Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment


Abstract
1. Introduction
Focus of This Review
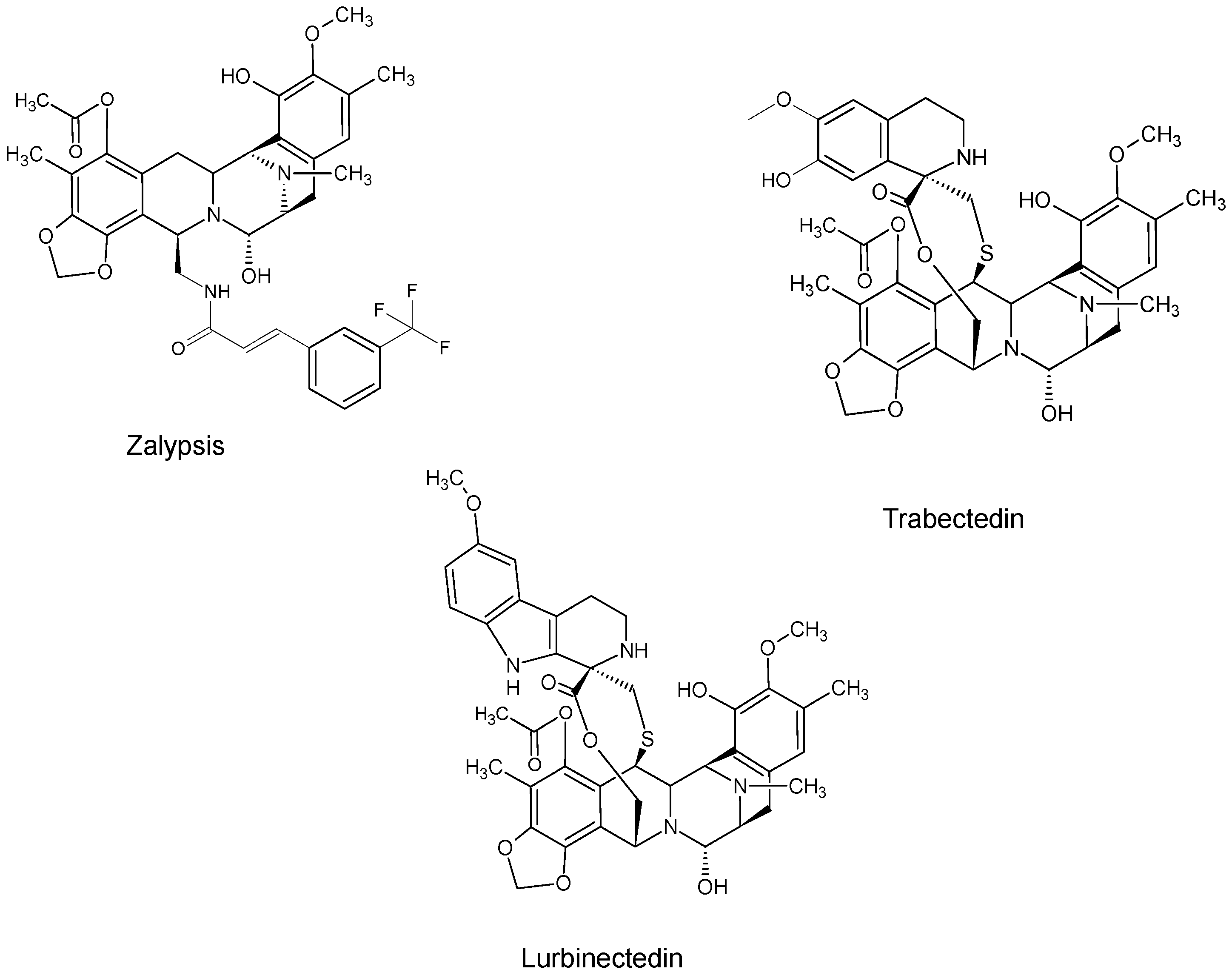
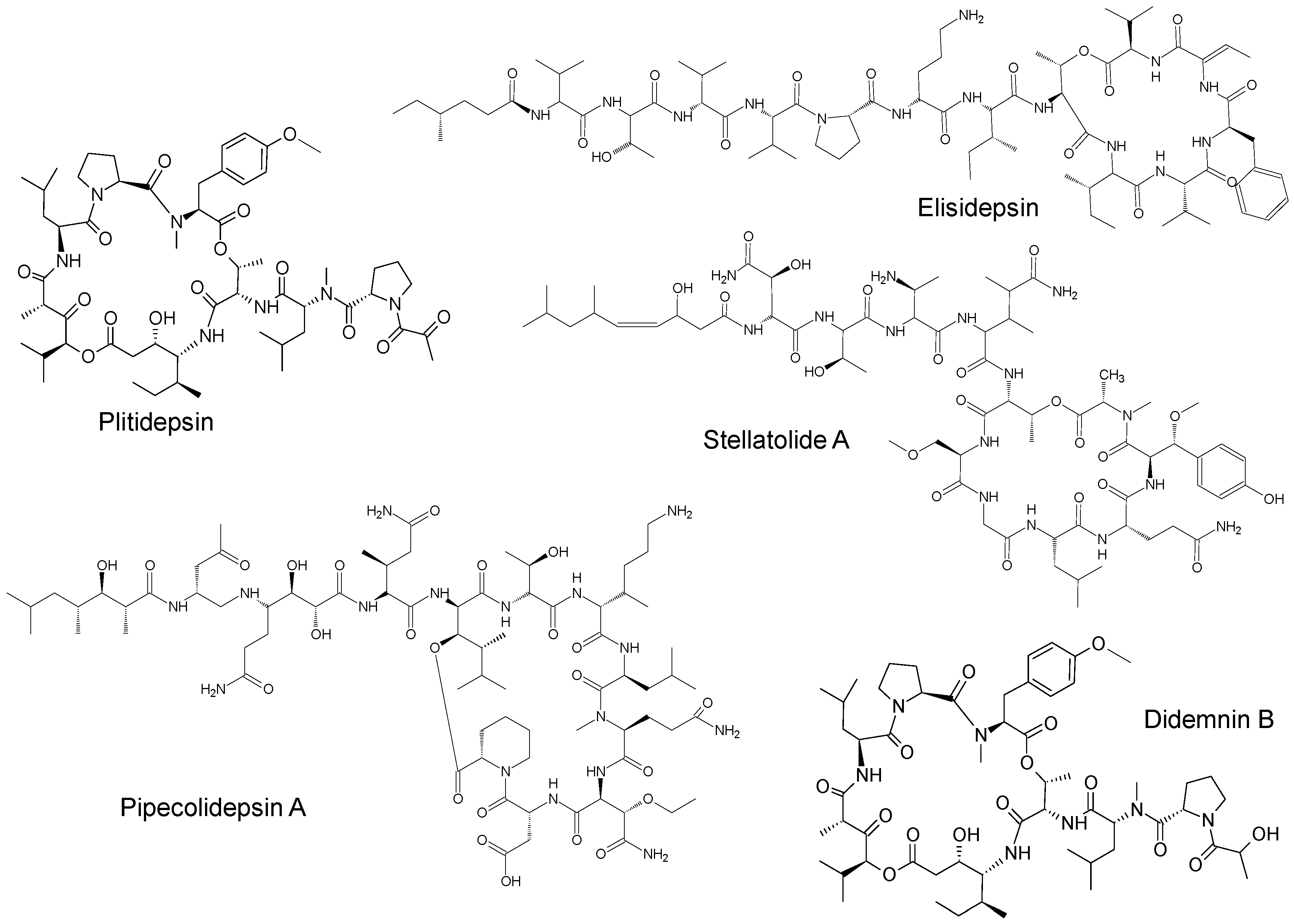
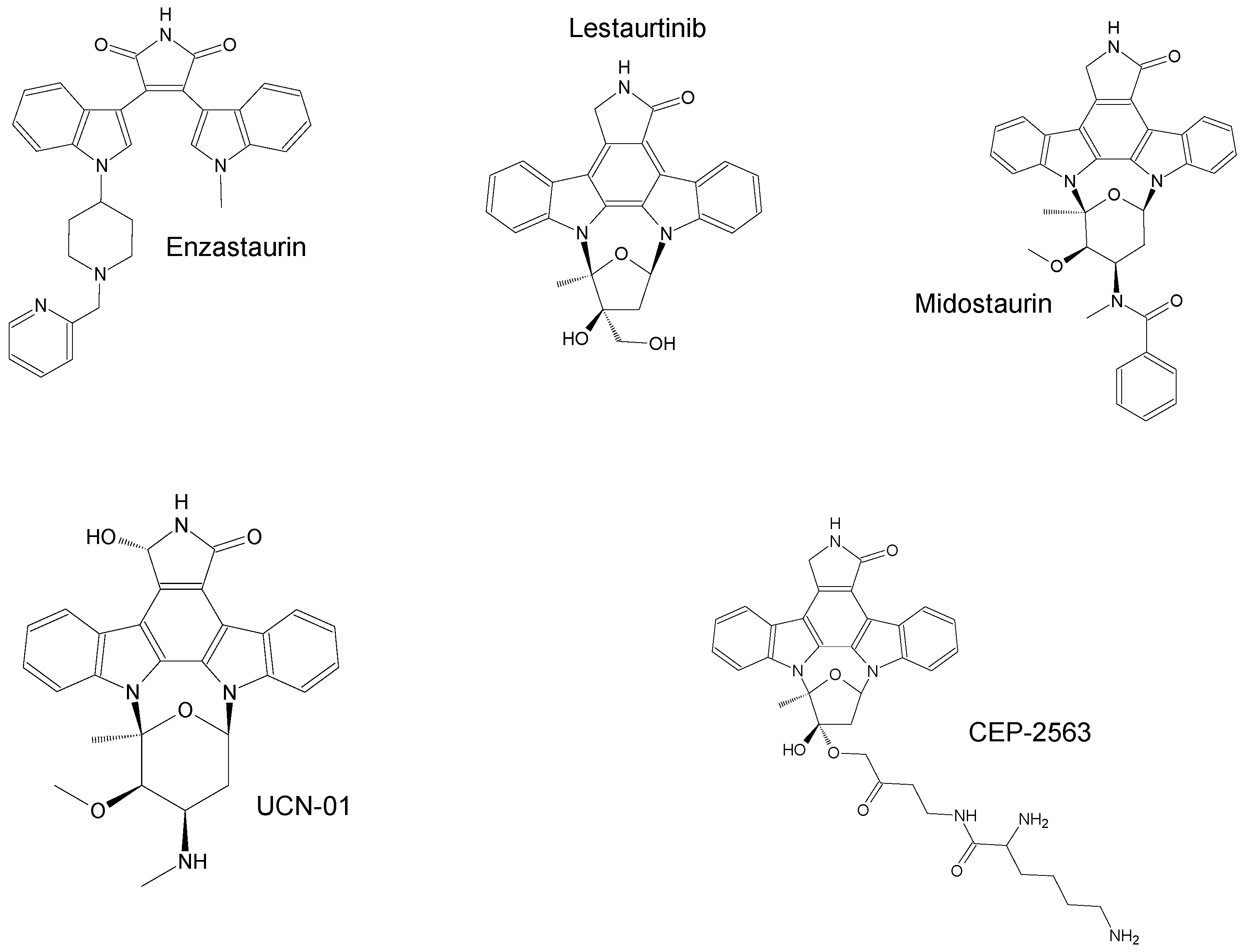
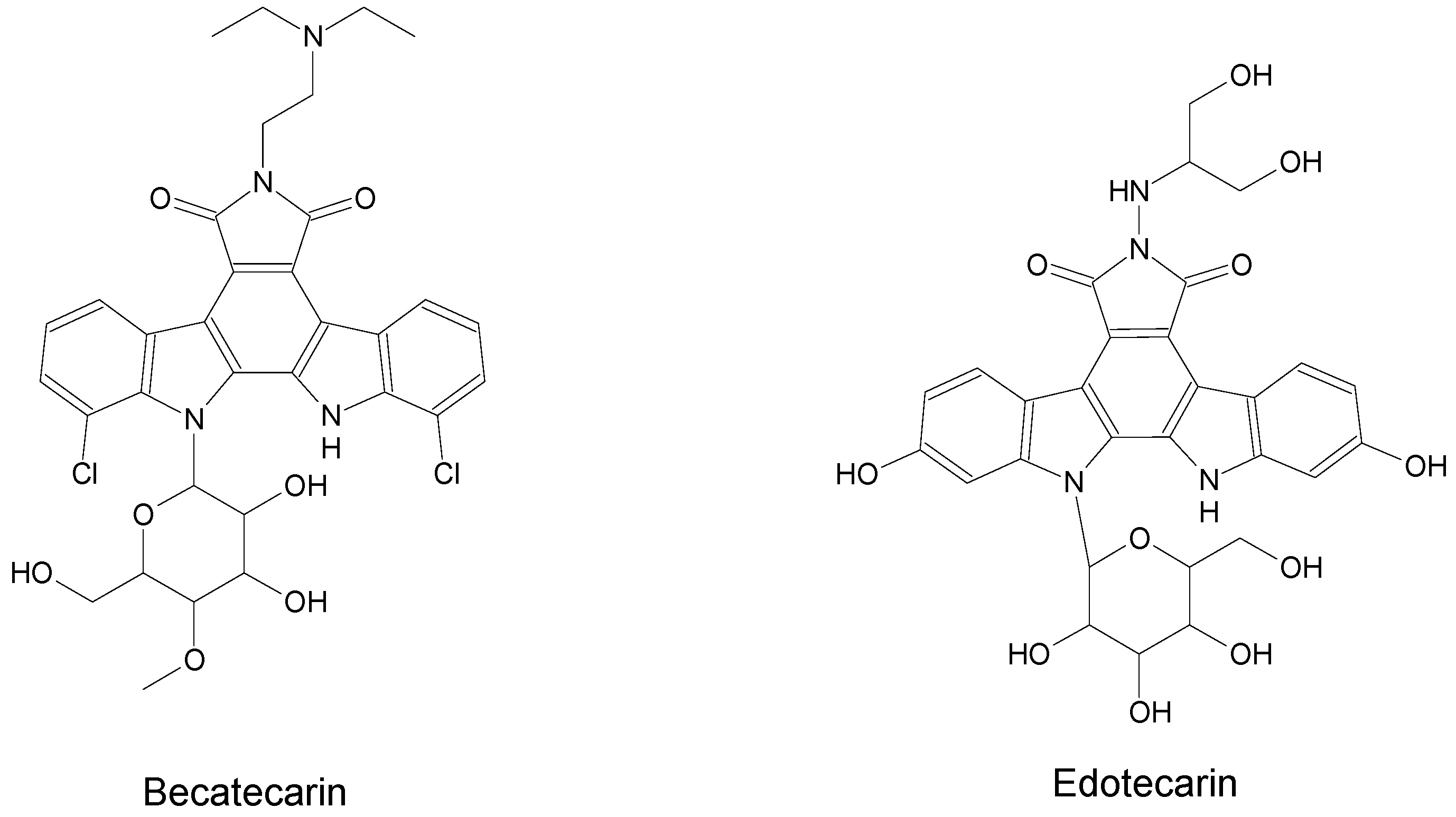
2. Marine-Derived Anticancer Drugs
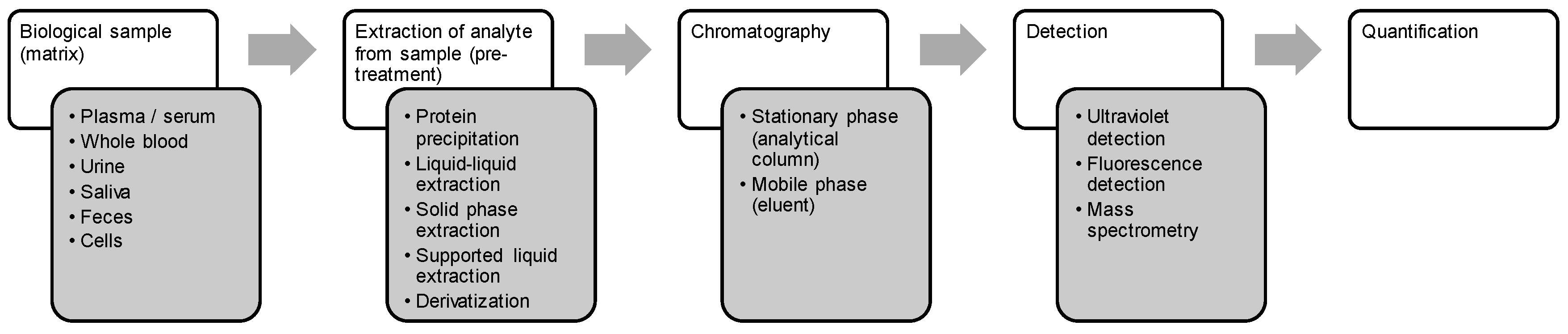
3. Bioanalysis of Marine-Derived Anticancer Drugs
3.1. Drugs
3.2. Matrices
3.2.1. Plasma and Serum
3.2.2. Whole Blood
3.2.3. Urine
3.2.4. Saliva
3.2.5. Feces
3.2.6. Cells
3.3. Sample Preparation and Recovery
3.3.1. Protein Precipitation
3.3.2. Liquid–Liquid Extraction
3.3.3. Solid Phase Extraction
3.3.4. Supported Liquid Extraction
3.3.5. Derivatization
3.4. Analytical Methods
3.4.1. Chromatography
3.4.2. Detection
UV Detection
Fluorescence Detection
Mass Spectrometry
3.5. Quantification
3.5.1. Analytical Range
3.5.2. Internal Standard
3.6. Metabolites
