Whole Genome Sequencing of the Blue Tilapia (Oreochromis aureus) Provides a Valuable Genetic Resource for Biomedical Research on Tilapias

 , ,
, ,
Abstract
:1. Introduction
2. Results
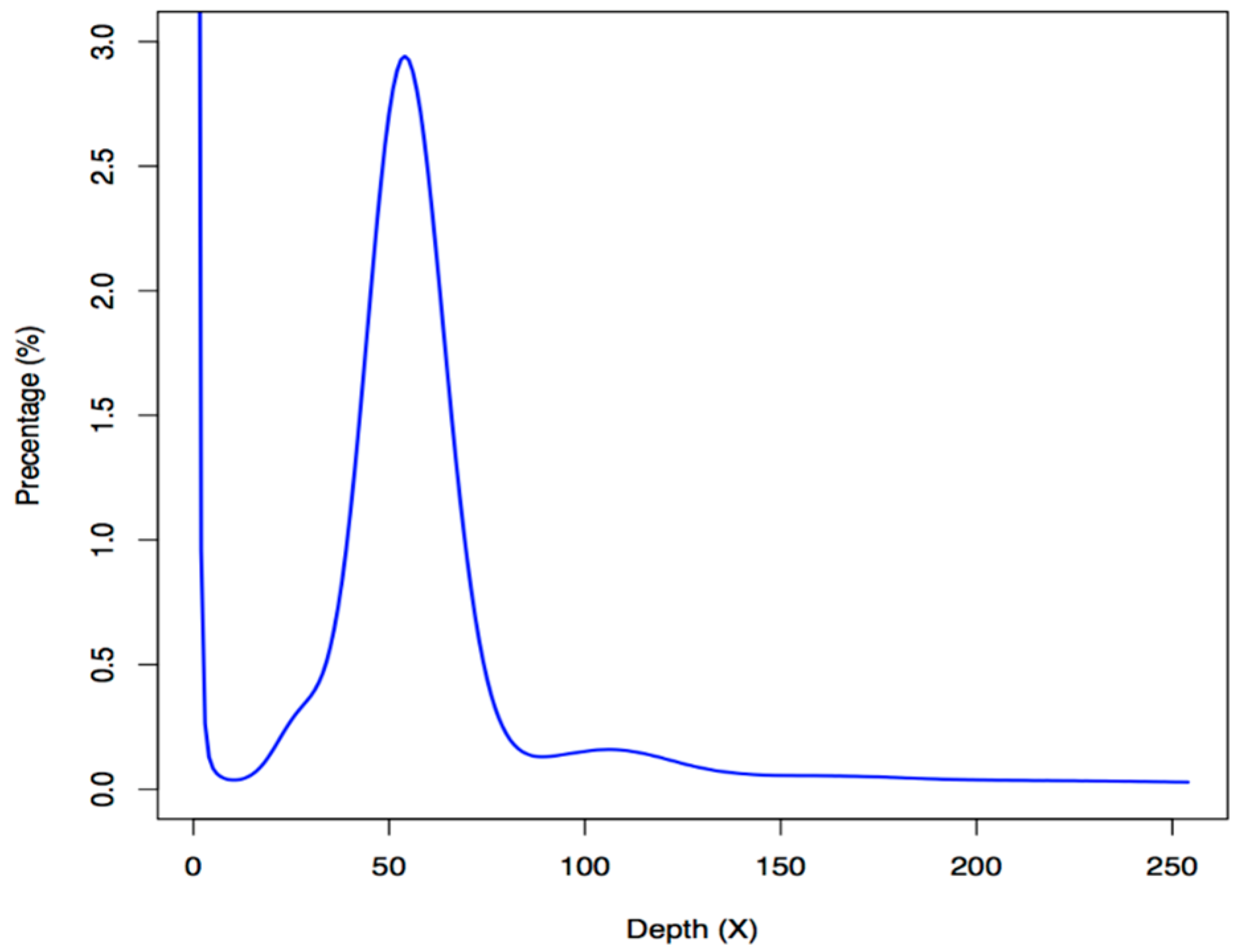
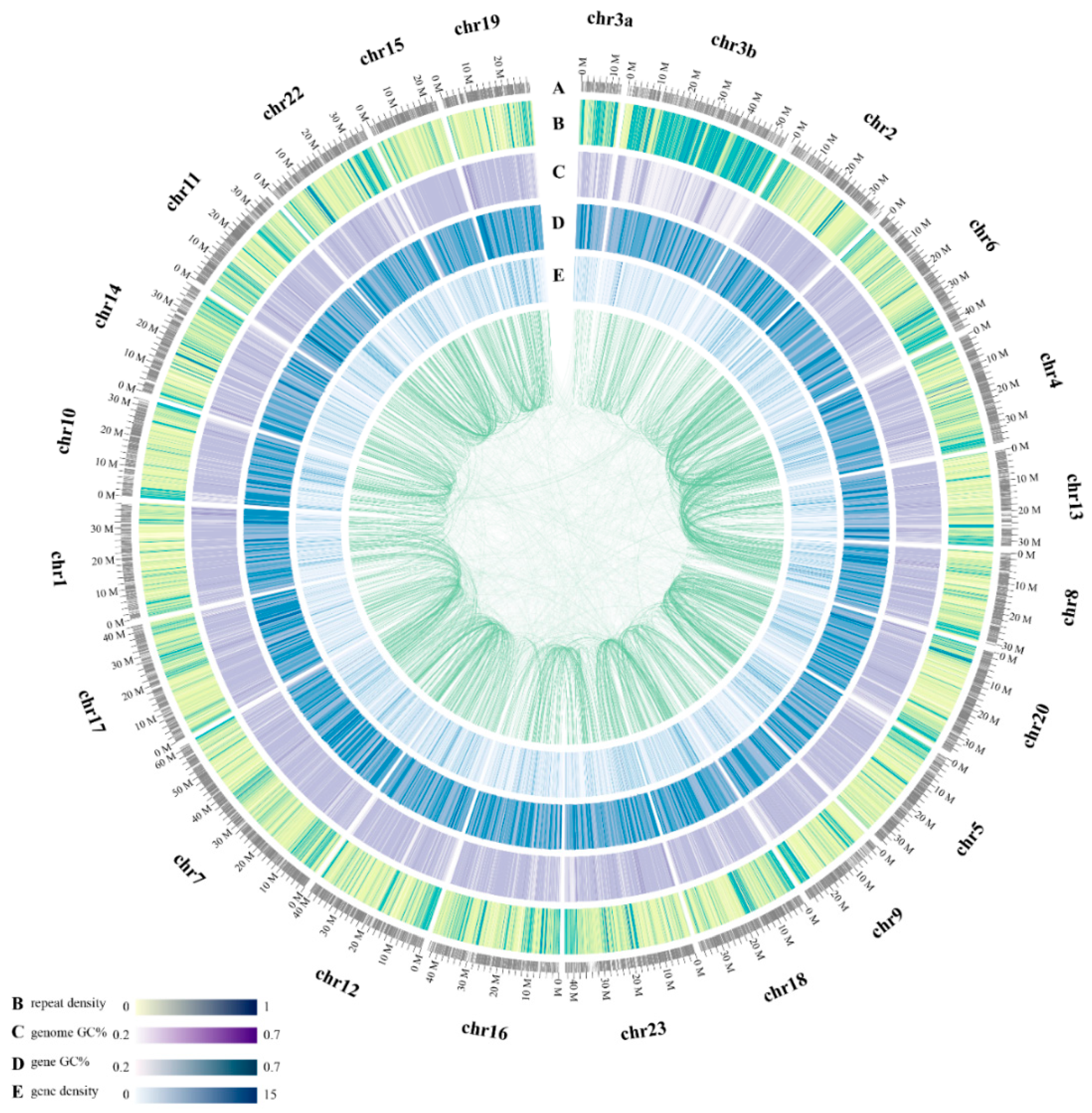
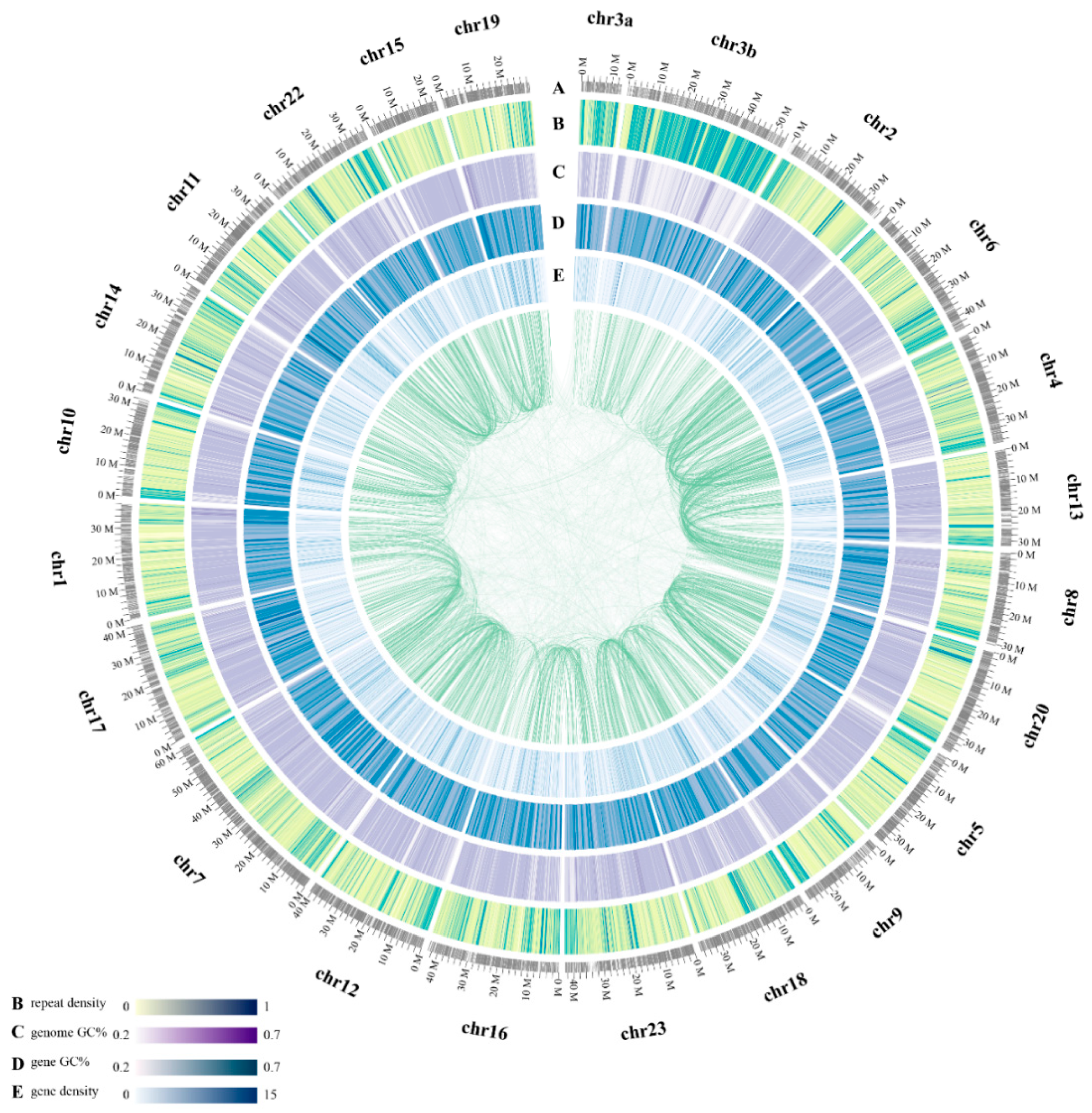
2.1. Statistics of Genome Assembly and Annotation
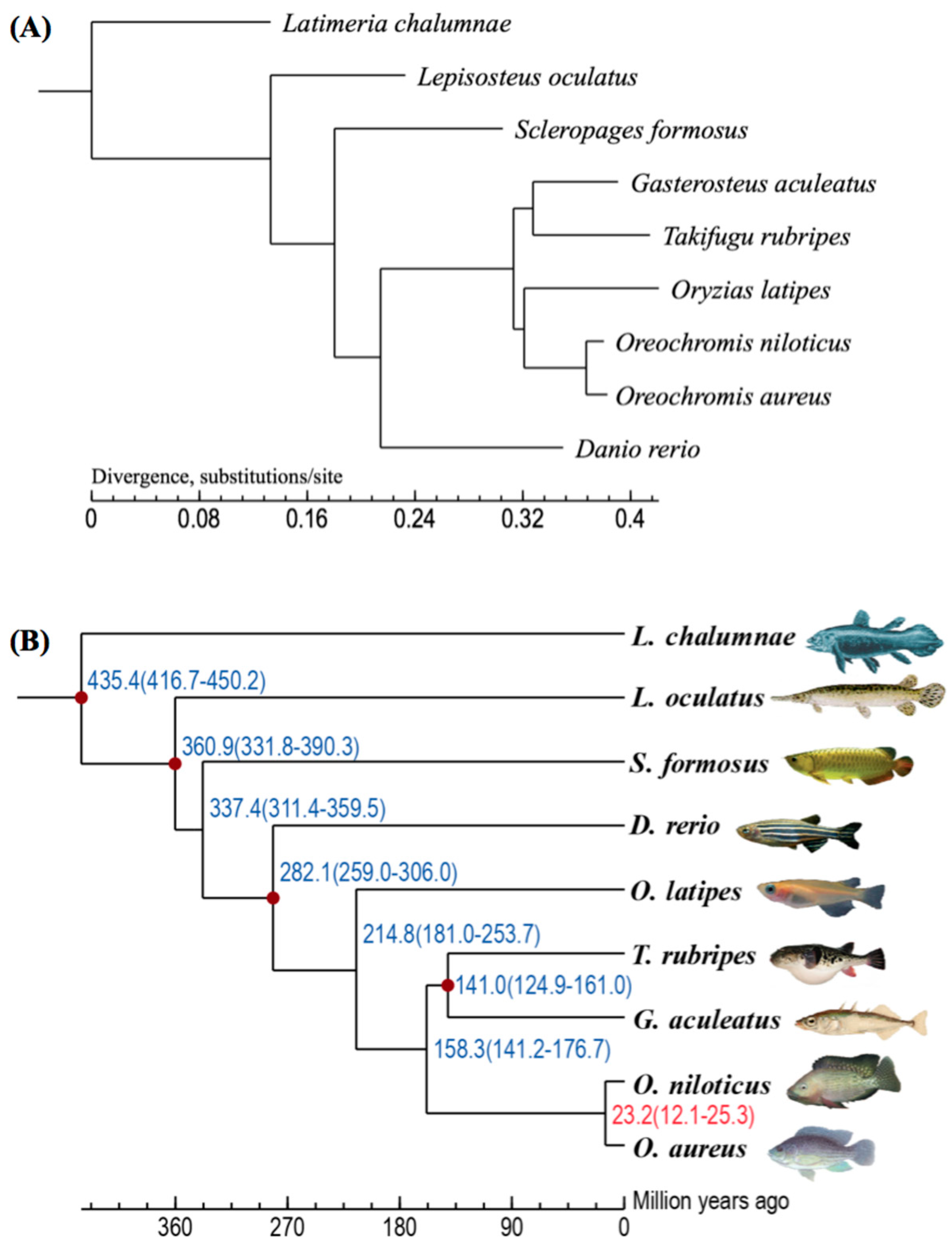
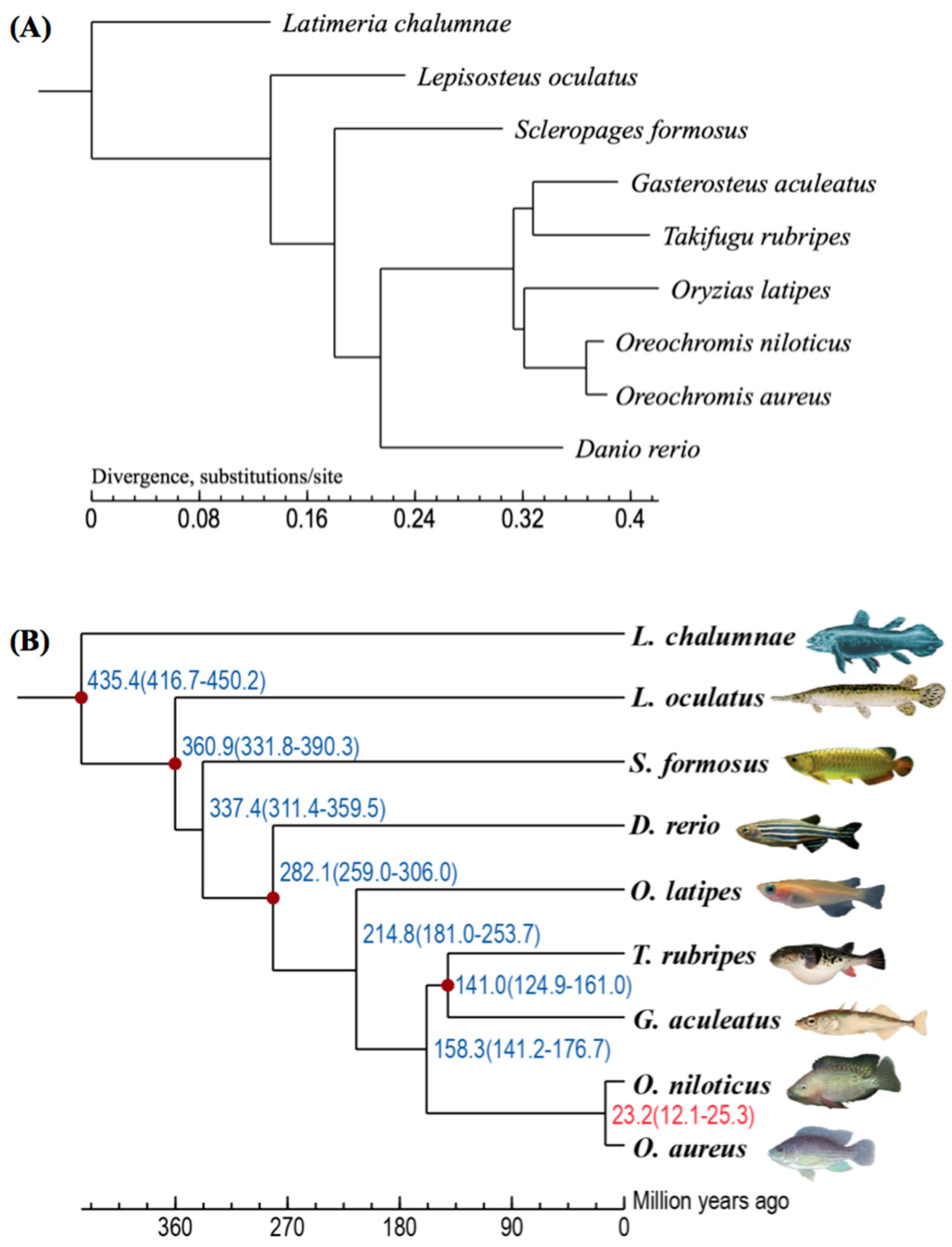
2.2. Summary of Gene Clustering and Phylogeny
2.3. Whole-Genome Chromosomal Evolution
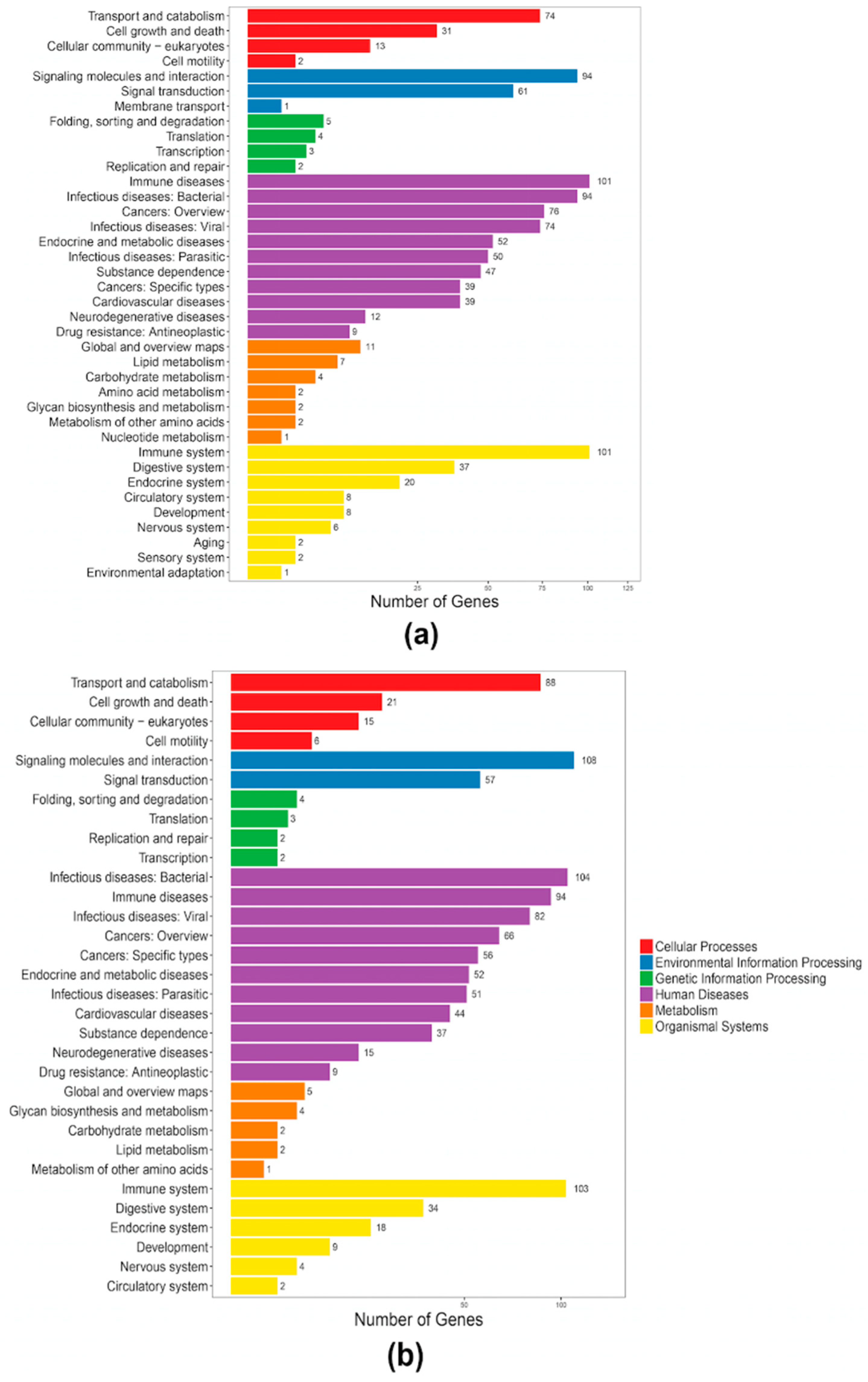
2.4. Antimicrobial Peptides in Both the Blue and Nile Tilapias
3. Discussion
3.1. High-Throughput Screening of AMPs from Our High-Quality Genome Assembly
3.2. Comparisons of AMPs between the Blue and Nile Tilapias
4. Materials and Methods
4.1. Sample Preparation and Sequencing
4.2. Estimation of Genome Size
4.3. Genome Assembly and Annotation
4.4. Constructions of the Phylogenetic and Divergence Time Trees
4.5. Chromosomal Localization of the Blue Tilapia Sequences
4.6. Reconstruction of the Ancestral Genome for Examination of Whole-Genome Chromosomal Evolution
4.7. High-Throughput Identification of Antimicrobial Peptides
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | antimicrobial peptide |
| BUSCO | Benchmarking Universal Single-copy Orthologs |
