Flaccidoxide-13-Acetate-Induced Apoptosis in Human Bladder Cancer Cells is through Activation of p38/JNK, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress Regulated Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
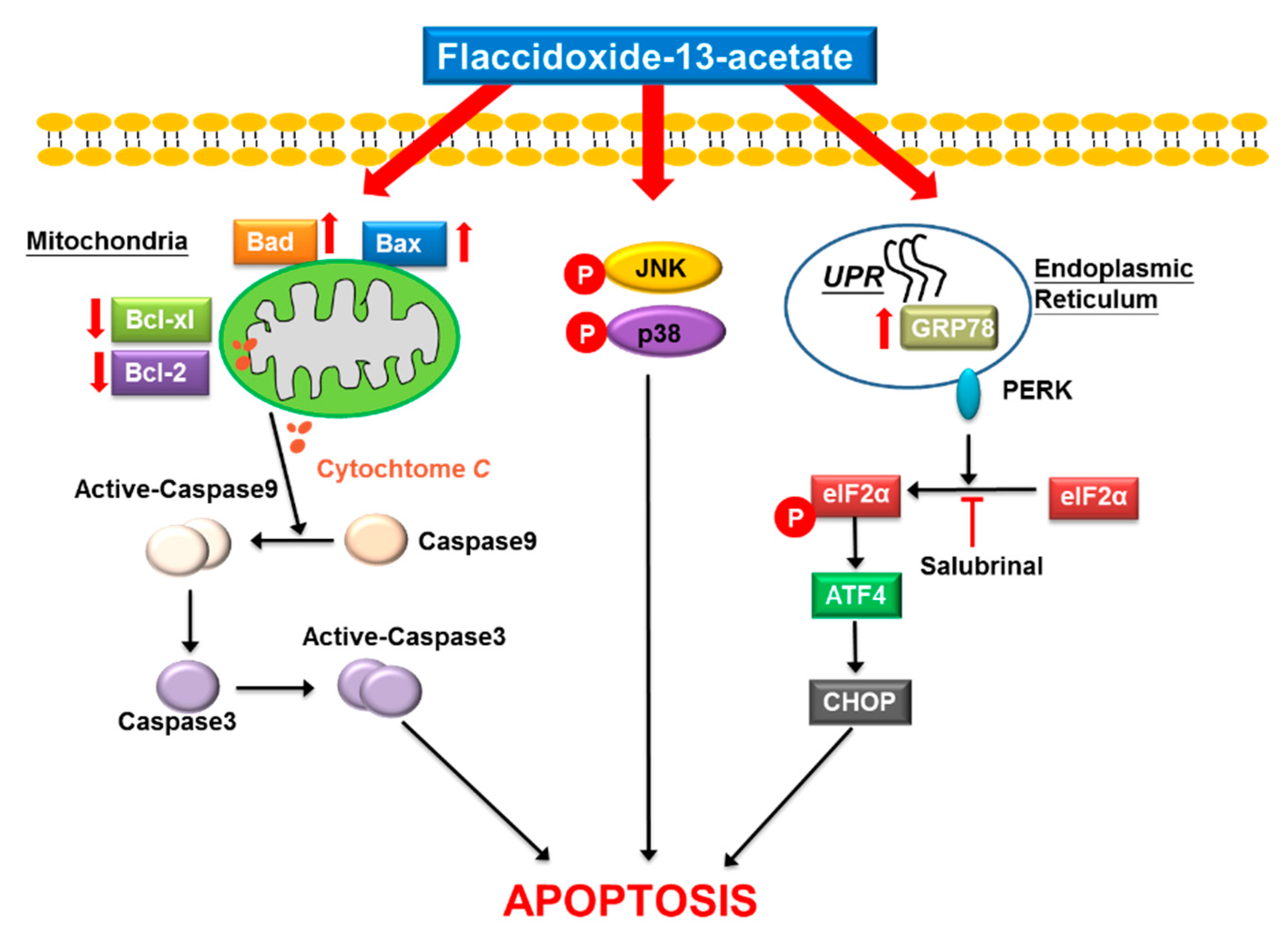
Abstract
1. Introduction
2. Results
2.1. Flaccidoxide-13-Acetate Inhibited Human Bladder Cancer Cell Proliferation
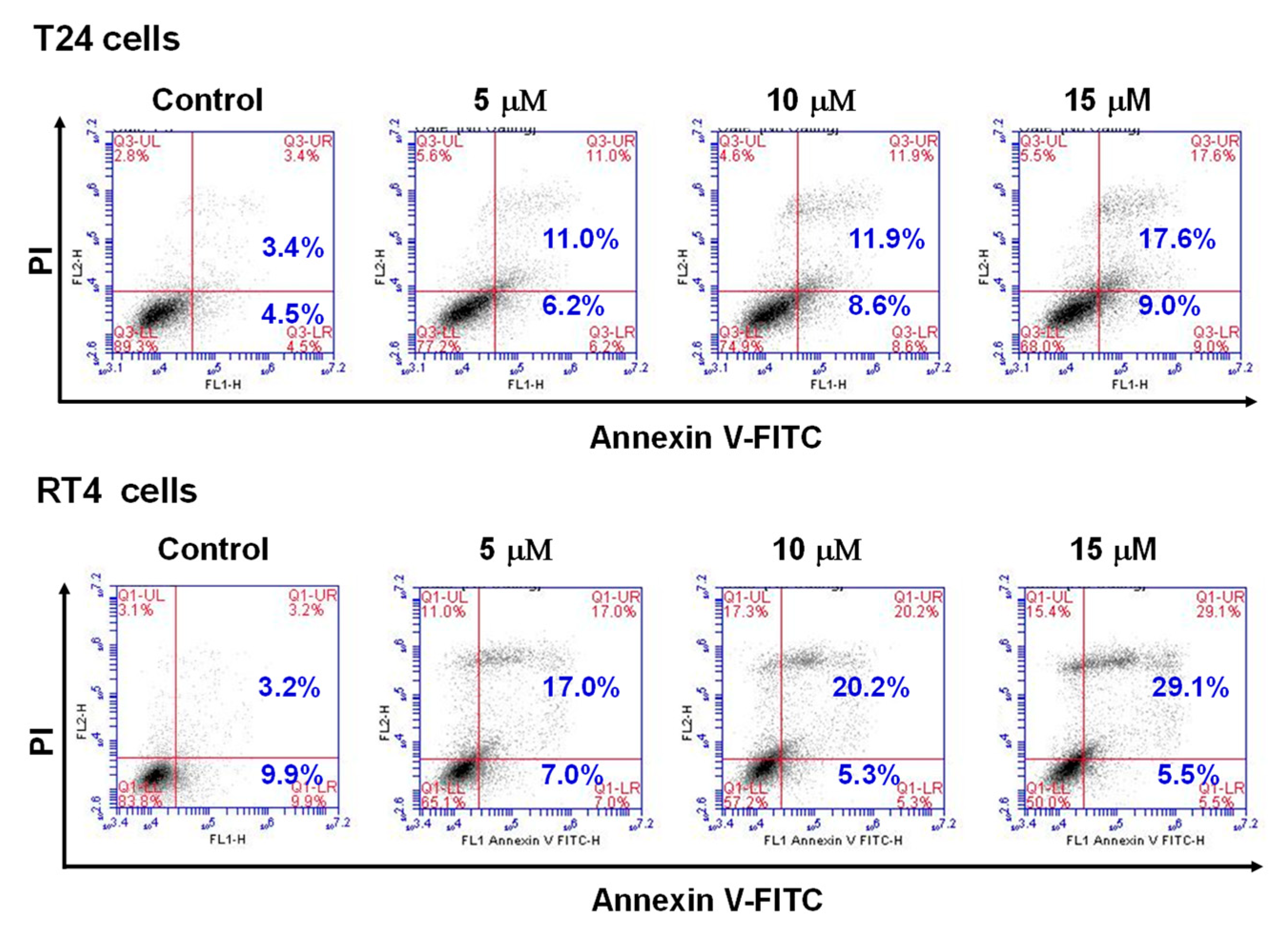
2.2. Flaccidoxide-13-Acetate Induced Apoptosis in RT4 and T24 Cells
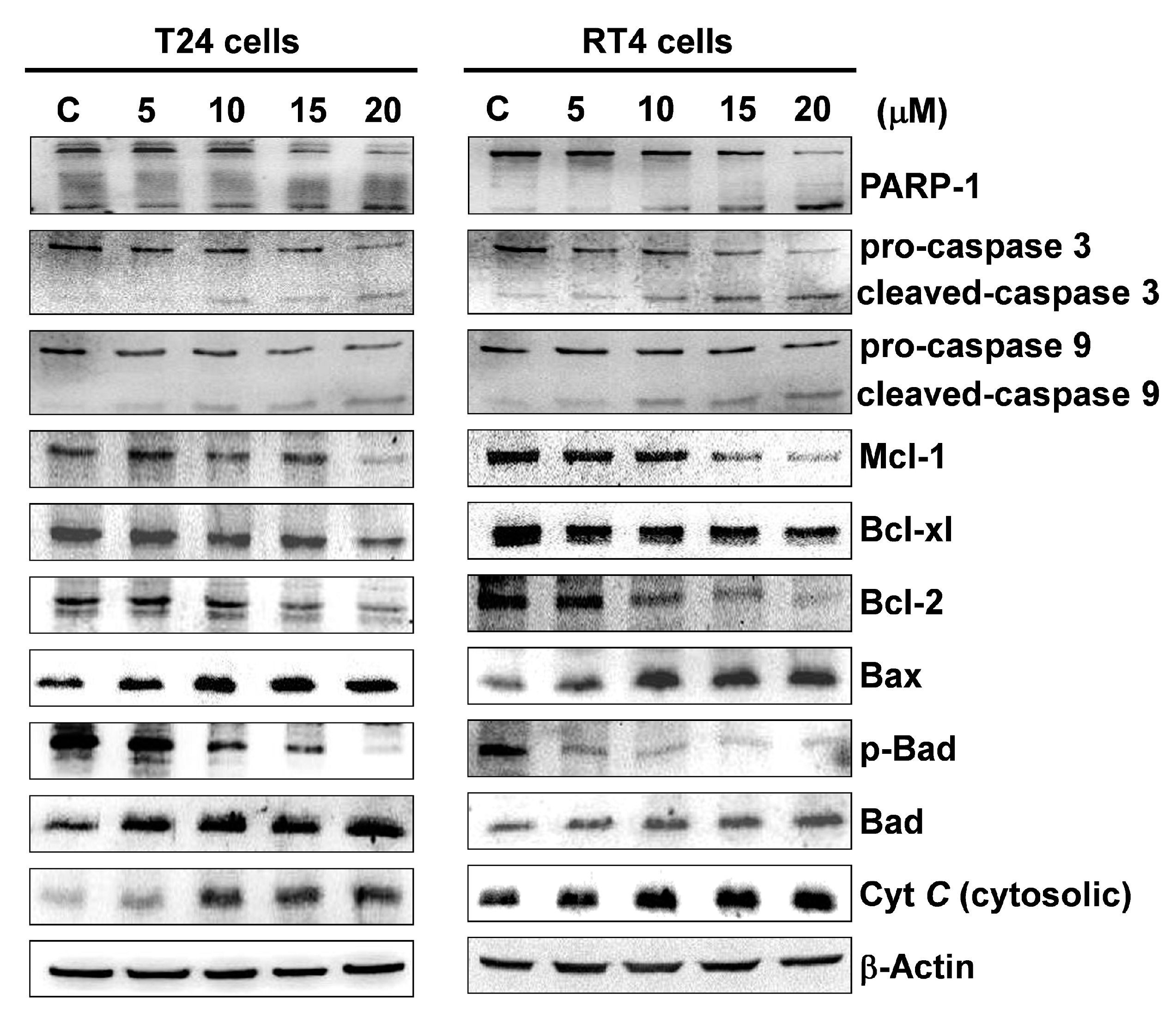
2.3. Flaccidoxide-13-Acetate Initiated Mitochondrial Dysfunction in T24 and RT4 Cells
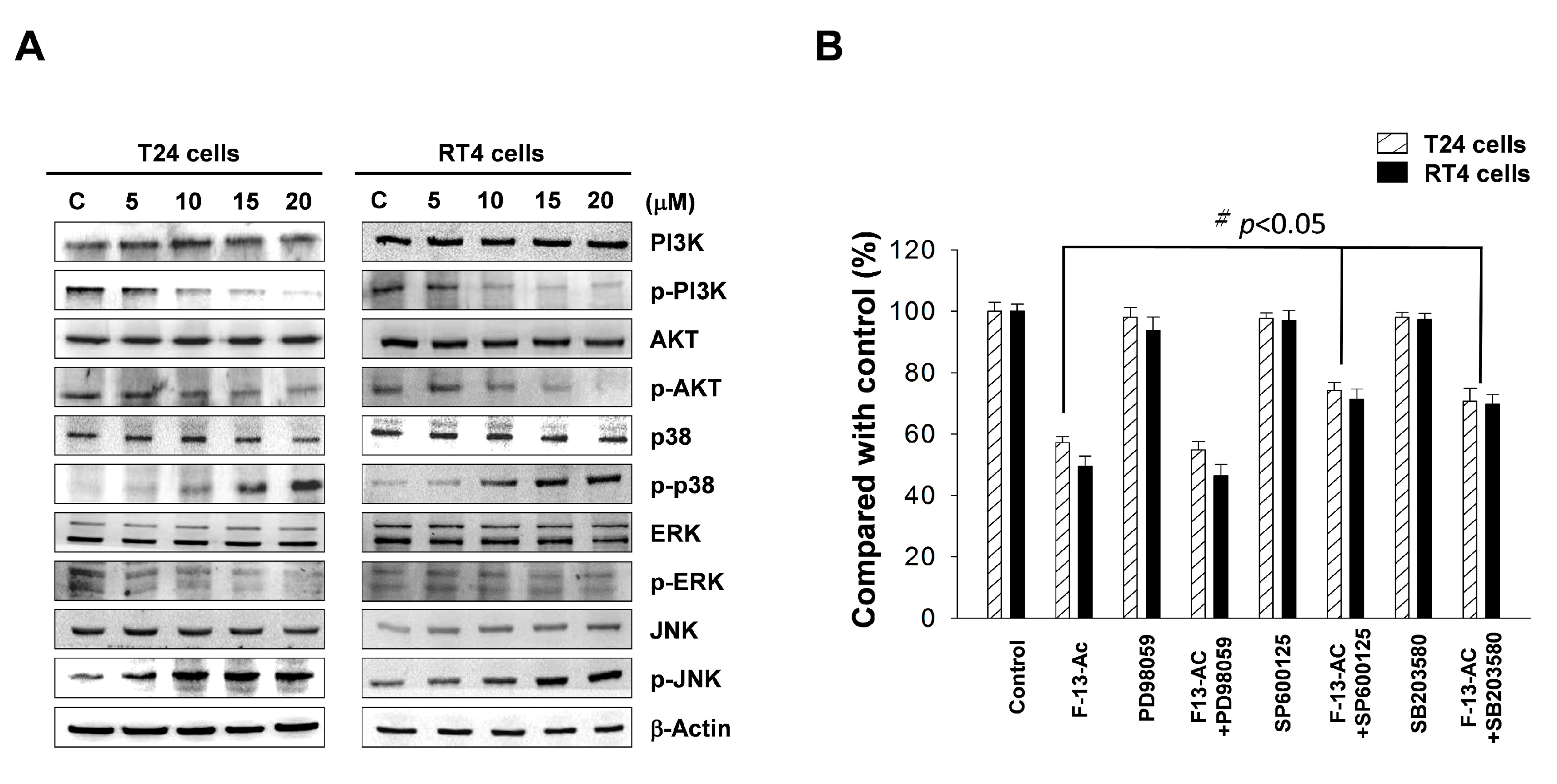
2.4. Flaccidoxide-13-Acetate Activated the p38 and JNK Pathways and Inhibited the PI3K/AKT Pathway
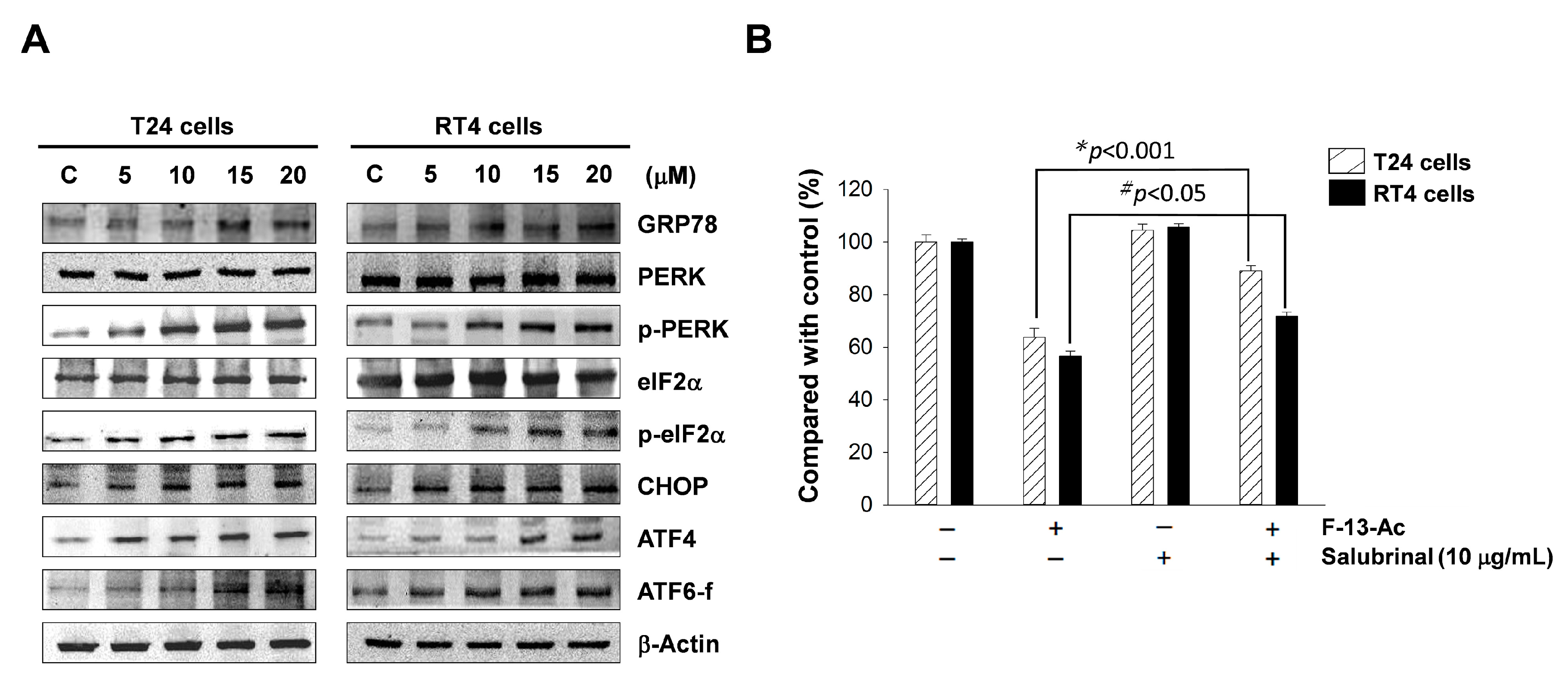
2.5. Endoplasmic Reticulum Stress is Involved in Flaccidoxide-13-Acetate-Induced Apoptosis
3. Discussion
3.1. Flaccidoxide-13-Acetate Triggers Mitochondrial Dysfunction in T24 and RT4 Bladder Cancer Cells
3.2. Flaccidoxide-13-Acetate Induces Apoptosis through Activation of the p38 and JNK Signaling Pathways and Inhibition of the PI3K/AKT Pathway
3.3. Flaccidoxide-13-Acetate-Induced Apoptosis Occurs Partially via Initiation of ER Stress in T24 and RT4 Cells
4. Material and Methods
4.1. Reagents
4.2. Cell Culture and Drug Treatment
4.3. Cell Viability Assay
4.4. Flow Cytometric Assay
4.5. Colony Formation Assay
4.6. Antibody and Western Blot Assay
4.7. Inhibitor Assessment
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carneiro, B.A.; Meeks, J.J.; Kuzel, T.M.; Scaranti, M.; Abdulkadir, S.A.; Giles, F.J. Emerging therapeutic targets in bladder cancer. Cancer Treat. Rev. 2015, 41, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Babjuk, M.; Burger, M.; Zigeuner, R.; Shariat, S.F.; van