Tersone A-G, New Pyridone Alkaloids from the Deep-Sea Fungus Phomopsis tersa


Abstract
:
1. Introduction
2. Results and Discussion
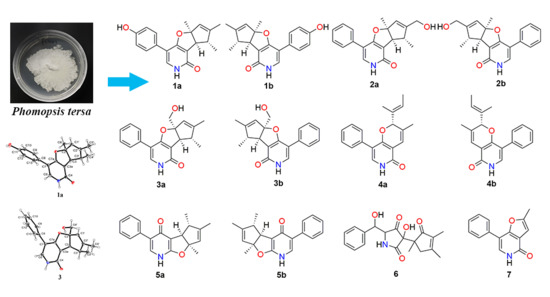
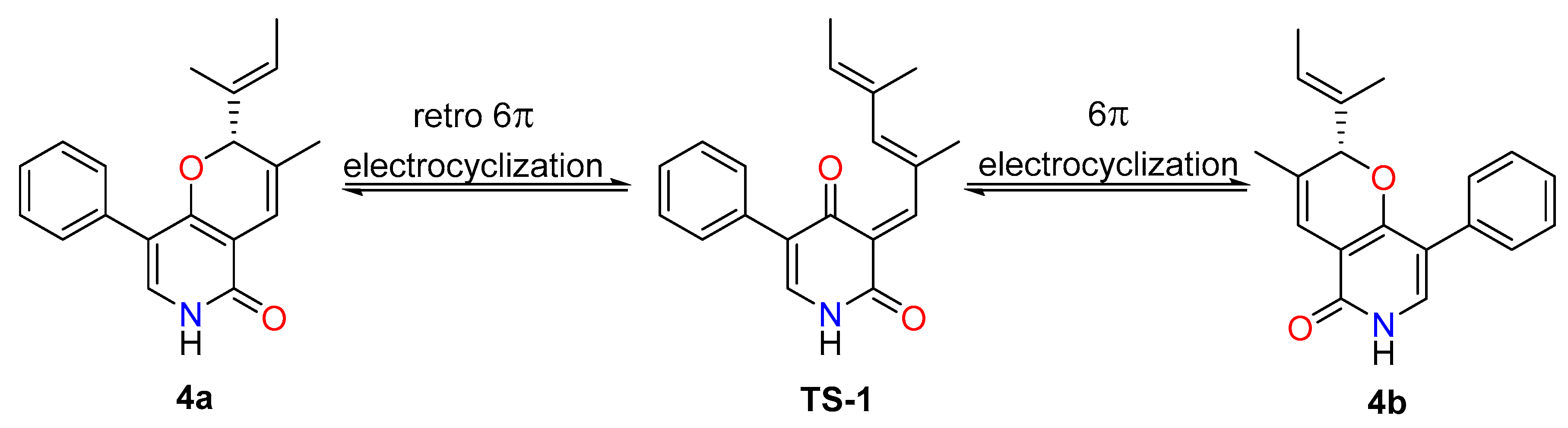
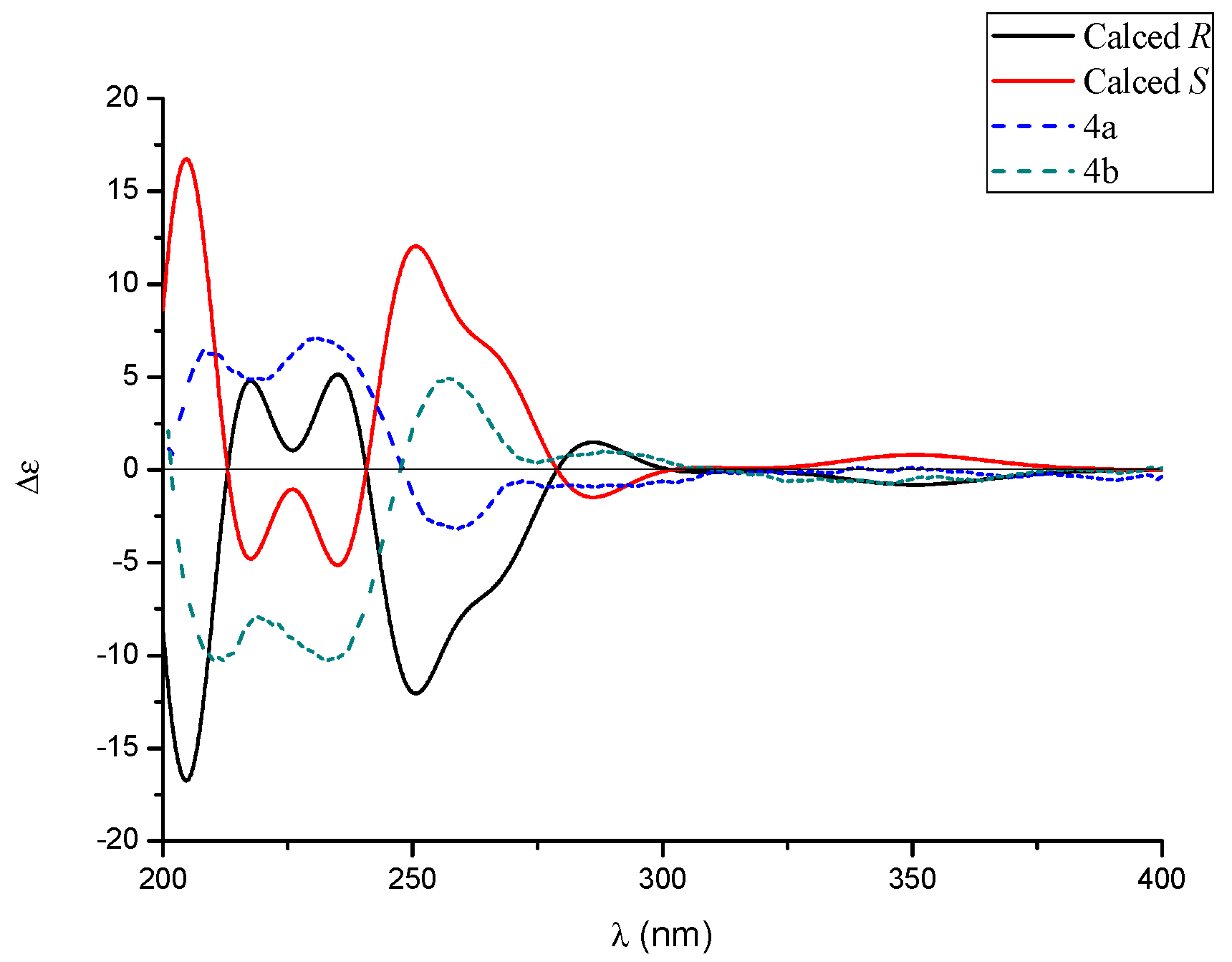
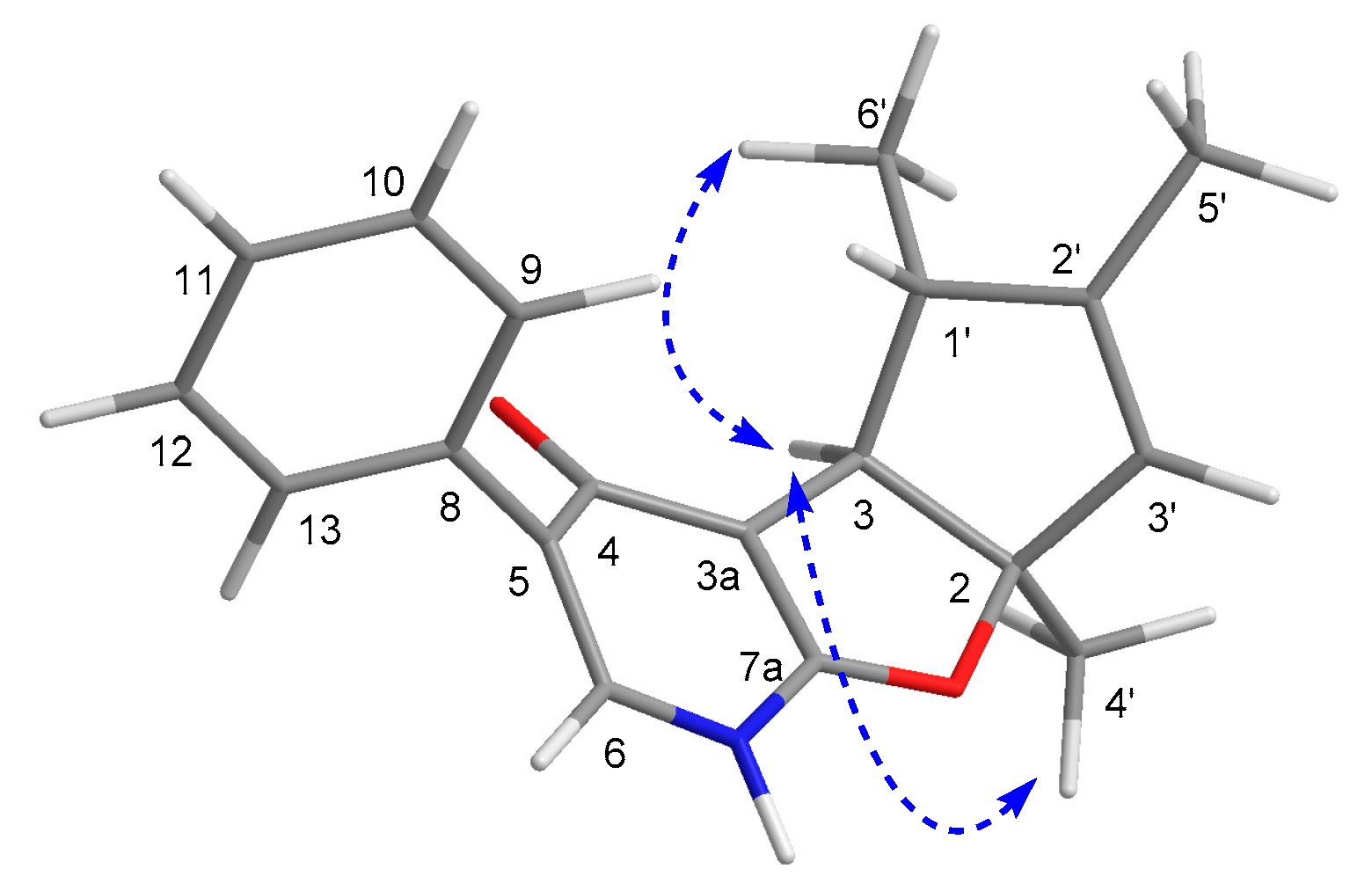
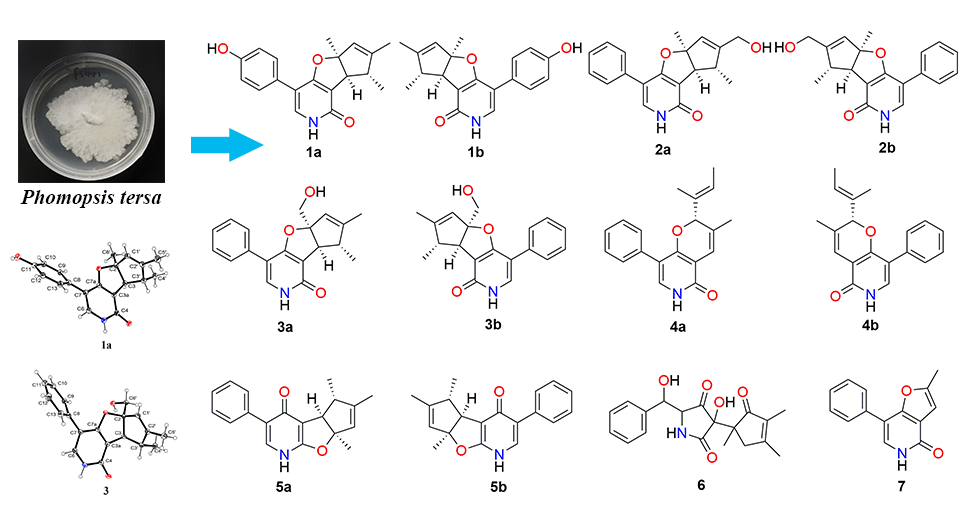
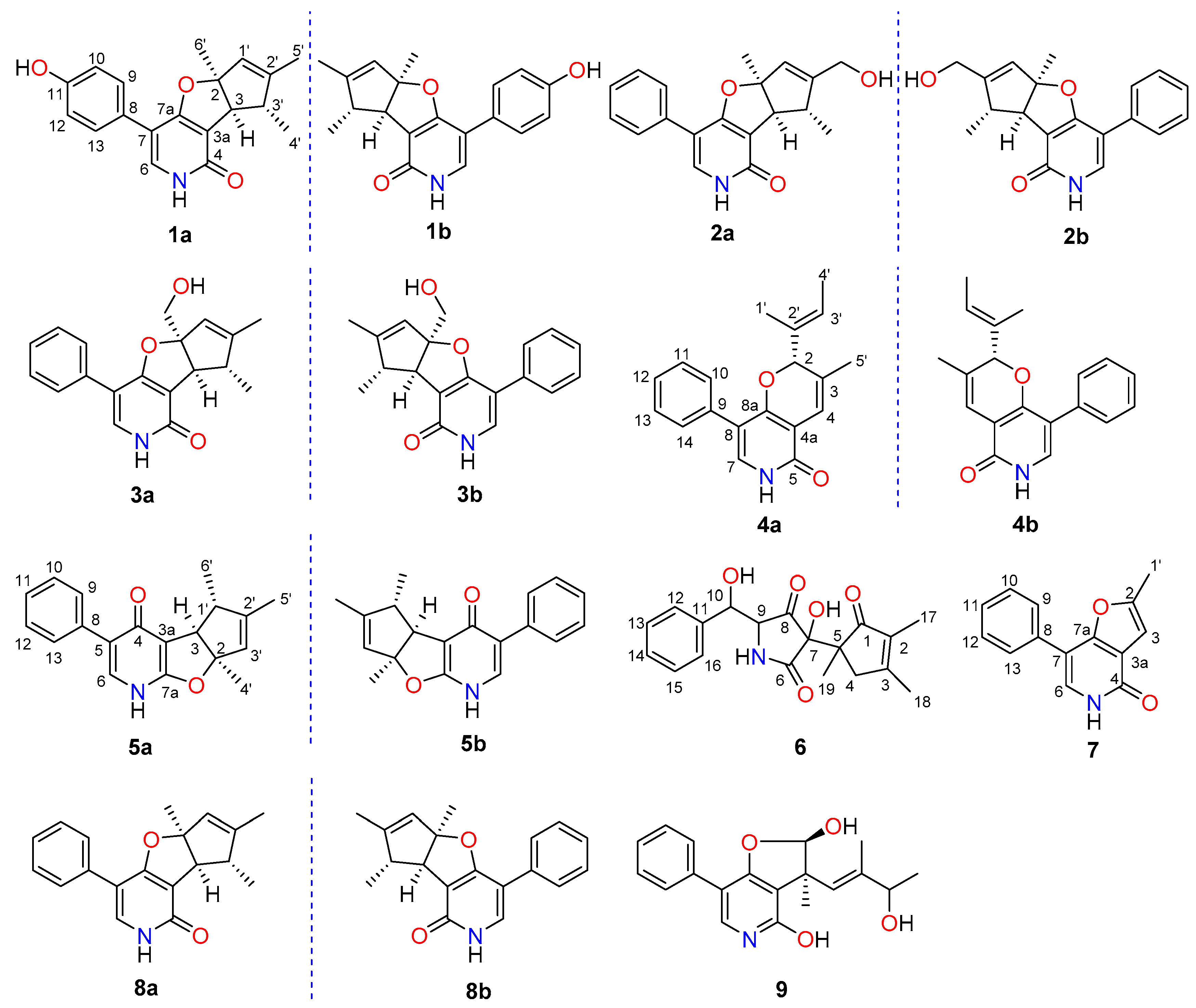
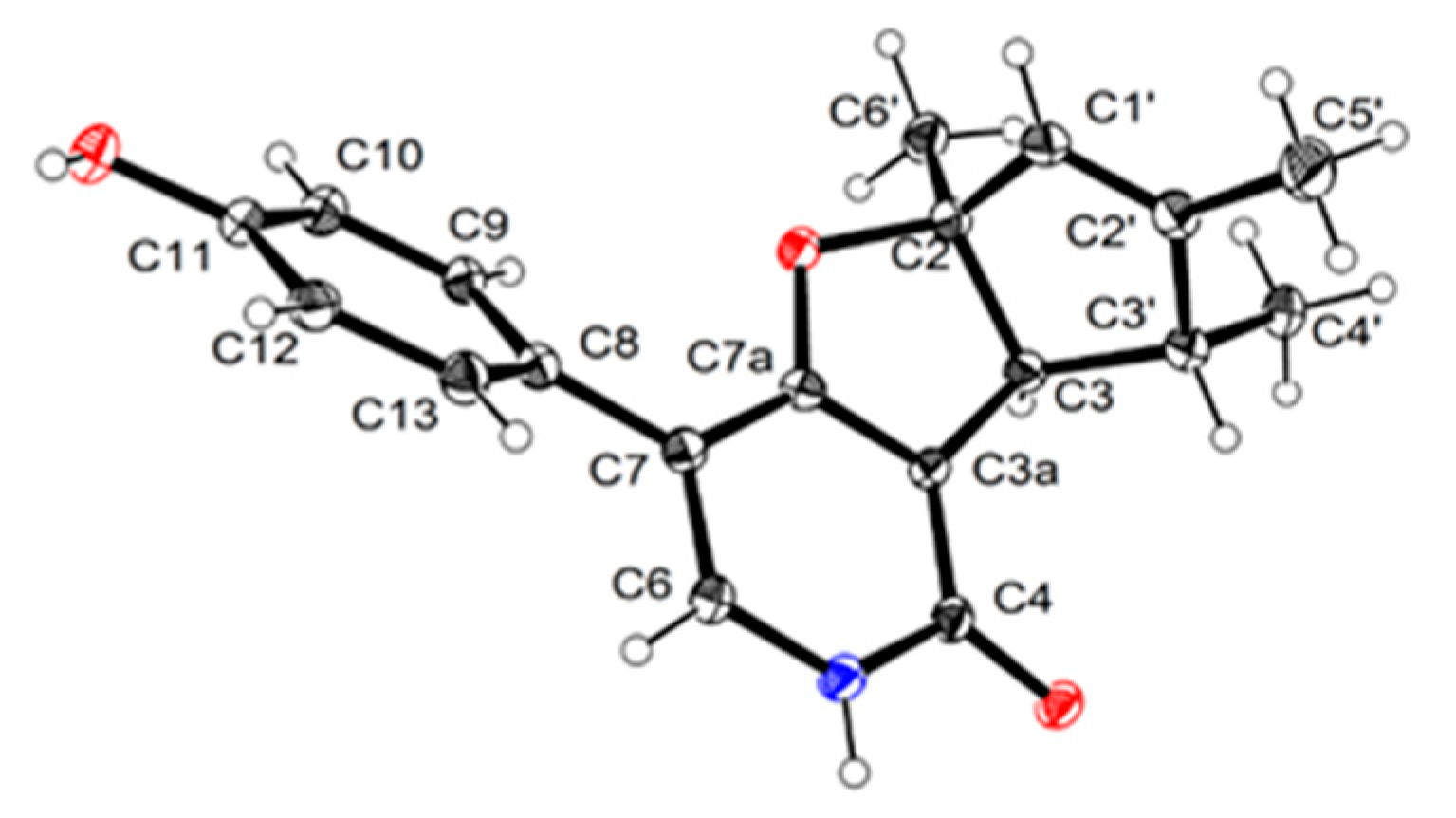
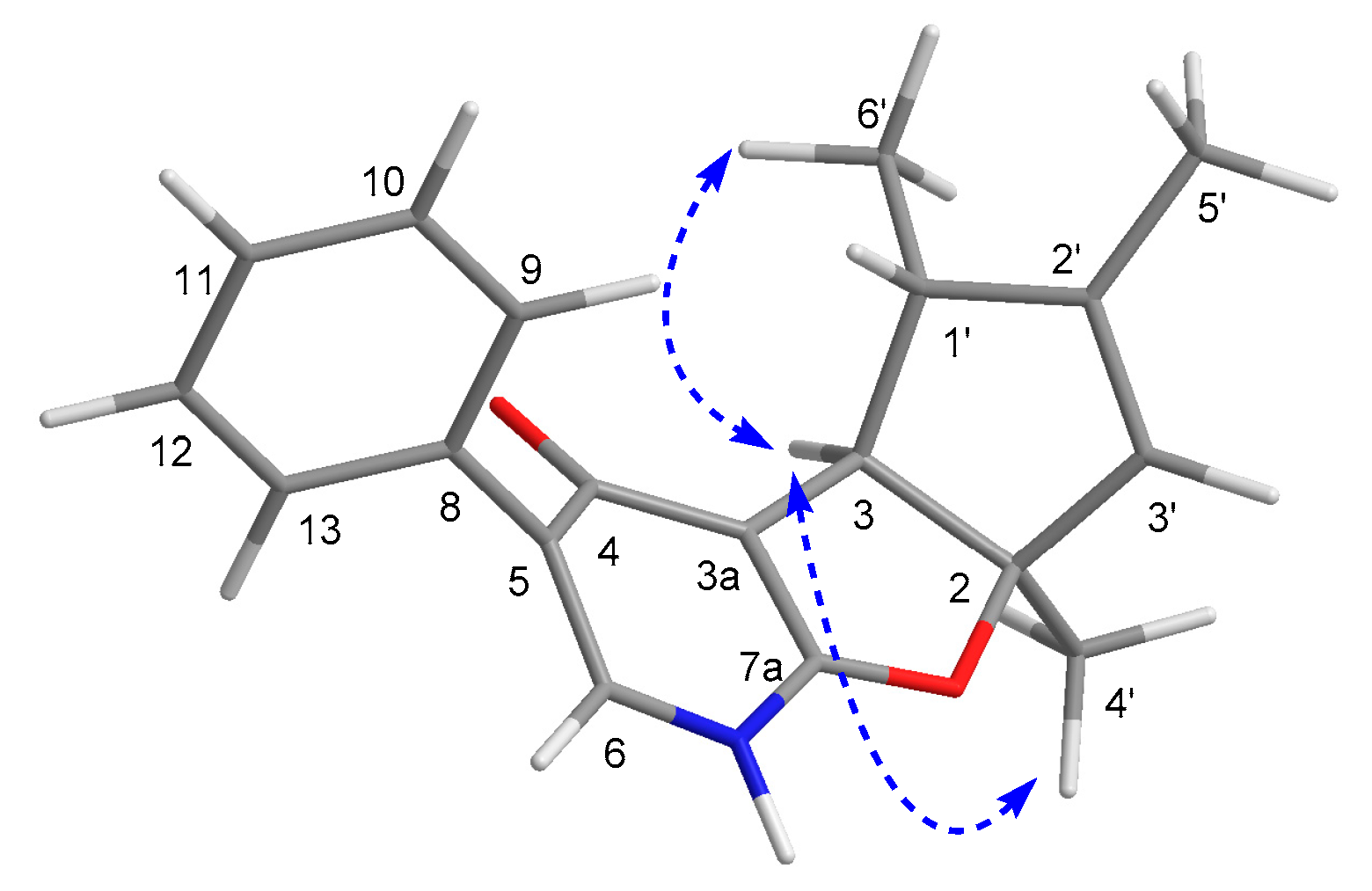
2.1. Structure Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
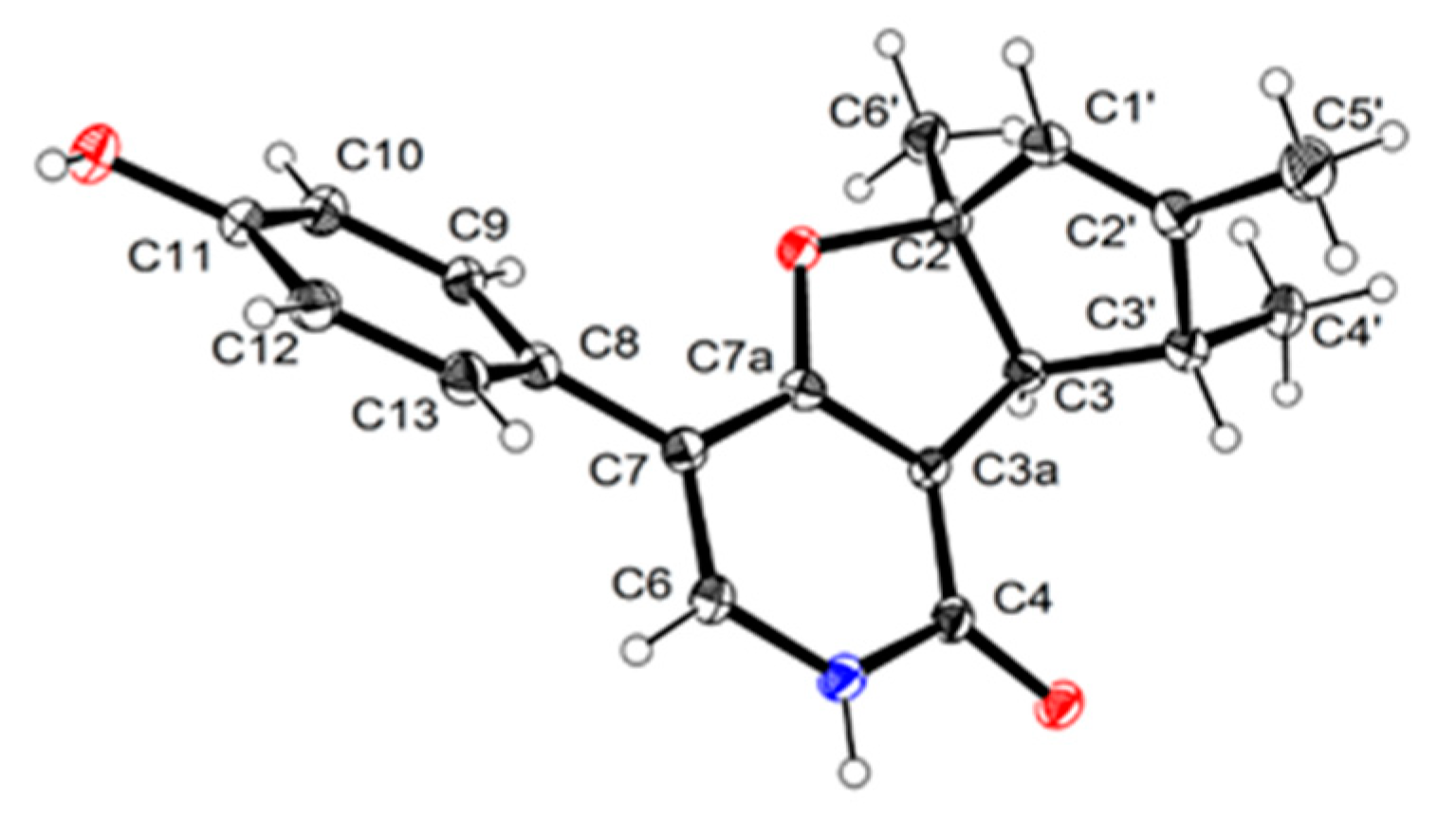
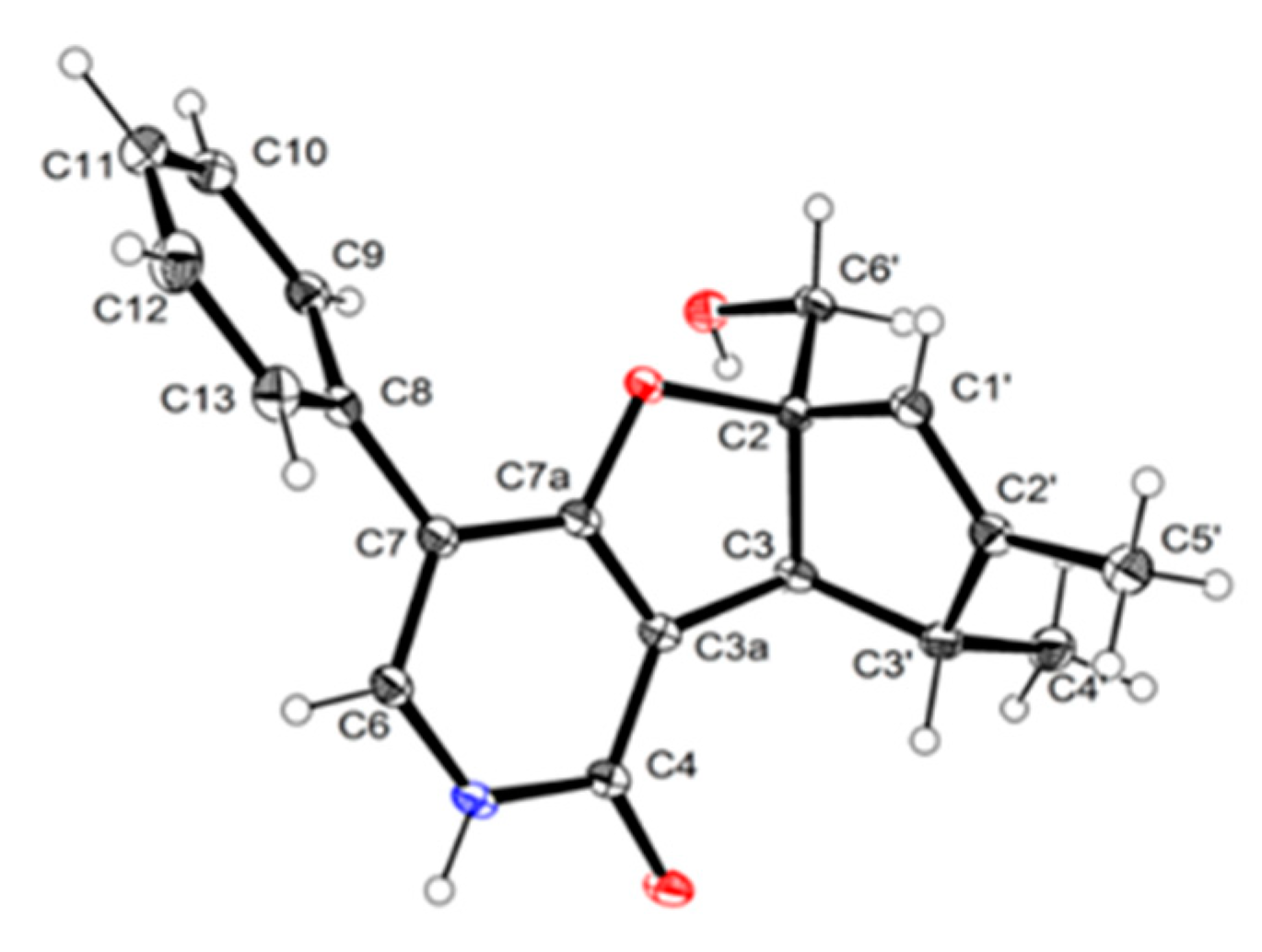
3.4. X-ray Crytallographic Data of Compounds 1a and 3
3.5. Antibacterial Assay
3.6. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.B.; Li, L.; Zhu, R.X.; Li, W.; Chang, W.Q.; Zhang, L.L.; Wang, X.N.; Zhao, Z.T.; Lou, H.X. Tetramic acids and pyridone alkaloids from the endolichenic fungus Tolypocladium cylindrosporum. J. Nat. Prod. 2015, 78, 2155–2160. [Google Scholar] [CrossRef]
- Shang, Z.; Li, L.; Esposito, B.P.; Salim, A.A.; Khalil, Z.G.; Quezada, M.; Bernhardt, P.V.; Capon, R.J. New PKS-NRPS tetramic acids and pyridinone from an Australian marine-derived fungus, Chaunopycnis sp. Org. Biomol. Chem. 2015, 13, 7795–7802. [Google Scholar] [CrossRef]