| Gb | gigabase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| N50 | 50% of the genome is in fragments of this length or longer |
| WGD | whole genome duplication |
References
- Gupta, M.V.; Acosta, B.O. A review of global tilapia farming practices. Aquac. Asia 2004, 9, 7–16. [Google Scholar]
- Chapman, F.A. Culture of Hybrid Tilapia: A Reference Profile. University of Florida IFAS Extension. Available online: http://edis.ifas.ufl.edu/pdffiles/FA/FA01200.pdf (accessed on 29 April 2019).
- Kirk, R.G. A review of recent developments in tilapia culture, with special reference to fish farming in the heated effluents of power stations. Aquaculture 1972, 1, 45–60. [Google Scholar] [CrossRef]
- Schramm, H.L.; Zale, A.V. Effects of cover and prey size on preferences of juvenile largemouth bass for blue tilapias and bluegills in tanks. T. Am. Fish. Soc. 1985, 114, 725–731. [Google Scholar] [CrossRef]
- Suresh, A.V.; Lin, C.K. Tilapia culture in saline waters: A review. Aquaculture 1992, 106, 201–226. [Google Scholar] [CrossRef]
- Peterson, M.S.; Slack, W.T.; Waggy, G.L.; Finley, J.; Woodley, C.M.; Partyka, M.L. Foraging in non-native environments: Comparison of Nile tilapia and three co-occurring native centrarchids in invaded coastal Mississippi watersheds. Environ. Biol. Fish 2006, 76, 283–301. [Google Scholar] [CrossRef]
- Hickling, C.F. The Malacca tilapia hybrid. J. Genet. 1960, 57, 1–10. [Google Scholar] [CrossRef]