Rhijn, B.W.; Compérat, E.; Sylvester, R.J.; Kaasinen, E.; Böhle, A.; Redorta, J.P. Eau guidelines on non–muscle-invasive urothelial carcinoma of the bladder: Update 2013. Eur. Urol. 2013, 64, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.J.; Bochner, B.H.; Dalbagni, G. Superficial and muscle-invasive bladder cancer: Principles of management for outcomes assessments. J. Clin. Oncol. 2006, 24, 5519–5527. [Google Scholar] [CrossRef] [PubMed]
- Chamie, K.; Litwin, M.S.; Bassett, J.C.; Daskivich, T.J.; Lai, J.; Hanley, J.M.; Konety, B.R.; Saigal, C.S. Urologic Diseases in America Project. Recurrence of high-risk bladder cancer: A population-based analysis. Cancer 2013, 119, 3219–3227. [Google Scholar] [CrossRef]
- Radhika, P. Chemical constituents and biological activities of the soft corals of genus Cladiella: A review. Biochem. Syst. Ecol. 2006, 34, 781–789. [Google Scholar] [CrossRef]
- Fan, M.; Nath, A.; Tang, Y.; Choi, Y.-J.; Debnath, T.; Choi, E.-J.; Kim, E.-K. Investigation of the anti-prostate cancer properties of marine-derived compounds. Mar. Drugs 2018, 16, 160. [Google Scholar] [CrossRef]
- Santacruz, L.; Thomas, O.P.; Duque, C.; Puyana, M.; Tello, E. Comparative analyses of metabolomic fingerprints and cytotoxic activities of soft corals from the colombian caribbean. Mar. Drugs 2019, 17, 37. [Google Scholar] [CrossRef]
- Kamada, T.; Kang, M.-C.; Phan, C.-S.; Zanil, I.; Jeon, Y.-J.; Vairappan, C. Bioactive cembranoids from the soft coral genus Sinularia sp. In Borneo. Mar. Drugs 2018, 16, 99. [Google Scholar] [CrossRef]
- Hou, P.; Zeng, Y.; Ma, B.; Bi, K.; Chen, X. A new cytotoxic cembrane diterpene from the roots of euphorbia pekinensis rupr. Fitoterapia 2013, 90, 10–13. [Google Scholar] [CrossRef]