- Sakemi, S.; Bordner, J.; Decosta, D.L.; Dekker, K.A.; Hirai, H.; Inagaki, T.; Kim, Y.J.; Kojima, N.; Sims, J.C.; Sugie, Y. CJ-15, 696 and its analogs, new furopyridine antibiotics from the fungus Cladobotryum varium: Fermentation, isolation, structural elucidation, biotransformation and antibacterial activities. J. Antibiot. 2002, 55, 6–18. [Google Scholar] [CrossRef]
- Breinholt, J.; Jensen, H.C.; Kjær, A.; Olsen, C.E.; Rassing, B.R.; Rosendahl, C.N.; Søtofte, I. Cladobotryal: A fungal metabolite with a novel ring system. Acta Chem. Scand. 1998, 52, 631–634. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, L.; Zhu, G.; Zuo, M.; Gong, Q.; He, W.; Li, M.; Yuan, C.; Hao, X.; Zhu, W. New phenylpyridone derivatives from the Penicillium sumatrense GZWMJZ-313, a fungal endophyte of Garcinia multiflora. Chin. Chem. Lett. 2019, 30, 431–434. [Google Scholar] [CrossRef]
- Fukuda, T.; Yamaguchi, Y.; Masuma, R.; Tomoda, H.; Omura, S. Citridones, new potentiators of antifungal miconazole activity, produced by Penicillium sp. FKI-1938. I. taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2005, 58, 309–314. [Google Scholar] [CrossRef]
- Fukuda, T.; Sakabe, Y.; Tomoda, H.; Omura, S. Fungal citridone D having a novel phenylfuropyridine skeleton. Chem. Pharm. Bull. 2006, 54, 1659–1661. [Google Scholar] [CrossRef]
- Fotiadou, A.D.; Zografos, A.L. Accessing the structural diversity of pyridone alkaloids: Concise total synthesis of rac-citridone A. Org. Lett. 2011, 13, 4592–4595. [Google Scholar] [CrossRef]