- Pruginin, Y.; Rothbard, S.; Wohlfarth, G.; Halevy, A.; Moav, R.; Hulata, G. All-male broods of Tilapia nilotica × T. aurea hybrids. Aquaculture 1975, 6, 11–21. [Google Scholar] [CrossRef]
- Mair, G.C.; Scott, A.G.; Penman, D.J.; Beardmore, J.A.; Skibinski, D.O. Sex determination in the genus Oreochromis: 1. Sex reversal, gynogenesis and triploidy in O. niloticus (L.). Theor. Appl. Genet. 1991, 82, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Müller-Belecke, A.; Hörstgen-Schwark, G. Sex determination in tilapia (Oreochromis niloticus) sex ratios in homozygous gynogenetic progeny and their offspring. Aquaculture 1995, 137, 57–65. [Google Scholar]
- Myers, J.M.; Penman, D.J.; Basavaraju, Y.; Powell, S.F.; Baoprasertkul, P.; Rana, K.J.; Bromage, N.; McAndrew, B.J. Induction of diploid androgenetic and mitotic gynogenetic Nile tilapia (Oreochromis niloticus L.). Theor. Appl. Genet. 1995, 90, 205–210. [Google Scholar] [CrossRef]
- Baroiller, J.F.; Chourrout, D.; Fostier, A.; Jalabert, B. Temperature and sex chromosomes govern sex ratios of the mouthbrooding Cichlid fish Oreochromis niloticus. J. Exp. Zool. 1995, 273, 216–223. [Google Scholar] [CrossRef]