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Indumathy, S.; Dass, C.R. Finding chemo: The search for marine-based pharmaceutical drugs active against cancer. J. Pharm. Pharmacol. 2013, 65, 1280–1301. [Google Scholar] [CrossRef] [PubMed]
- Mudit, M.; El Sayed, K.A. Cancer control potential of marine natural product scaffolds through inhibition of tumor cell migration and invasion. Drug Discov. Today 2016, 21, 1745–1760. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.U.; Anjum, K.; Abbas, S.Q.; Akhter, N.; Shagufta, B.I.; Shah, S.A.A.; Tasneem, U. Emerging biopharmaceuticals from marine actinobacteria. Environ. Toxicol. Pharmacol. 2017, 49, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Advanced preclinical and clinical trials of natural products and related compounds from marine sources. Curr. Med. Chem. 2004, 11, 1693–1713. [Google Scholar] [CrossRef] [PubMed]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the Past and Charting the Future of Marine Natural Products Drug Discovery and Chemical Biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Jimenez, G.M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.M.; Dasari, R.; Chandra, S.; Kiss, R.; Kornienko, A. Marine invertebrate metabolites with anticancer activities: Solutions to the “supply problem”. Mar. Drugs 2016, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhou, F.; Al-Kareef, A.M.Q.; Wang, H. Anticancer agents from marine sponges. J. Asian Nat. Prod. Res. 2015, 17, 64–88. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.P. How might we increase success in marine-based drug discovery? Expert Opin. Drug Discov. 2014, 9, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; García-Ramos, Y.; Jesús Martin, M.; Spengler, J.; Molina-Guijarro, J.M.; Munt, S.; Francesch, A.M.; Cuevas, C.; Tulla-Puche, J.; Albericio, F. The first total synthesis of the cyclodepsipeptide pipecolidepsin A. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Ocio, E.M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J.C.; Mitsiades, N.; McMullan, C.J.; et al. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008, 68, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, S.K.; Rajendran, N.M.; Marino, A. Natural products diversity of marine ascidians (tunicates; ascidiacea) and successful drugs in clinical development. Nat. Prod. Bioprospect. 2017, 7, 1–111. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Mass spectrometry in the quantitative analysis of therapeutic intracellular nucleotide analogs. Mass Spectrom. Rev. 2011, 30, 321–343. [Google Scholar] [CrossRef] [PubMed]
- Astellas R&D Pipeline (As of January 2017). Available online: https://www.astellas.com/en/ir/library/pdf/3q2017_rd_en.pdf (accessed on 20 November 2017).