- Snider, B.B.; Che, Q. Synthesis of cladobotryal, CJ16, 169, and CJ16, 170. Org. Lett. 2004, 6, 2877–2880. [Google Scholar] [CrossRef]
- Clive, D.L.; Huang, X. Model studies and first synthesis of the antifungal and antibacterial agent cladobotryal. J. Org. Chem. 2004, 69, 1872–1879. [Google Scholar] [CrossRef]
- Clive, D.L.; Huang, X. Studies related to furopyridinone antibiotics. Synthesis of 2-epi-CJ-16,170. Tetrahedron 2002, 58, 10243–10250. [Google Scholar] [CrossRef]
- Moore, M.C.; Cox, R.J.; Duffin, G.R.; O’Hagan, D. Synthesis and evaluation of a putative acyl tetramic acid intermediate in tenellin biosynthesis in Beauveria bassiana. A new role for tyrosine. Tetrahedron 1998, 54, 9195–9206. [Google Scholar] [CrossRef]
- Tomoda, H. New approaches to drug discovery for combating MRSA. Chem. Pharm. Bull. 2016, 64, 104–111. [Google Scholar] [CrossRef]
- Miyagawa, T.; Nagai, K.; Yamada, A.; Sugihara, Y.; Fukuda, T.; Fukuda, T.; Uchida, R.; Tomoda, H.; O̅mura, S.; Nagamitsu, T. Total synthesis of citridone A. Org. Lett. 2011, 13, 1158–1161. [Google Scholar] [CrossRef]
- Fukuda, T.; Shimoyama, K.; Nagamitsu, T.; Tomoda, H. Synthesis and biological activity of Citridone A and its derivatives. J. Antibiot. 2014, 67, 445–450. [Google Scholar] [CrossRef]
