Application of Bioactive Thermal Proteome Profiling to Decipher the Mechanism of Action of the Lipid Lowering 132-Hydroxy-pheophytin Isolated from a Marine Cyanobacteria

 ,
,  ,
,
Abstract
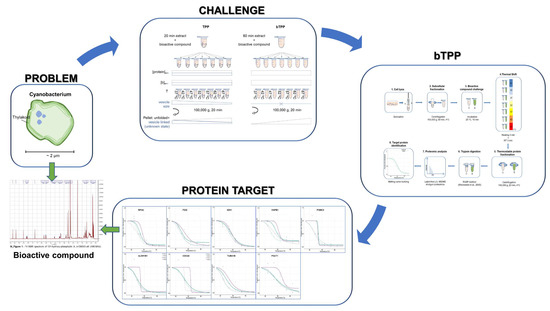
1. Introduction
2. Results
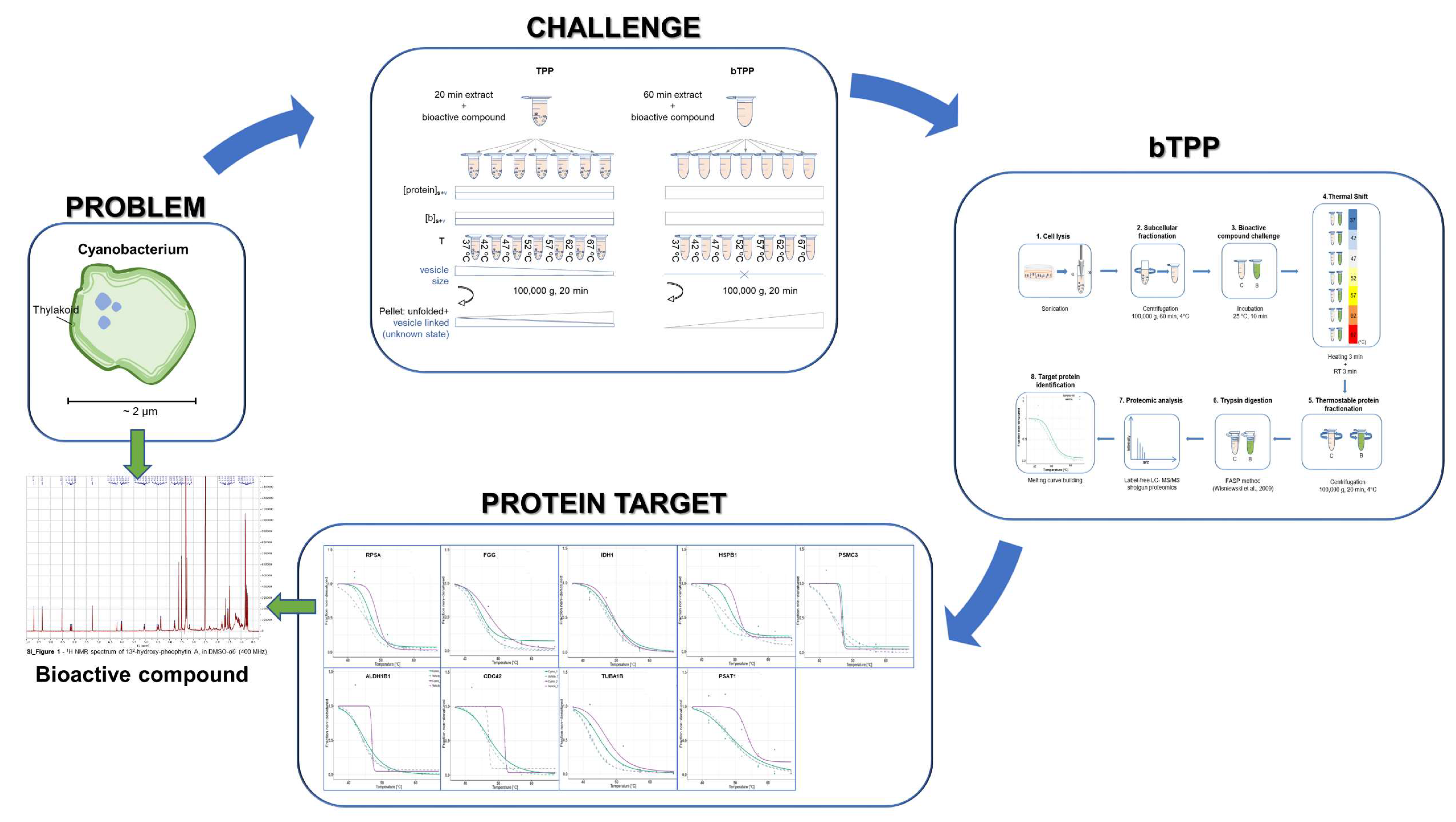
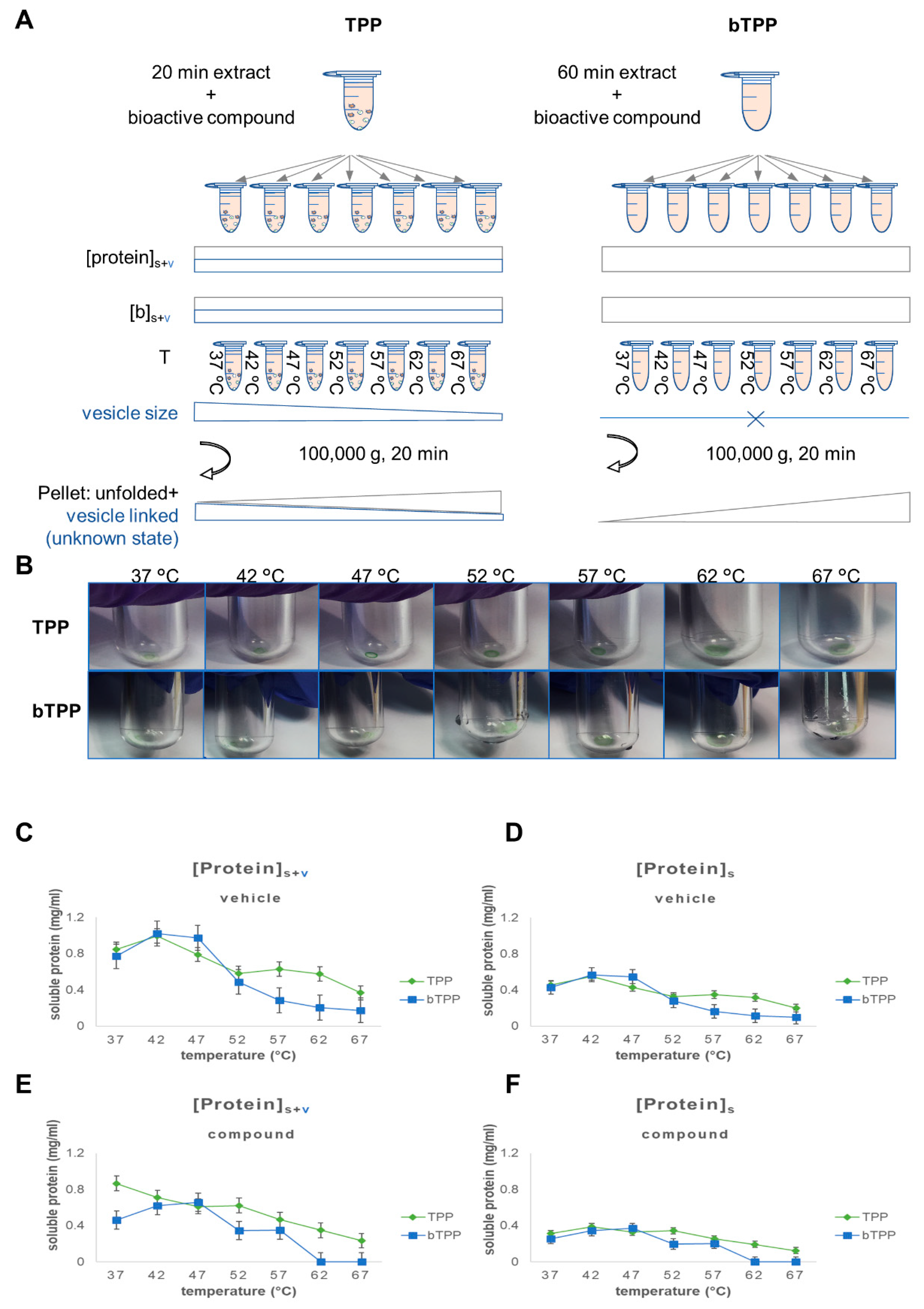
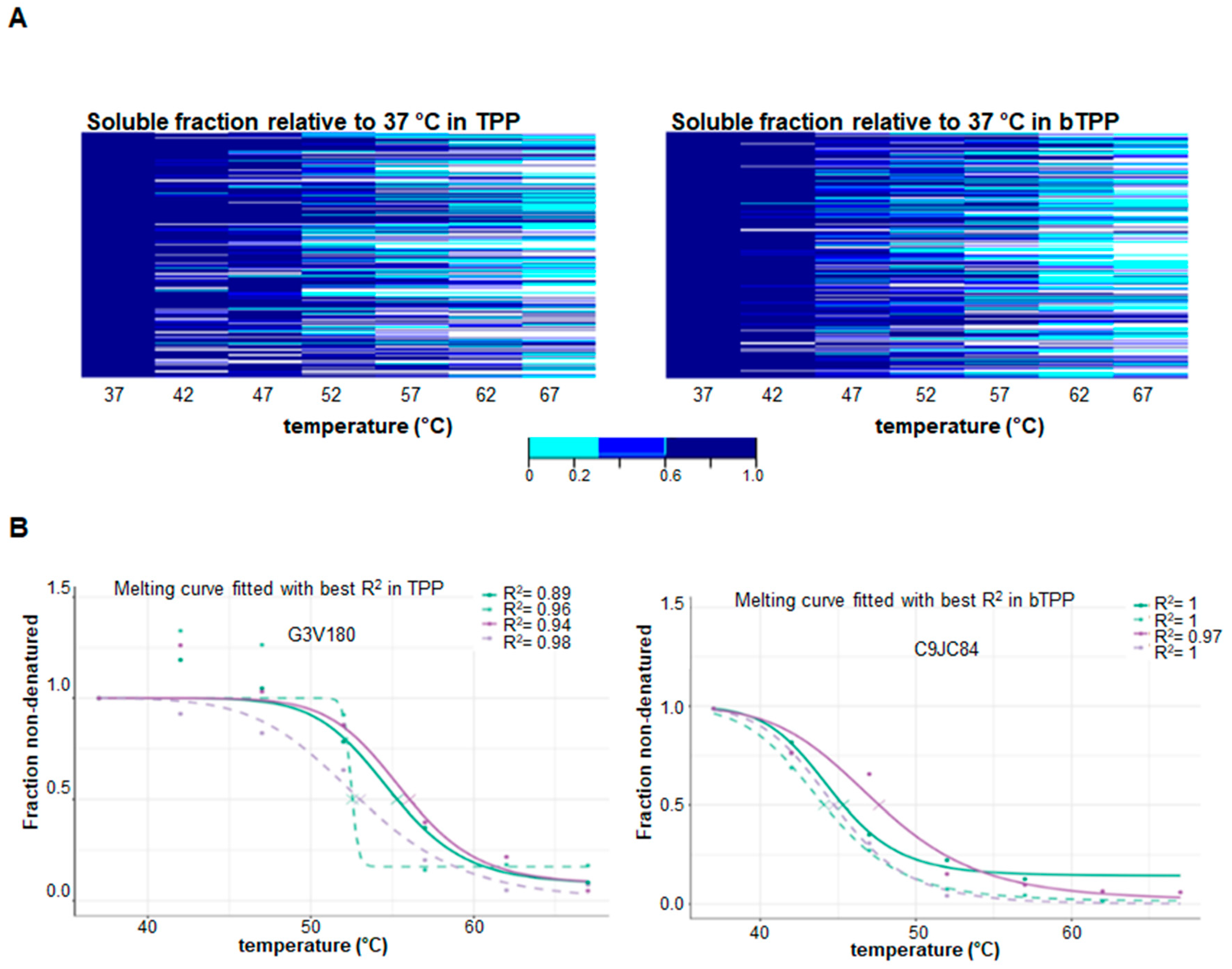
2.1. Implementation of the Novel Methodology, bTPP
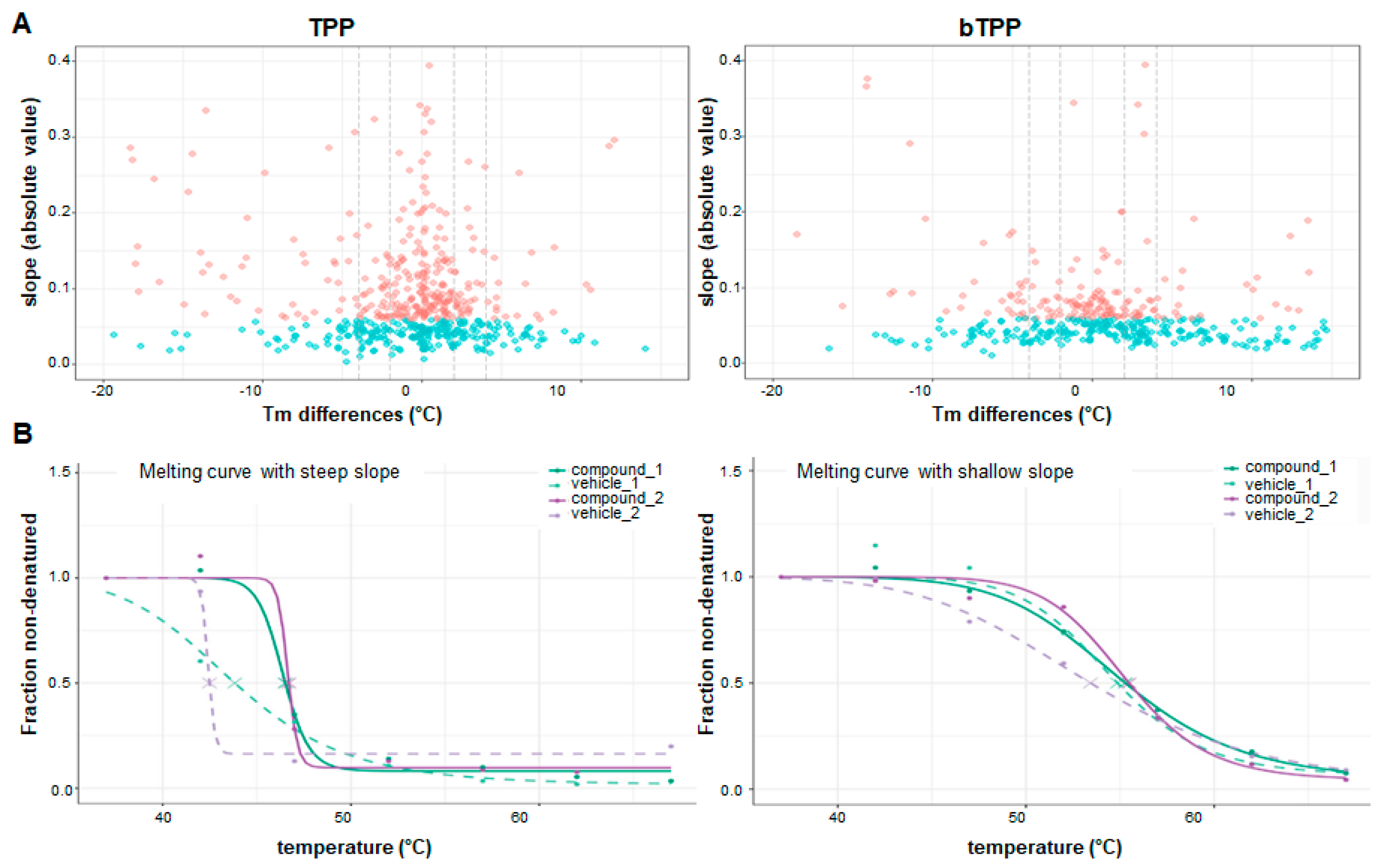
2.2. Comparative Analysis of Protein Targets Applying TPP and bTPP
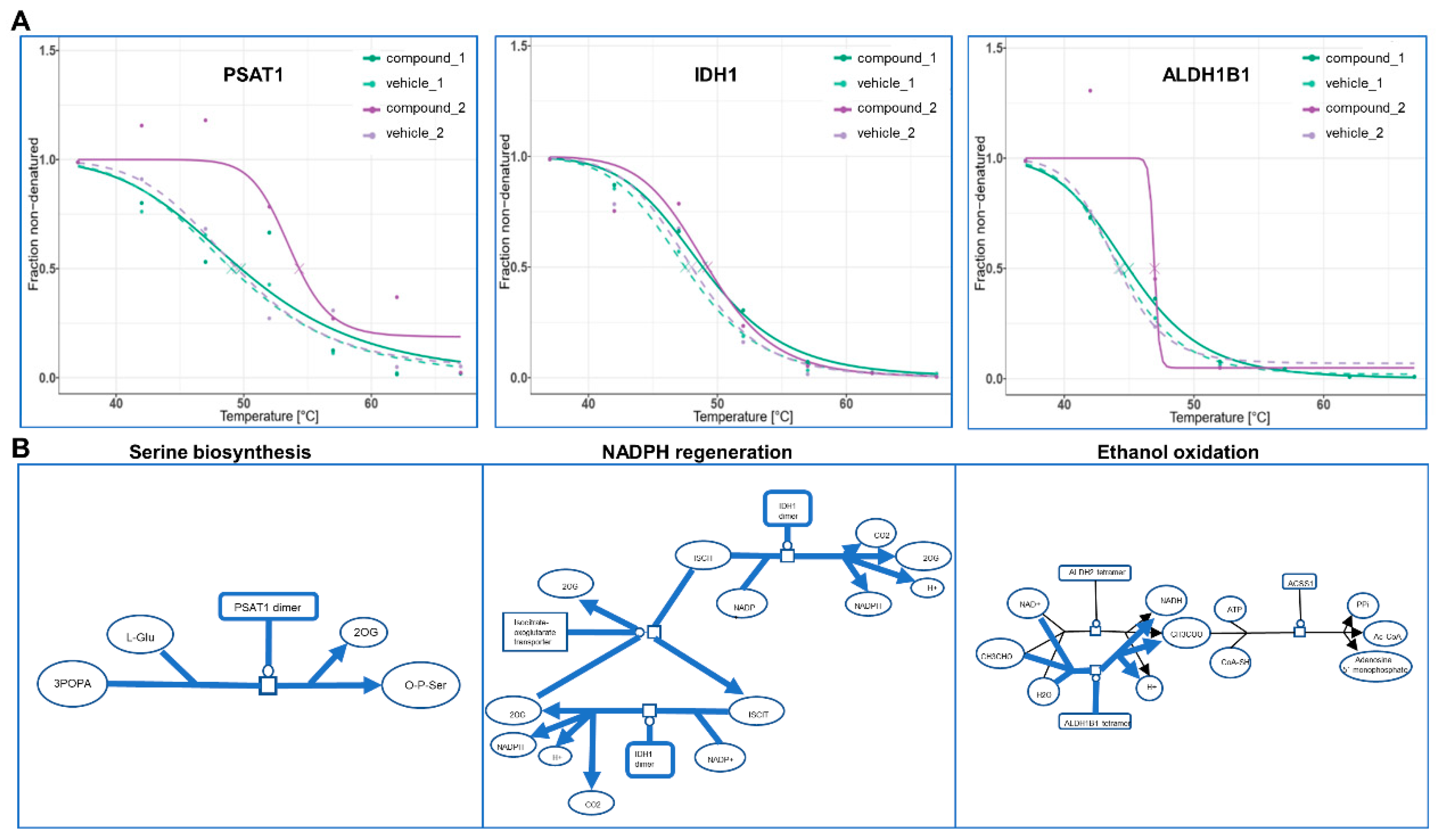
2.3. Deciphering the MOAs for 132-Hydroxypheophytine a by bTPP
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Culture
4.2. Selection of 132-Hydroxypheophytine a as Test Compound
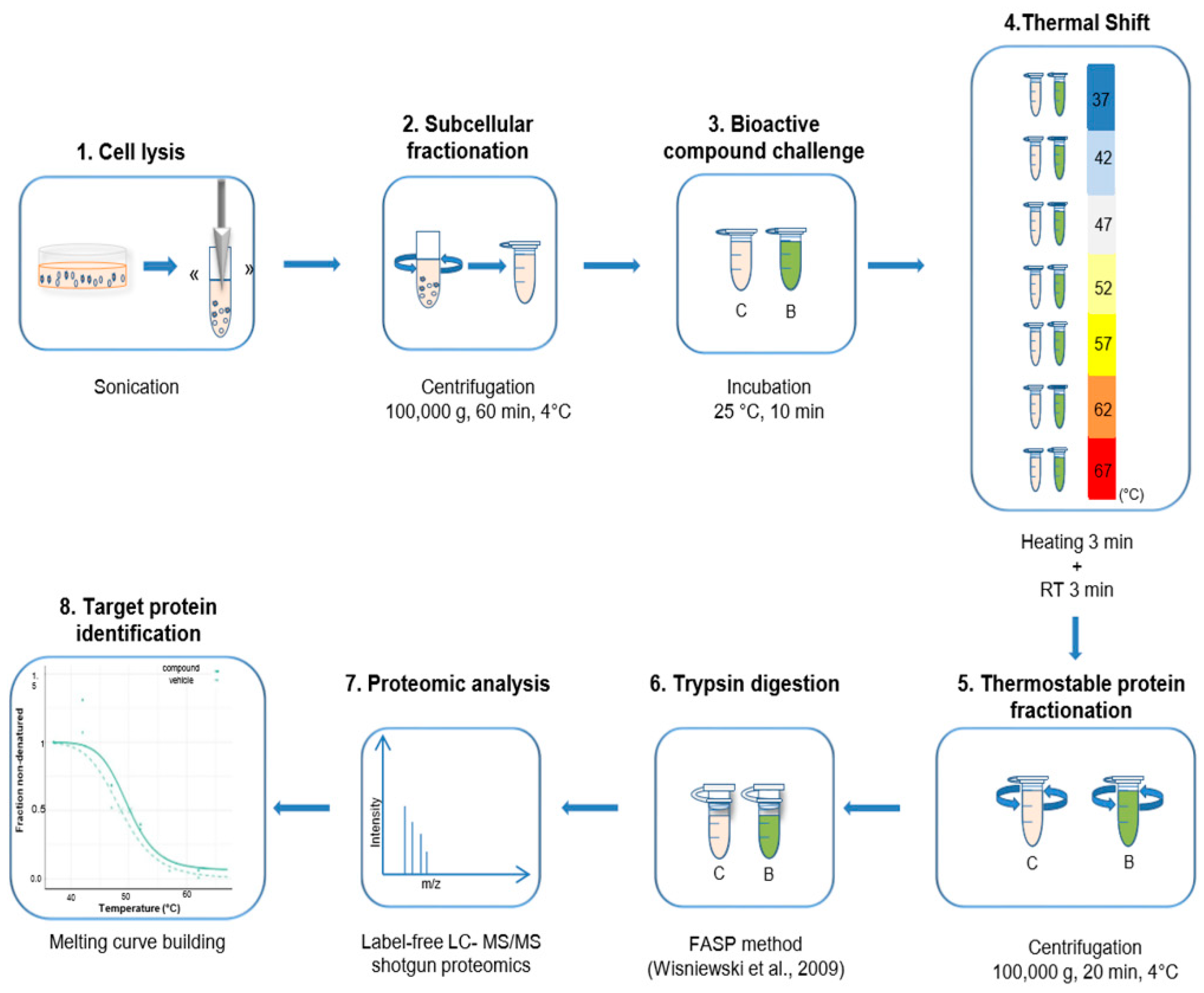
4.3. Thermal Proteome Profiling Experiments in Cellular Protein Extracts
4.4. Filter Aided Sample Preparation (FASP)
4.5. Nano LC-MS/MS Analysis
4.6. Peptide and Protein Identification and Quantification
4.7. Analysis of TPP Experiments
4.8. Pathway Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TPP | thermal proteome profiling |
| MOA | mechanisms of action |
| CETSA | cellular thermal shift assay |
| bTPP | bioactive thermal proteome profiling |
| Tm | melting temperature |
| hpa | 132-hydroxy-pheophytin a |
| DMSO | dimethyl sulfoxide |