- European Medicines Agency Summary of Product Characteristics Adcetris. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002455/WC500135055.pdf (accessed on 27 November 2017).
- European Medicines Agency Summary of Product Characteristics DepoCyte. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000317/WC500035649.pdf (accessed on 27 November 2017).
- Seattle Genetics Denintuzumab Mafodotin. Available online: http://www.seattlegenetics.com/pipeline/denintuzumab-mafodotin (accessed on 20 November 2017).
- Abbvie Depatuxizumab Mafodotin (ABT-414). Available online: https://www.abbvie.com/our-science/pipeline/depatuxizumab-mafodotin.html (accessed on 20 November 2017).
- Seattle Genetics Enfortumab Vedotin. Available online: http://www.seattlegenetics.com/pipeline/enfortumab-vedotin (accessed on 20 November 2017).
- European Medicines Agency Summary of Product Characteristics Halaven. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002084/WC500105112.pdf (accessed on 27 November 2017).
- Celldex Therapeutics Glembatumumab Vedotin—Antibody-Drug Conjugate Targeting gpNMB in Metastatic Breast Cancer and Metastatic Melanoma. Available online: http://www.celldex.com/pipeline/cdx-011.php (accessed on 20 November 2017).
- GSK Product Pipeline. Available online: https://www.gsk.com/en-gb/investors/product-pipeline/ (accessed on 12 April 2017).
- PharmaMar Oncology Pipeline. Available online: https://www.pharmamar.com/science-and-innovation/oncology-pipeline/ (accessed on 20 November 2017).
- Triphase Marizomib. Available online: http://triphaseco.com/marizomib/ (accessed on 20 November 2017).
- Gescher, A. Staurosporine analogues—Pharmacological toys or useful antitumour agents? Crit. Rev. Oncol. Hematol. 2000, 34, 127–135. [Google Scholar] [CrossRef]
- European Medicines Agency Summary of Product Characteristics Rydapt. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004095/WC500237581.pdf (accessed on 27 November 2017).
- BeyondSpring Broadening Our Developmental Pipeline. Available online: http://www.beyondspringpharma.com/en/pipeline/ (accessed on 20 November 2017).
- Genentech Pipeline. Available online: https://www.gene.com/medical-professionals/pipeline (accessed on 20 November 2017).
- Genmab Tisotumab Vedotin. Available online: http://www.genmab.com/product-pipeline/products-in-development/humax-tf-adc (accessed on 20 November 2017).
- European Medicines Agency Summary of Product Characteristics Yondelis. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000773/WC500045832.pdf (accessed on 27 November 2017).
- Astellas R&D Pipeline (As of October 2017). Available online: https://www.astellas.com/en/system/files/2q2018_rd_en.pdf (accessed on 20 November 2017).
- Merchant, J.; Tutsch, K.; Dresen, A.; Arzoomanian, R.; Alberti, D.; Feierabend, C.; Binger, K.; Marnoccha, R.; Thomas, J.; Cleary, J.; et al. Phase I clinical and pharmacokinetic study of NSC 655649, a rebeccamycin analogue, given in both single-dose and multiple-dose formats. Clin. Cancer Res. 2002, 8, 2193–2201. [Google Scholar] [PubMed]
- Exelixis Helsinn to Discontinue Becatecarin Trial Program. Available online: https://www.prnewswire.com/news-releases/helsinn-to-discontinue-becatecarin-trial-program-56441722.html (accessed on 26 October 2017).