- Liu, H.; Tan, H.; Chen, Y.; Guo, X.; Wang, W.; Guo, H.; Liu, Z.; Zhang, W. Cytorhizins A−D, four highly structure-combined benzophenones from the endophytic fungus Cytospora rhizophorae. Org. Lett. 2019, 21, 1063–1067. [Google Scholar] [CrossRef]
- Liu, H.; Tan, H.; Wang, W.; Zhang, W.; Chen, Y.; Li, S.; Liu, Z.; Li, H.; Zhang, W. Cytorhizophins A and B, benzophenone-hemiterpene adducts from the endophytic fungus Cytospora rhizophorae. Org. Chem. Front. 2019, 6, 591–596. [Google Scholar] [CrossRef]
- Xu, J.; Tan, H.; Chen, Y.; Li, S.; Huang, Z.; Guo, H.; Li, H.; Gao, X.; Liu, H.; Zhang, W. Lithocarpins A–D: Four tenellone-macrolide conjugated [4 + 2] hetero-adducts from the deep-sea derived fungus Phomopsis lithocarpus FS508. Org. Chem. Front. 2018, 5, 1792–1797. [Google Scholar] [CrossRef]
- Chen, S.; Li, H.; Chen, Y.; Li, S.; Xu, J.; Guo, H.; Liu, Z.; Zhu, S.; Liu, H.; Zhang, W. Three new diterpenes and two new sesquiterpenoids from the endophytic fungus Trichoderma koningiopsis A729. Bioorg. Chem. 2019, 86, 368–374. [Google Scholar] [CrossRef]
- Fukuda, T.; Tomoda, H.; Omura, S. Citridones, new potentiators of antifungal miconazole activity, produced by Penicillium sp. FKI-1938: II. structure elucidation. J. Antibiot. 2005, 58, 315–321. [Google Scholar] [CrossRef]