| PBS | phosphate buffered saline |
| nLC-MS/MS | nano liquid chromatography-tandem mass spectrometry |
| FASP | filter aided sample preparation |
| RPSA | 40S ribosomal protein SA |
| FGG | fibrinogen gamma chain |
| IDH1 | isocitrate dehydrogenase |
| HSPB1 | heat shock protein beta-1 |
| PSMC3 | 26S proteasome regulatory subunit 6A |
| ALDH1B1 | aldehyde dehydrogenase X |
| CDC42 | cell division control protein 42 homolog |
| TUBA1B | tubulin alpha-1B chain |
| PSAT1 | phosphoserine aminotransferase |
| NPARC | non-parametric analysis of response curves |
References
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.A. Target discovery. Nat. Rev. Drug Discov. 2003, 2, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, J.; Bayat, N.; Cristobal, S. Proteomic Analysis of Endothelial Cells Exposed to Ultrasmall Nanoparticles Reveals Disruption in Paracellular and Transcellular Transport. Proteomics 2019, 19, 1800228. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Kurganov, B.I.; Rafikova, E.R.; Dobrov, E.N. Kinetics of thermal aggregation of tobacco mosaic virus coat protein. Biochemistry 2002, 67, 525–533. [Google Scholar]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Martinez Molina, D.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef]