- Li, L.-M.; Li, G.-Y.; Pu, J.-X.; Xiao, W.-L.; Ding, L.-S.; Sun, H.-D. Ent-kaurane and cembrane diterpenoids from isodon sculponeatus and their cytotoxicity. J. Nat. Prod. 2009, 72, 1851–1856. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Chen, C.-H.; Liaw, C.-C.; Chen, Y.-C.; Kuo, Y.-H.; Shen, Y.-C. Cembrane diterpenoids from the taiwanese soft coral Sinularia flexibilis. Tetrahedron 2009, 65, 9157–9164. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Chien, C.-L.; Pan, S.-L.; Chen, W.-P.; Teng, C.-M.; Shen, Y.-C.; Guh, J.-H. Induction of endoplasmic reticulum stress and apoptosis by a marine prostanoid in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 679–686. [Google Scholar] [CrossRef]
- Kamel, H.N.; Ferreira, D.; Garcia-Fernandez, L.F.; Slattery, M. Cytotoxic diterpenoids from the hybrid soft coral Sinularia maxima × Sinularia polydactyla. J. Nat. Prod. 2007, 70, 1223–1227. [Google Scholar] [CrossRef]
- Liu, C.-I.; Chen, C.-C.; Chen, J.-C.; Su, J.-H.; Huang, H.H.; Chen, J.Y.-F.; Wu, Y.-J. Proteomic analysis of anti-tumor effects of 11-dehydrosinulariolide on cal-27 cells. Mar. Drugs 2011, 9, 1254–1272. [Google Scholar] [CrossRef]
- Neoh, C.-A.; Wang, R.Y.-L.; Din, Z.-H.; Su, J.-H.; Chen, Y.-K.; Tsai, F.-J.; Weng, S.-H.; Wu, Y.-J. Induction of apoptosis by sinulariolide from soft coral through mitochondrial-related and p38mapk pathways on human bladder carcinoma cells. Mar. Drugs 2012, 10, 2893–2911. [Google Scholar] [CrossRef]
- Pachycladins, A. Prostate cancer invasion and migration inhibitory eunicellin-based diterpenoids from the red sea soft coral cladiella pachyclados hassan. Hossam M 2010, 73, 848–853. [Google Scholar]
- Poza, J.J.; Fernandez, R.; Reyes, F.; Rodriguez, J.; Jimenez, C. Isolation, biological significance, synthesis, and cytotoxic evaluation of new natural parathiosteroids a−c and analogues from the soft coral Paragorgia sp. J. Organ. Chem. 2008, 73, 7978–7984. [Google Scholar] [CrossRef]