- Drummond, A.J.; Waigh, R.D. Recent Research Developments in Phytochemistry; Pandalai, S.G., Ed.; Research Signpost: Thiruvananthapuram, Kerala, India, 2000; Volume 4, pp. 143–152. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer. Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | No. | 2 | ||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 105.1, C | 2 | 104.7, C | ||
| 3 | 3.22 (d, 1.8) | 57.9, CH | 3 | 3.28 (br s) | 58.0, CH |
| 3a | 114.8, C | 3a | 114.8, C | ||
| 4 | 163.2, C | 4 | 163.3, C | ||
| 6 | 7.34 (s) | 134.1, CH | 6 | 7.44 (s) | 135.1, CH |
| 7 | 113.3, C | 7 | 113.0, C | ||
| 7a | 166.7, C | 7a | 166.5, C | ||
| 8 | 125.6, C | 8 | 134.5, C | ||
| 9 | 7.32 (d, 8.7) | 130.0, CH | 9 | 7.50 (d, 7.7) | 128.7, CH |
| 10 | 6.79 (d, 8.6) | 116.3, CH | 10 | 7.37 (t, 7.7) | 129.5, CH |
| 11 | 158.2, C | 11 | 7.29 (t, 7.7) | 128.5, CH | |
| 12 | 6.79 (d, 8.6) | 116.3, CH | 12 | 7.37 (t, 7.7) | 129.5, CH |
| 13 | 7.32 (d, 8.7) | 130.0, CH | 13 | 7.50 (d, 7.7) | 128.7, CH |
| 1′ | 5.40 (s) | 127.4, CH | 1′ | 5.65 (s) | 126.3, CH |
| 2′ | 151.7, C | 2′ | 155.4, C | ||
| 3′ | 2.80 (q, 7.4) | 50.6, CH | 3′ | 3.00 (q, 7.4) | 47.0, CH |
| 4′ | 1.27 (d, 7.2) | 20.5, CH3 | 4′ | 1.29 (d, 6.9) | 20.8, CH3 |
| 5′ | 1.74 (s) | 14.8, CH3 | 5′α 5′β | 4.10 (d, 15.3) 4.18 (d, 15.3) | 60.2, CH2 |
| 6′ | 1.63 (s) | 26.5, CH3 | 6′ | 1.68 (s) | 26.3, CH3 |
| No. | 3 | No. | 4 | ||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 108.2, C | 2 | 5.23 (s) | 87.3, CH | |