- Stewart, J.A.; Low, J.B.; Roberts, J.D.; Blow, A. A phase I clinical trial of didemnin B. Cancer 1991, 68, 2550–2554. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, J.; Shi, G.; Miyauchi, S.; Murakami, S.; Yang, Y. Cyclic depsipeptides as potential cancer therapeutics. Anti-Cancer Drugs 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Discontinued drugs in 2012: Oncology drugs. Expert Opin. Investig. Drugs 2013, 22, 1627–1644. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Discontinued in 2013: Oncology drugs. Expert Opin. Investig. Drugs 2015, 24, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Almhanna, K.; Miron, M.L.L.; Wright, D.; Gracian, A.C.; Hubner, R.A.; Van Laethem, J.L.; López, C.M.; Alsina, M.; Muñoz, F.L.; Bendell, J.; et al. Phase II study of the antibody-drug conjugate TAK-264 (MLN0264) in patients with metastatic or recurrent adenocarcinoma of the stomach or gastroesophageal junction expressing guanylyl cyclase C. Investig. New Drugs 2017, 35, 235–241. [Google Scholar] [CrossRef] [PubMed]
- InPress Media Group LLC Lifastuzumab Vedotin is Well-tolerated + Improves Objective Response Rate in Phase II Trial in Platinum-resistant Ovarian Cancer. Available online: https://adcreview.com/tag/lifastuzumab-vedotin/ (accessed on 13 April 2018).
- Martin, M.J.; Rodriguez-Acebes, R.; Garcia-Ramos, Y.; Martinez, V.; Murcia, C.; Digon, I.; Marco, I.; Pelay-Gimeno, M.; Fernández, R.; Reyes, F.; et al. Stellatolides, a new cyclodepsipeptide family from the sponge Ecionemia acervus: Isolation, solid-phase total synthesis, and full structural assignment of stellatolide A. J. Am. Chem. Soc. 2014, 136, 6754–6762. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Eckhardt, S.G.; Kuhn, J.; Hammond, L.; Weiss, G.; Rizzo, J.; Aylesworth, C.; Hidalgo, M.; Patnaik, A.; Schwartz, G.; et al. Phase I and pharmacokinetic study of NSC 655649, a rebeccamycin analog with topoisomerase inhibitory properties. J. Clin. Oncol. 2001, 19, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, J.A.; Newman, R.A.; Bignami, G.S.; Raybould, T.J.G.; Raber, M.N.; Esparza, L.; Walters, R.S. Phase II clinical and pharmacological study of didemnin B in patients with metastatic breast cancer. Investig. New Drugs 1992, 10, 113–117. [Google Scholar] [CrossRef]
- Yamada, Y.; Tamura, T.; Yamamoto, N.; Shimoyama, T.; Ueda, Y.; Murakami, H.; Kusaba, H.; Kamiya, Y.; Saka, H.; Tanigawara, Y.; et al. Phase I and pharmacokinetic study of edotecarin, a novel topoisomerase I inhibitor, administered once every 3 weeks in patients with solid tumors. Cancer Chemother. Pharmacol. 2006, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Sellers, S.; Diasio, R.B.; Douillard, J.-Y. A phase I dose-escalation study of edotecarin (J-107088) combined with infusional 5-fluorouracil and leucovorin in patients with advanced/metastatic solid tumors. Anti-Cancer Drugs 2010, 21, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Jones, R.J.; Oaknin, A.; Crawford, D.; Cuadra, C.; Hopkins, C.; Gil, M.; Coronado, C.; Soto-Matos, A.; Cullell-Young, M.; et al. A phase I and pharmacokinetic study of elisidepsin (PM02734) in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Ratain, M.J.; Geary, D.; Undevia, S.D.; Coronado, C.; Alfaro, V.; Iglesias, J.L.; Schilsky, R.L.; Miguel-Lillo, B. First-in-human, phase I study of elisidepsin (PM02734) administered as a 30-min or as a 3-hour intravenous infusion every three weeks in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Welch, P.A.; Sinha, V.P.; Cleverly, A.L.; Darstein, C.; Flanagan, S.D.; Musib, L.C. Safety, tolerability, QTc evaluation, and pharmacokinetics of single and multiple doses of enzastaurin HCl (LY317615), a protein kinase C-beta inhibitor, in healthy subjects. J. Clin. Pharmacol. 2007, 47, 1138–1151. [Google Scholar] [CrossRef] [PubMed]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase I clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Heath, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase I first-in-human trial of the vascular disrupting agent plinabulin (NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Nalda-Molina, R.; Valenzuela, B.; Ramon-Lopez, A.; Miguel-Lillo, B.; Soto-Matos, A.; Perez-Ruixo, J.J. Population pharmacokinetics meta-analysis of plitidepsin (Aplidin) in cancer subjects. Cancer Chemother. Pharmacol. 2009, 64, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Sharma, M.R.; Szyldergemajn, S.; Gore, L.; Geary, D.; Diamond, J.R.; Fernandez Teruel, C.; Soto Matos-Pita, A.; Iglesias, J.L.; Cullell-Young, M.; et al. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly× 2 every-3-week schedule. Investig. New Drugs 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Forster, M.; Boni, V.; Szyldergemajn, S.; Corral, J.; Turnbull, S.; Cubillo, A.; Teruel, C.F.; Calderero, I.L.; Siguero, M.; et al. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Clive, S.; Casado, E.; Michie, C.; Piera, A.; Sicart, E.; Carreras, M.J.; Coronado, C.; Kahatt, C.; Soto Matos-Pita, A.; et al. A phase I pharmacokinetic study of PM00104 (Zalypsis®) administered as a 24-h intravenous infusion every 3 weeks in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Dees, E.C.; Baker, S.D.; O’Reilly, S.; Rudek, M.A.; Davidson, S.B.; Aylesworth, C.; Elza-Brown, K.; Carducci, M.A.; Donehower, R.C. A phase I and pharmacokinetic study of short infusions of UCN-01 in patients with refractory solid tumors. Clin. Cancer Res. 2005, 11, 664–671. [Google Scholar] [PubMed]
- Jimeno, A.; Rudek, M.A.; Purcell, T.; Laheru, D.A.; Messersmith, W.A.; Dancey, J.; Carducci, M.A.; Baker, S.D.; Hidalgo, M.; Donehower, R.C. Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother. Pharmacol. 2008, 61, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Undevia, S.D.; Vogelzang, N.J.; Mauer, A.M.; Janisch, L.; Mani, S.; Ratain, M.J. Phase I clinical trial of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, in patients with refractory solid tumors. Investig. New Drugs 2004, 22, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Avilés, P.; Lee, W.; Ly, C.; Guillen, M.J.; Munt, S.; Cuevas, C.; Faircloth, G. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM02734, a novel antineoplastic agent, in dog plasma. Rapid Commun. Mass Spectrom. 2006, 20, 2535–2740. [Google Scholar] [CrossRef] [PubMed]
- DesJardins, C.; Saxton, P.; Lu, S.X.; Li, X.; Rowbottom, C.; Wong, Y.N. A high-performance liquid chromatography-tandem mass spectrometry method for the clinical combination study of carboplatin and anti-tumor agent eribulin mesylate (E7389) in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 875, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Dubbelman, A.C.; Rosing, H.; Thijssen, B.; Lucas, L.; Copalu, W.; Wanders, J.; Schellens, J.H.M.; Beijnen, J.H. Validation of high-performance liquid chromatography-tandem mass spectrometry assays for the quantification of eribulin (E7389) in various biological matrices. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Pernice, T.; Bishop, A.G.; Guillen, M.J.; Cuevas, C.; Avilés, P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (Lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J. Pharm. Biomed. Anal. 2016, 123, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Van Gijn, R.; van Tellingen, O.; de Clippeleir, J.J.M.; Hillebrand, M.J.X.; Boven, E.; Vermorken, J.B.; ten Bokkel Huinink, W.W.; Schwertz, S.; Graf, P.; Beijnen, J.H. Analytical procedure for the determination of the new antitumour drug N-benzoylstaurosporine and three potential metabolites in human plasma by reversed-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1995, 667, 269–276. [Google Scholar] [CrossRef]
- Van Gijn, R.; Havik, E.; Boven, E.; Vermorken, J.B.; ten Bokkel Huinink, W.W.; van Tellingen, O.; Beijnen, J.H. High-performance liquid chromatographic analysis of the new four potential metabolites in micro-volumes of plasma. J. Pharm. Biomed. Anal. 1995, 14, 165–174. [Google Scholar] [CrossRef]
- Illmer, T.; Thiede, H.-M.; Thiede, C.; Bornhauser, M.; Schaich, M.; Schleyer, E.; Ehninger, G. A highly sensitive method for the detection of PKC412 (CGP41251) and its metabolites by high-performance liquid chromatography. J. Pharmacol. Toxicol. Methods 2007, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Bourget, P.; Amin, A.; Chandesris, M.-O.; Vidal, F.; Merlette, C.; Hirsch, I.; Cabaret, L.; Carvalhosa, A.; Mogenet, A.; Frenzel, L.; et al. Liquid chromatography–tandem mass spectrometry assay for therapeutic drug monitoring of the tyrosine kinase inhibitor, midostaurin, in plasma from patients with advanced systemic mastocytosis. J. Chromatogr. B 2014, 944, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Celli, N.; Gallardo, A.M.; Rossi, C.; Zucchetti, M.; D’Incalci, M.; Rotilio, D. Analysis of aplidine (dehydrodidemnin B), a new marine-derived depsipeptide, in rat biological fluids by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 335–343. [Google Scholar] [CrossRef]