- Becher, I.; Werner, T.; Doce, C.; Zaal, E.A.; Togel, I.; Khan, C.A.; Rueger, A.; Muelbaier, M.; Salzer, E.; Berkers, C.R.; et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 2016, 12, 908–910. [Google Scholar] [CrossRef]
- Azimi, A.; Caramuta, S.; Seashore-Ludlow, B.; Bostrom, J.; Robinson, J.L.; Edfors, F.; Tuominen, R.; Kemper, K.; Krijgsman, O.; Peeper, D.S.; et al. Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors. Mol. Syst. Biol. 2018, 14, e7858. [Google Scholar] [CrossRef]
- Turkowsky, D.; Lohmann, P.; Muhlenbrink, M.; Schubert, T.; Adrian, L.; Goris, T.; Jehmlich, N.; von Bergen, M. Thermal proteome profiling allows quantitative assessment of interactions between tetrachloroethene reductive dehalogenase and trichloroethene. J. Proteom. 2019, 192, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Zinn, N.; Faelth-Savitski, M.; Poeckel, D.; Gade, S.; Becher, I.; Muelbaier, M.; Wagner, A.J.; Strohmer, K.; Werner, T.; et al. Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 2018, 173, 260–274.e25. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Zhang, J.; He, S.; Yan, X. New Peptides Isolated from Marine Cyanobacteria, an Overview over the Past Decade. Mar. Drugs 2017, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Silva, N.G.; Sousa, M.L.; Ribeiro, T.; Rosa, F.; Leao, P.N.; Vasconcelos, V.; Reis, M.A.; Urbatzka, R. Chlorophyll Derivatives from Marine Cyanobacteria with Lipid-Reducing Activities. Mar. Drugs 2019, 17, 229. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, S.; Ochoa, B.; Fresnedo, O. Purification and properties of a cholesteryl ester hydrolase from rat liver microsomes. J. Lipid Res. 1999, 40, 715–725. [Google Scholar] [PubMed]
- Morgenstern, R.; Guthenberg, C.; Depierre, J.W. Microsomal glutathione S-transferase. Purification, initial characterization and demonstration that it is not identical to the cytosolic glutathione S-transferases A, B and C. Eur. J. Biochem. 1982, 128, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Duve, C. Exploring cells with a centrifuge. Science 1975, 189, 186–194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Franken, H.; Mathieson, T.; Childs, D.; Sweetman, G.M.; Werner, T.; Togel, I.; Doce, C.; Gade, S.; Bantscheff, M.; Drewes, G.; et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jia, Y.; Cao, Y.; Wu, S.; Jiang, H.; Sun, X.; Ma, J.; Yin, X.; Mao, A.; Shang, M. Overexpression of Phosphoserine Aminotransferase 1 (PSAT1) Predicts Poor Prognosis and Associates with Tumor Progression in Human Esophageal Squamous Cell Carcinoma. Cell. Physiol. Biochem. 2016, 39, 395–406. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Gao, S.; Ge, A.; Xu, S.; You, Z.; Ning, S.; Zhao, Y.; Pang, D. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3beta/beta-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 179. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xiao, F.; Guo, Y.; Deng, J.; Liu, B.; Zhang, Q.; Li, K.; Wang, C.; Chen, S.; Guo, F. Hepatic Phosphoserine Aminotransferase 1 Regulates Insulin Sensitivity in Mice via Tribbles Homolog 3. Diabetes 2015, 64, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Babcock, S.A.; Hu, N.; Maris, J.R.; Wang, H.; Ren, J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: Role of GSK3beta and mitochondrial function. BMC Med. 2012, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H.; Liao, P.R.; Guo, C.J.; Chen, C.H.; Mochly-Rosen, D.; Chuang, L.M. PKC-ALDH2 Pathway Plays a Novel Role in Adipocyte Differentiation. PLoS ONE 2016, 11, e0161993. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Toth, E.; Cherrington, N.J. Alcohol Metabolism in the Progression of Human Nonalcoholic Steatohepatitis. Toxicol. Sci. 2018, 164, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, S.H.; Park, K.M.; Lee, J.H.; Park, J.W. Increased obesity resistance and insulin sensitivity in mice lacking the isocitrate dehydrogenase 2 gene. Free Radic. Biol. Med. 2016, 99, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.W.; Park, D.C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Name | Protein Name | Tm Control 1 (°C) | Tm Treatment 1 (°C) | Tm Control 2 (°C) | Tm Treatment 2 (°C) | p-Value |