- Baroiller, J.F.; D’Cotta, H.; Bezault, E.; Wessels, S.; Hoerstgen-Schwark, G. Tilapia sex determination: Where temperature and genetics meet. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2009, 153, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Cnaani, A.; Lee, B.Y.; Zilberman, N.; Ozouf-Costaz, C.; Hulata, G.; Ron, M.; D’Hont, A.; Baroiller, J.F.; D’Cotta, H.; Penman, D.J.; et al. Genetics of sex determination in tilapiine species. Sex. Devel. 2008, 2, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.A.; Gammerdinger, W.J.; Bartie, K.L.; Penman, D.J.; Kocher, T.D. A high quality assembly of the Nile Tilapia (Oreochromis niloticus) genome reveals the structure of two sex determination regions. BMC Genom. 2017, 18, 341. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Erratum: SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2015, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.M.; Brunet, F.; Petit, J.L.; Stange-Thomann, N.; Mauceli, E.; Bouneau, L.; Fischer, C.; Ozouf-Costaz, C.; Bernot, A.; et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 2004, 431, 946–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, C.; Hu, Y.; Ravi, V.; Kuznetsova, I.S.; Shen, X.; Mu, X.; Sun, Y.; You, X.; Li, J.; Li, X.; et al. The Asian arowana (Scleropages formosus) genome provides new insights into the evolution of an early lineage of teleosts. Sci. Rep. 2016, 6, 24501. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; You, X.; Bian, C.; Chen, S.; Lv, Z.; Qiu, L.; Shi, Q. High-throughput identification of antimicrobial peptides from amphibious mudskippers. Mar. Drugs 2017, 15, 364. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-Q.; Li, S.-F.; Ma, J.-Y. Diseases resistance of Nile tilapia (Oreochromis niloticus), blue tilapia (Oreochromis aureus) and their hybrid (female Nile tilapia×male blue tilapia) to Aeromonas sobria. Aquaculture 2004, 229, 79–87. [Google Scholar] [CrossRef]
- Nair, D.G.; Fry, B.G.; Alewood, P.; Kumar, P.P.; Kini, R.M. Antimicrobial activity of omwaprin, a new member of the waprin family of snake venom proteins. Biochem. J. 2007, 402, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Prajanban, B.O.; Jangpromma, N.; Araki, T.; Klaynongsruang, S. Antimicrobial effects of novel peptides cOT2 and sOT2 derived from Crocodylus siamensis and Pelodiscus sinensis ovotransferrins. Biochim. Biophys. Acta Biomembr. 2017, 1859, 860–869. [Google Scholar] [CrossRef]
- Iordache, F.; Ionita, M.; Mitrea, L.I.; Fafaneata, C.; Pop, A. Antimicrobial and antiparasitic activity of lectins. Curr. Pharm. Biotechnol. 2015, 16, 152–161. [Google Scholar] [CrossRef]
- Takahashi, K.G.; Kuroda, T.; Muroga, K. Purification and antibacterial characterization of a novel isoform of the Manila clam lectin (MCL-4) from the plasma of the Manila clam, Ruditapes philippinarum. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 45–52. [Google Scholar] [CrossRef]