| 3 | 3.49 (d, 1.8) | 53.2, CH | 3 | 129.7, C | |
| 3a | 115.3, C | 4 | 6.50 (s) | 115.1, CH | |
| 4 | 163.2, C | 4a | 108.3, C | ||
| 6 | 7.44 (s) | 135.0, CH | 5 | 162.5, C | |
| 7 | 113.1, C | 7 | 7.21 (s) | 134.6, CH | |
| 7a | 167.1, C | 8 | 116.6, C | ||
| 8 | 134.6, C | 8a | 160.8, C | ||
| 9 | 7.54 (d, 7.1) | 128.9, CH | 9 | 134.9, C | |
| 10 | 7.37 (t, 7.7) | 129.5, CH | 10 | 7.36 (overlapped) | 129.3, CH |
| 11 | 7.29 (t, 7.4) | 128.4, CH | 11 | 7.36 (overlapped) | 130.0, CH |
| 12 | 7.37 (t, 7.7) | 129.5, CH | 12 | 7.30 (t, 7.5) | 128.5, CH |
| 13 | 7.54 (d, 7.1) | 128.9, CH | 13 | 7.36 (overlapped) | 130.0, CH |
| 1′ | 5.39 (s) | 123.7, CH | 14 | 7.36 (overlapped) | 129.3, CH |
| 2′ | 154.4, C | 1′ | 1.64 (overlapped) | 11.7, CH3 | |
| 3′ | 2.83 (q, 7.3) | 50.5, CH | 2′ | 133.9, C | |
| 4′ | 1.29 (d, 7.2) | 20.4, CH3 | 3′ | 5.61 (q, 6.2) | 126.2, CH |
| 5′ | 1.78 (s) | 15.1, CH3 | 4′ | 1.64 (overlapped) | 13.3, CH3 |
| 6′α 6′β | 3.67 (d, 12.2) 3.89 (d, 12.2) | 66.7, CH2 | 5′ | 1.72 (s) | 19.6, CH3 |
| No. | 5 | No. | 6 | ||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 105.3, C | 1 | 211.7, C | ||
| 3 | 3.23 (d, 1.8) | 57.8, CH | 2 | 134.3, C | |
| 3a | 114.9, C | 3 | 174.6, C | ||
| 4 | 166.6, C | 4 | 2.14 (d, 18.1) 3.68 (d, 18.1) | 43.8, CH2 | |
| 5 | 113.0, C | 5 | 57.4, C | ||