|---|---|---|---|---|---|---|
| P08865 * | 40S Ribosomal protein SA | 45.74 | 47.16 | 45.32 | 49.00 | 0.0385 |
| C9JC84 | Fibrinogen gamma chain | 44.09 | 45.32 | 44.74 | 47.63 | 0.1800 |
| O75874 | Isocitrate dehydrogenase [NADP] cytoplasmic | 47.49 | 48.86 | 48.00 | 49.23 | 0.6307 |
| P04792 * | Heat shock protein beta-1 | 45.23 | 49.82 | 47.94 | 51.40 | 0.0099 |
| P17980 | 26S proteasome regulatory subunit 6A | 44.67 | 47.01 | 44.18 | 46.65 | 0.1239 |
| P30837 | Aldehyde dehydrogenase X, mitochondrial | 44.34 | 44.97 | 44.19 | 46.96 | 0.6788 |
| P60953 | Cell division control protein 42 homolog | 46.93 | 47.70 | 47.09 | 51.92 | 0.6179 |
| P68363 * | Tubulin alpha-1B chain | 42.59 | 45.12 | 42.85 | 47.43 | 0.0242 |
| Q9Y617 | Phosphoserine aminotransferase | 49.03 | 49.78 | 49.49 | 54.35 | 0.2253 |
| Accession Name | Protein Name | Tm Control 1 (°C) | Tm Treatment 1 (°C) | Tm Control 2 (°C) | Tm Treatment 2 (°C) | p-Value |
|---|---|---|---|---|---|---|
| Q00341 | Vigilin | 47.04 | 50.62 | 46.91 | 47.60 | 0.2204 |
| P42330 * | Aldo-keto reductase family 1-member C3 | 47.64 | 52.02 | 48.57 | 52.08 | 0.0001 |
| G3V180 | Dipeptidyl peptidase 3 | 52.55 | 55.30 | 53.01 | 56.06 | 0.6632 |
| O14980 | Exportin-1 | 47.84 | 49.29 | 49.28 | 51.48 | 0.1985 |