- Zhao, L.L.; Wang, Y.Q.; Dai, Y.J.; Zhao, L.J.; Qin, Q.; Lin, L.; Ren, Q.; Lan, J.F. A novel C-type lectin with four CRDs is involved in the regulation of antimicrobial peptide gene expression in Hyriopsis cumingii. Fish Shellfish Immunol. 2016, 55, 339–347. [Google Scholar] [CrossRef]
- Ullal, A.J.; Noga, E.J. Antiparasitic activity of the antimicrobial peptide HbbetaP-1, a member of the beta-haemoglobin peptide family. J. Fish Dis. 2010, 33, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.K.; Lee, M.J.; Jung, H.G.; Go, H.J.; Kim, Y.J.; Park, N.G. Antimicrobial function of SHbetaAP, a novel hemoglobin beta chain-related antimicrobial peptide, isolated from the liver of skipjack tuna, Katsuwonus pelamis. Fish Shellfish Immunol. 2014, 37, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.G.; Wei, J.G.; Xu, D.; Cui, H.C.; Yan, Y.; Ou-Yang, Z.L.; Huang, X.H.; Huang, Y.H.; Qin, Q.W. Molecular cloning and characterization of two novel hepcidins from orange-spotted grouper, Epinephelus coioides. Fish Shellfish Immunol. 2011, 30, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Chen, J.Y.; Kuo, C.M. Three different hepcidins from tilapia, Oreochromis mossambicus: Analysis of their expressions and biological functions. Mol. Immunol. 