Taylor, C.W. A novel Ca2+-induced Ca2+ release mechanism mediated by neither inositol trisphosphate nor ryanodine receptors. Biochem. J. 2002, 361, 605–611. [Google Scholar] [CrossRef]
- Guaragna, A.; Roviello, G.N.; D’Errico, S.; Paolella, C.; Palumbo, G.; D’Alonzo, D. Solid phase synthesis of a novel folate-conjugated 5-aminolevulinic acid methyl ester based photosensitizer for selective photodynamic therapy. Tetrahedron Lett. 2015, 56, 775–778. [Google Scholar] [CrossRef]
- D’Errico, S.; Piccialli, V.; Oliviero, G.; Borbone, N.; Amato, J.; D’Atri, V.; Piccialli, G. Probing the reactivity of nebularine N1-oxide. A novel approach to C-6 C-substituted purine nucleosides. Tetrahedron 2011, 67, 6138–6144. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Errico, S.; Basso, E.; Falanga, A.P.; Marzano, M.; Pozzan, T.; Piccialli, V.; Piccialli, G.; Oliviero, G.; Borbone, N. New Linear Precursors of cIDPR Derivatives as Stable Analogs of cADPR: A Potent Second Messenger with Ca2+-Modulating Activity Isolated from Sea Urchin Eggs. Mar. Drugs 2019, 17, 476. https://doi.org/10.3390/md17080476
D’Errico S, Basso E, Falanga AP, Marzano M, Pozzan T, Piccialli V, Piccialli G, Oliviero G, Borbone N. New Linear Precursors of cIDPR Derivatives as Stable Analogs of cADPR: A Potent Second Messenger with Ca2+-Modulating Activity Isolated from Sea Urchin Eggs. Marine Drugs. 2019; 17(8):476. https://doi.org/10.3390/md17080476
Chicago/Turabian StyleD’Errico, Stefano, Emy Basso, Andrea Patrizia Falanga, Maria Marzano, Tullio Pozzan, Vincenzo Piccialli, Gennaro Piccialli, Giorgia Oliviero, and Nicola Borbone. 2019. "New Linear Precursors of cIDPR Derivatives as Stable Analogs of cADPR: A Potent Second Messenger with Ca2+-Modulating Activity Isolated from Sea Urchin Eggs" Marine Drugs 17, no. 8: 476. https://doi.org/10.3390/md17080476
APA StyleD’Errico, S., Basso, E., Falanga, A. P., Marzano, M., Pozzan, T., Piccialli, V., Piccialli, G., Oliviero, G., & Borbone, N. (2019). New Linear Precursors of cIDPR Derivatives as Stable Analogs of cADPR: A Potent Second Messenger with Ca2+-Modulating Activity Isolated from Sea Urchin Eggs. Marine Drugs, 17(8), 476. https://doi.org/10.3390/md17080476