| P00558 | Phosphoglycerate kinase 1 | 53.07 | 53.91 | 53.03 | 54.06 | 0.9584 |
| P07339 * | Cathepsin D | 50.68 | 51.78 | 50.47 | 51.73 | 0.0045 |
| P07814 | Bifunctional glutamate/proline--tRNA ligase | 43.83 | 46.51 | 42.48 | 46.70 | 0.2816 |
| P08133 | Annexin A6 | 52.45 | 53.03 | 52.99 | 55.30 | 0.9961 |
| P09327 | Villin-1 | 49.20 | 51.33 | 48.06 | 50.58 | 0.3429 |
| P13674 | Prolyl 4-hydroxylase subunit alpha-1 | 51.29 | 54.34 | 52.04 | 54.96 | 0.0954 |
| P15559 | NAD(P)H dehydrogenase [quinone] 1 | 49.21 | 50.48 | 49.08 | 49.30 | 0.8192 |
| P30038 | Delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial | 42.97 | 44.66 | 42.77 | 43.22 | 0.9284 |
| P45954 | Short/branched chain specific acyl-CoA dehydrogenase, mitochondrial | 45.27 | 47.28 | 44.85 | 46.52 | 0.7475 |
| P60709 * | Actin, cytoplasmic 1 | 44.10 | 48.42 | 41.84 | 45.14 | 0.0455 |
| Q06210 | Glutamine--fructose-6-phosphate aminotransferase [isomerizing] 1 | 47.31 | 48.12 | 47.14 | 47.55 | 0.9119 |
| Q13347 | Eukaryotic translation initiation factor 3 subunit I | 46.89 | 48.21 | 46.64 | 46.91 | 0.6253 |
| Q9NR45 | Sialic acid synthase | 53.17 | 53.41 | 53.22 | 54.67 | 0.4993 |
| Q9Y490 | Talin-1 | 49.93 | 51.85 | 51.75 | 53.92 | 0.1579 |