2007, 44, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bian, C.; Luo, Y.; Wang, L.; You, X.; Li, J.; Qiu, Y.; Ma, X.; Zhu, Z.; Ma, L.; et al. Draft genome of the Chinese mitten crab, Eriocheir sinensis. GigaScience 2016, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, K.; Chen, S.; Zhang, Z.; Zhang, J.; You, X.; Bian, C.; Xu, J.; Jia, C.; Qiang, J.; et al. Draft genome of the protandrous Chinese black porgy, Acanthopagrus schlegelii. Gigascience 2018, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 2013, 35, 62–67. [Google Scholar]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Mackey, A.J.; Reese, J.T.; Milshina, N.V.; Roos, D.S.; Weinstock, G.M. Creating a honey bee consensus gene set. Genome Biol. 2007, 8, R13. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006, 23, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Kent, W.J.; Smit, A.; Zhang, Z.; Baertsch, R.; Hardison, R.C.; Haussler, D.; Miller, W. Human-mouse alignments with BLASTZ. Genome Res. 2003, 13, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Beitz, E. TEXshade: Shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics 2000, 16, 135–139. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Blue Tilapia | Nile Tilapia [15] |
|---|---|---|
| Genome Assembly | ||
| Contig N50 size (kb) | 53.2 | 3.11 |
| Scaffold N50 size (Mb) | 1.10 | - |
| Estimated genome size (Gb) | 1.02 | 1.20 |
| Assembled genome size (Gb) | 0.92 | 1.01 |
| Genome annotation | ||