- Su, C.-C.; Su, J.-H.; Lin, J.-J.; Chen, C.-C.; Hwang, W.-I.; Huang, H.H.; Wu, Y.-J. An investigation into the cytotoxic effects of 13-acetoxysarcocrassolide from the soft coral sarcophyton crassocaule on bladder cancer cells. Mar. Drugs 2011, 9, 2622–2642. [Google Scholar] [CrossRef]
- Denicourt, C.; Dowdy, S.F. Targeting apoptotic pathways in cancer cells. Science 2004, 305, 1411–1413. [Google Scholar] [CrossRef]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Zielinski, R.R.; Eigl, B.J.; Chi, K.N. Targeting the apoptosis pathway in prostate cancer. Cancer J. 2013, 19, 79–89. [Google Scholar] [CrossRef]
- Campbell, N.A.; Williamson, B.; Heyden, R.J. Biology: Exploring Life; Pearson Prentice Hall: Boston, MA, USA, 2006; ISBN 0-13-250882-6. [Google Scholar]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Basañez, G.; Soane, L.; Hardwick, J.M. A new view of the lethal apoptotic pore. PLoS Biol. 2012, 10, e1001399. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Ballarin, L.; Burighel, P.; Cima, F. A tale of death and life: Natural apoptosis in the colonial ascidian botryllus schlosseri (Urochordata, Ascidiacea). Curr. Pharm. Des. 2008, 14, 138–147. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116, 1861–1862. [Google Scholar] [CrossRef]
- Sato, A.; Asano, T.; Okubo, K.; Isono, M.; Asano, T. Nelfinavir and ritonavir kill bladder cancer cells synergistically by inducing endoplasmic reticulum stress. Oncol. Res. Feat. Preclin. Clin. Cancer Ther. 2018, 26, 323–332. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, Y.; Ying, J.; Lei, Z.; Wan, L.; Zhu, X.; Ye, F.; Mao, P.; Wu, X.; Pan, R. Chrysin induces cell growth arrest, apoptosis, and er stress and inhibits the activation of stat3 through the generation of ros in bladder cancer cells. Oncol. Lett. 2018, 15, 9117–9125. [Google Scholar] [CrossRef]