| Q9Y696 | Chloride intracellular channel protein 4 | 56.78 | 57.32 | 56.63 | 56.85 | 0.7306 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco del Amor, A.; Freitas, S.; Urbatzka, R.; Fresnedo, O.; Cristobal, S. Application of Bioactive Thermal Proteome Profiling to Decipher the Mechanism of Action of the Lipid Lowering 132-Hydroxy-pheophytin Isolated from a Marine Cyanobacteria. Mar. Drugs 2019, 17, 371. https://doi.org/10.3390/md17060371
Carrasco del Amor A, Freitas S, Urbatzka R, Fresnedo O, Cristobal S. Application of Bioactive Thermal Proteome Profiling to Decipher the Mechanism of Action of the Lipid Lowering 132-Hydroxy-pheophytin Isolated from a Marine Cyanobacteria. Marine Drugs. 2019; 17(6):371. https://doi.org/10.3390/md17060371
Chicago/Turabian StyleCarrasco del Amor, Ana, Sara Freitas, Ralph Urbatzka, Olatz Fresnedo, and Susana Cristobal. 2019. "Application of Bioactive Thermal Proteome Profiling to Decipher the Mechanism of Action of the Lipid Lowering 132-Hydroxy-pheophytin Isolated from a Marine Cyanobacteria" Marine Drugs 17, no. 6: 371. https://doi.org/10.3390/md17060371
APA StyleCarrasco del Amor, A., Freitas, S., Urbatzka, R., Fresnedo, O., & Cristobal, S. (2019). Application of Bioactive Thermal Proteome Profiling to Decipher the Mechanism of Action of the Lipid Lowering 132-Hydroxy-pheophytin Isolated from a Marine Cyanobacteria. Marine Drugs, 17(6), 371. https://doi.org/10.3390/md17060371