| Protein-coding gene number | 23,117 | 29,249 |
| Annotated functional gene number | 22,573 | - |
| Unannotated functional gene number | 544 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, C.; Li, J.; Lin, X.; Chen, X.; Yi, Y.; You, X.; Zhang, Y.; Lv, Y.; Shi, Q. Whole Genome Sequencing of the Blue Tilapia (Oreochromis aureus) Provides a Valuable Genetic Resource for Biomedical Research on Tilapias. Mar. Drugs 2019, 17, 386. https://doi.org/10.3390/md17070386
Bian C, Li J, Lin X, Chen X, Yi Y, You X, Zhang Y, Lv Y, Shi Q. Whole Genome Sequencing of the Blue Tilapia (Oreochromis aureus) Provides a Valuable Genetic Resource for Biomedical Research on Tilapias. Marine Drugs. 2019; 17(7):386. https://doi.org/10.3390/md17070386
Chicago/Turabian StyleBian, Chao, Jia Li, Xueqiang Lin, Xiyang Chen, Yunhai Yi, Xinxin You, Yiping Zhang, Yunyun Lv, and Qiong Shi. 2019. "Whole Genome Sequencing of the Blue Tilapia (Oreochromis aureus) Provides a Valuable Genetic Resource for Biomedical Research on Tilapias" Marine Drugs 17, no. 7: 386. https://doi.org/10.3390/md17070386
APA StyleBian, C., Li, J., Lin, X., Chen, X., Yi, Y., You, X., Zhang, Y., Lv, Y., & Shi, Q. (2019). Whole Genome Sequencing of the Blue Tilapia (Oreochromis aureus) Provides a Valuable Genetic Resource for Biomedical Research on Tilapias. Marine Drugs, 17(7), 386. https://doi.org/10.3390/md17070386