| 6 | 7.43 (s) | 135.0, CH | 6 | 175.0, C | |
| 7a | 163.3, C | 7 | 76.1, C | ||
| 8 | 134.5, C | 8 | 209.0, C | ||
| 9 | 7.49 (d, 7.0) | 128.7, CH | 9 | 4.13 (d, 2.9) | 68.7, CH |
| 10 | 7.36 (t, 7.7) | 129.5, CH | 10 | 5.14 (d, 2.9) | 72.4, CH |
| 11 | 7.29 (t, 7.4) | 128.4, CH | 11 | 142.3, C | |
| 12 | 7.36 (t, 7.7) | 129.5, CH | 12 | 7.39 (d, 6.9) | 127.1, CH |
| 13 | 7.49 (d, 7.0) | 128.7, CH | 13 | 7.37 (t, 6.9) | 129.6, CH |
| 1′ | 2.80 (q, 7.2) | 50.5, CH | 14 | 7.28 (t, 6.9) | 128.7, CH |
| 2′ | 151.8, C | 15 | 7.37 (t, 6.9) | 129.6, CH | |
| 3′ | 5.40 (s) | 127.3, CH | 16 | 7.39 (d, 6.9) | 127.1, CH |
| 4′ | 1.63 (s) | 26.4, CH3 | 17 | 1.57 (s) | 7.8, CH3 |
| 5′ | 1.74 (s) | 14.8, CH3 | 18 | 2.04 (s) | 17.2, CH3 |
| 6′ | 1.27 (d, 7.2) | 20.5, CH3 | 19 | 1.25 (s) | 19.0, CH3 |
| No. | 7 | No. | 7 | ||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 156.4, C | 8 | 133.9, C | ||
| 3 | 6.66 (s) | 103.3, CH | 9 | 7.70 (d, 7.5) | 128.6, CH |
| 3a | 118.4, C | 10 | 7.46 (t, 7.5) | 129.9, CH | |
| 4 | 159.6, C | 11 | 7.38 (t, 7.5) | 128.9, CH | |
| 6 | 7.43 (s) | 128.6, CH | 12 | 7.46 (t, 7.5) | 129.9, CH |
| 7 | 113.2, C | 13 | 7.70 (d, 7.5) | 128.6, CH | |
| 7a | 161.6, C | 1′ | 2.48 (s) | 13.6, CH3 | |
| Compounds | MIC (μg/mL) | Compounds | MIC (μg/mL) | ||
|---|---|---|---|---|---|
| S. aureus (ATCC 29213) | E. coli (ATCC 8739) | S. aureus (ATCC 29213) | E. coli (ATCC 8739) | ||