- Zhang, M.; Du, H.; Huang, Z.; Zhang, P.; Yue, Y.; Wang, W.; Liu, W.; Zeng, J.; Ma, J.; Chen, G. Thymoquinone induces apoptosis in bladder cancer cell via endoplasmic reticulum stress-dependent mitochondrial pathway. Chem. Biol. Interact. 2018, 292, 65–75. [Google Scholar] [CrossRef]
- Neoh, C.-A.; Wu, W.-T.; Dai, G.-F.; Su, J.-H.; Liu, C.-I.; Su, T.-R.; Wu, Y.-J. Flaccidoxide-13-acetate extracted from the soft coral cladiella kashmani reduces human bladder cancer cell migration and invasion through reducing activation of the fak/pi3k/akt/mtor signaling pathway. Molecules 2017, 23, 58. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Pich, M.M.; Paolucci, U.; Castelli, G.P. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.-E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (map) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.; Kominami, E.; Momoi, T. Er stress (perk/eif2α phosphorylation) mediates the polyglutamine-induced lc3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of chop/gadd153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Shimizu, S.; Tsujimoto, Y. Proapoptotic bh3-only bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. USA 2000, 97, 577–582. [Google Scholar] [CrossRef]
- González-Gironès, D.M.; Moncunill-Massaguer, C.; Iglesias-Serret, D.; Cosialls, A.M.; Pérez-Perarnau, A.; Palmeri, C.M.; Rubio-Patiño, C.; Villunger, A.; Pons, G.; Gil, J. Aicar induces bax/bak-dependent apoptosis through upregulation of the bh3-only proteins bim and noxa in mouse embryonic fibroblasts. Apoptosis 2013, 18, 1008–1016. [Google Scholar] [CrossRef]
- Gotoh, M.; Sano-Maeda, K.; Murofushi, H.; Murakami-Murofushi, K. Protection of neuroblastoma neuro2a cells from hypoxia-induced apoptosis by cyclic phosphatidic acid (cpa). PLoS ONE 2012, 7, e51093. [Google Scholar] [CrossRef]
- Hoshyar, R.; Bathaie, S.Z.; Sadeghizadeh, M. Crocin triggers the apoptosis through increasing the bax/bcl-2 ratio and caspase activation in human gastric adenocarcinoma, ags, cells. DNA Cell Biol. 2013, 32, 50–57. [Google Scholar] [CrossRef]
- Kaparou, M.; Choumerianou, D.; Perdikogianni, C.; Martimianaki, G.; Kalmanti, M.; Stiakaki, E. Enhanced levels of the apoptotic bax/bcl-2 ratio in children with acute lymphoblastic leukemia and high-risk features. Genet. Mol. Biol. 2013, 36, 7–11. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Thornberry, N.A. Life and death decisions. Science 2003, 299, 214–215. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian map kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the mapk–ras–raf signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38mapk: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Masmoudi-Kouki, O.; Douiri, S.; Hamdi, Y.; Kaddour, H.; Bahdoudi, S.; Vaudry, D.; Basille, M.; Leprince, J.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide protects astroglial cells against oxidative stress-induced apoptosis. J. Neurochem. 2011, 117, 403–411. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Sweatt, J.D. The neuronal map kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. Map kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I. Pi3k/akt/mtor inhibitors in patients with breast and gynecologic malignancies harboring pik3ca mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Cantley, L.C.; Auger, K.R.; Carpenter, C.; Duckworth, B.; Graziani, A.; Kapeller, R.; Soltoff, S. Oncogenes and signal transduction. Cell 1991, 64, 281–302. [Google Scholar] [CrossRef]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the wnt signaling pathway, forms a complex with gsk-3β and β-catenin and promotes gsk-3β-dependent phosphorylation of β-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for bcl-xl and bcl-2, displaces bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist bad in response to survival factor results in binding to 14-3-3 not bcl-xl. Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Hong, S.-W.; Shin, J.-S.; Moon, J.-H.; Kim, Y.-S.; Lee, J.; Choi, E.K.; Ha, S.-H.; Lee, D.H.; Chung, H.N.; Kim, J.E. Nvp-bez235, a dual pi3k/mtor inhibitor, induces cell death through alternate routes in prostate cancer cells depending on the pten genotype. Apoptosis 2014, 19, 895–904. [Google Scholar] [CrossRef]
- Liu, M.; Li, C.-M.; Chen, Z.-F.; Ji, R.; Guo, Q.-H.; Li, Q.; Zhang, H.-L.; Zhou, Y.-N. Celecoxib regulates apoptosis and autophagy via the pi3k/akt signaling pathway in sgc-7901 gastric cancer cells. Int. J. Mol. Med. 2014, 33, 1451–1458. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef]
- Saito, A.; Ochiai, K.; Kondo, S.; Tsumagari, K.; Murakami, T.; Cavener, D.R.; Imaizumi, K. Endoplasmic reticulum stress response mediated by the perk-eif2α-atf4 pathway is involved in osteoblast differentiation induced by bmp2. J. Biol. Chem. 2011, 286, 4809–4818. [Google Scholar] [CrossRef]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of perk eif2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone p58ipk. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of er and oxidative stress signaling: The perk/nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase perk. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef]
- Liu, Z.-W.; Zhu, H.-T.; Chen, K.-L.; Dong, X.; Wei, J.; Qiu, C.; Xue, J.-H. Protein kinase rna-like endoplasmic reticulum kinase (perk) signaling pathway plays a major role in reactive oxygen species (ros)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc. Diabetol. 2013, 12, 158. [Google Scholar] [CrossRef]