| 1–3 | >500 | >500 | 6–7 | >500 | >500 |
| (+)-4a | 125 | >500 | (−)-8a | 62.5 | >500 |
| (−)-4b | 125 | >500 | (+)-8b | 31.5 | >500 |
| (−)-5a | 62.5 | >500 | 9 | 250 | >500 |
| (+)-5b | 31.2 | >500 | Van | 1.8 | 125 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-C.; Liu, Z.-M.; Tan, H.-B.; Chen, Y.-C.; Li, S.-N.; Li, H.-H.; Guo, H.; Zhu, S.; Liu, H.-X.; Zhang, W.-M. Tersone A-G, New Pyridone Alkaloids from the Deep-Sea Fungus Phomopsis tersa. Mar. Drugs 2019, 17, 394. https://doi.org/10.3390/md17070394
Chen S-C, Liu Z-M, Tan H-B, Chen Y-C, Li S-N, Li H-H, Guo H, Zhu S, Liu H-X, Zhang W-M. Tersone A-G, New Pyridone Alkaloids from the Deep-Sea Fungus Phomopsis tersa. Marine Drugs. 2019; 17(7):394. https://doi.org/10.3390/md17070394
Chicago/Turabian StyleChen, Shan-Chong, Zhao-Ming Liu, Hai-Bo Tan, Yu-Chan Chen, Sai-Ni Li, Hao-Hua Li, Heng Guo, Shuang Zhu, Hong-Xin Liu, and Wei-Min Zhang. 2019. "Tersone A-G, New Pyridone Alkaloids from the Deep-Sea Fungus Phomopsis tersa" Marine Drugs 17, no. 7: 394. https://doi.org/10.3390/md17070394
APA StyleChen, S.-C., Liu, Z.-M., Tan, H.-B., Chen, Y.-C., Li, S.-N., Li, H.-H., Guo, H., Zhu, S., Liu, H.-X., & Zhang, W.-M. (2019). Tersone A-G, New Pyridone Alkaloids from the Deep-Sea Fungus Phomopsis tersa. Marine Drugs, 17(7), 394. https://doi.org/10.3390/md17070394