- Matsumoto, H.; Miyazaki, S.; Matsuyama, S.; Takeda, M.; Kawano, M.; Nakagawa, H.; Nishimura, K.; Matsuo, S. Selection of autophagy or apoptosis in cells exposed to er-stress depends on atf4 expression pattern with or without chop expression. Biol. Open 2013, 2, 1084–1090. [Google Scholar] [CrossRef]
- Toth, A.; Nickson, P.; Mandl, A.; Bannister, M.L.; Toth, K.; Erhardt, P. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc. Haematol. Disord. Drug Targets 2007, 7, 205–218. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-J.; Su, T.-R.; Dai, G.-F.; Su, J.-H.; Liu, C.-I. Flaccidoxide-13-Acetate-Induced Apoptosis in Human Bladder Cancer Cells is through Activation of p38/JNK, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress Regulated Pathway. Mar. Drugs 2019, 17, 287. https://doi.org/10.3390/md17050287
Wu Y-J, Su T-R, Dai G-F, Su J-H, Liu C-I. Flaccidoxide-13-Acetate-Induced Apoptosis in Human Bladder Cancer Cells is through Activation of p38/JNK, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress Regulated Pathway. Marine Drugs. 2019; 17(5):287. https://doi.org/10.3390/md17050287
Chicago/Turabian StyleWu, Yu-Jen, Tzu-Rong Su, Guo-Fong Dai, Jui-Hsin Su, and Chih-I Liu. 2019. "Flaccidoxide-13-Acetate-Induced Apoptosis in Human Bladder Cancer Cells is through Activation of p38/JNK, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress Regulated Pathway" Marine Drugs 17, no. 5: 287. https://doi.org/10.3390/md17050287
APA StyleWu, Y.-J., Su, T.-R., Dai, G.-F., Su, J.-H., & Liu, C.-I. (2019). Flaccidoxide-13-Acetate-Induced Apoptosis in Human Bladder Cancer Cells is through Activation of p38/JNK, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress Regulated Pathway. Marine Drugs, 17(5), 287. https://doi.org/10.3390/md17050287