- Sparidans, R.W.; Kettenes-Van Den Bosch, J.J.; Van Tellingen, O.; Nuijen, B.; Henrar, R.E.C.; Jimeno, J.M.; Faircloth, G.; Floriano, P.; Rinehart, K.L.; Beijnen, J.H. Bioanalysis of aplidine, a new marine antitumoral depsipeptide, in plasma by high-performance liquid chromatography after derivatization with trans-4′-hydrazino-2-stilbazole. J. Chromatogr. B Biomed. Sci. Appl. 1999, 729, 43–53. [Google Scholar] [CrossRef]
- Yin, J.; Avilés, P.; Lee, W.; Ly, C.; Floriano, P.; Ignacio, M.; Faircloth, G. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of Aplidin, a novel marine-derived antineoplastic agent, in human plasma. Rapid Commun. Mass Spectrom. 2003, 17, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Celli, N.; Mariani, B.; Di Carlo, F.; Zucchetti, M.; Lopez-Lazaro, L.; D’Incalci, M.; Rotilio, D. Determination of Aplidin®, a marine-derived anticancer drug, in human plasma, whole blood and urine by liquid chromatography with electrospray ionisation tandem mass spectrometric detection. J. Pharm. Biomed. Anal. 2004, 34, 619–630. [Google Scholar] [CrossRef]
- Sparidans, R.W.; Schellens, J.H.M.; López-Lázaro, L.; Jimeno, J.M.; Beijnen, J.H. Liquid chromatographic assay for the cyclic depsipeptide aplidine, a new marine antitumor drug, in whole blood using derivatization with trans-4′-hydrazino-2-stilbazole. Biomed. Chromatogr. 2004, 18, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Van Andel, L.; Rosing, H.; Fudio, S.; Avilés, P.; Tibben, M.M.; Gebretensae, A.; Schellens, J.H.M.; Beijnen, J.H. Liquid chromatography-tandem mass spectrometry assay to quantify plitidepsin in human plasma, whole blood and urine. J. Pharm. Biomed. Anal. 2017, 145, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Rosing, H.; Hillebrand, M.J.X.; Jimeno, J.M.; Gómez, A.; Floriano, P.; Faircloth, G.; Cameron, L.; Henrar, R.E.C.; Vermorken, J.B.; Bult, A.; et al. Analysis of ecteinascidin 743, a new potent marine-derived anticancer drug, in human plasma by high-performance liquid chromatography in combination with solid-phase extraction. J. Chromatogr. B Biomed. Appl. 1998, 710, 183–189. [Google Scholar] [CrossRef]
- Rosing, H.; Hillebrand, M.J.X.; Jimeno, J.M.; Gómez, A.; Floriano, P.; Faircloth, G.; Henrar, R.E.C.; Vermorken, J.B.; Cvitkovic, E.; Bult, A.; et al. Quantitative determination of Ecteinascidin 743 in human plasma by miniaturized high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 1998, 33, 1134–1140. [Google Scholar] [CrossRef]
- Stokvis, E.; Rosing, H.; López-Lázaro, L.; Beijnen, J.H. Simple and sensitive liquid chromatographic quantitative analysis of the novel marine anticancer drug YondelisTM (ET-743, trabectedin) in human plasma using column switching and tandem mass spectrometric detection. J. Mass Spectrom. 2004, 39, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Zangarini, M.; Ceriani, L.; Sala, F.; Marangon, E.; Bagnati, R.; D’Incalci, M.; Grosso, F.; Zucchetti, M. Quantification of trabectedin in human plasma: Validation of a high-performance liquid chromatography–mass spectrometry method and its application in a clinical pharmacokinetic study. J. Pharm. Biomed. Anal. J. Pharm. Biomed. 2014, 95, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Ceriani, L.; Ferrari, M.; Zangarini, M.; Licandro, S.A.; Bello, E.; Frapolli, R.; Falcetta, F.; D’Incalci, M.; Libener, R.; Grosso, F.; et al. HPLC–MS/MS method to measure trabectedin in tumors: Preliminary PK study in a mesothelioma xenograft model. Bioanalysis 2015, 7, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Kurata, N.; Kuramitsu, T.; Tanii, H.; Fuse, E.; Kuwabara, T.; Kobayashi, H.; Kobayashi, S. Development of a highly sensitive high-performance liquid chromatographic method for measuring an anticancer drug, UCN-01, in human plasma or urine. J. Chromatogr. B 1998, 708, 223–227. [Google Scholar] [CrossRef]
- Bauer, K.S.; Lush, R.M.; Rudek, M.A.; Shih, C.; Sausville, E.; Figg, W.D. A high-performance liquid chromatography method using ultraviolet and fluorescence detection for the quantitation of UCN-01, 7-hydroxystaurosporine, from human plasma and saliva. Biomed. Chromatogr. 2000, 14, 338–343. [Google Scholar] [CrossRef]
- Smith, J.A.; Cortes, J.; Newman, R.A.; Madden, T.L. Development of a simplified, sensitive high-performance liquid chromatographic method using fluorescence detection to determine the concentration of UCN-01 in human plasma. J. Chromatogr. B Biomed. Sci. Appl. 2001, 760, 247–253. [Google Scholar] [CrossRef]
- Yin, J.; Avilés, P.; Lee, W.; Ly, C.; Guillen, M.J.; Munt, S.; Cuevas, C.; Faircloth, G. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM00104, a novel antineoplastic agent, in mouse, rat, dog, and human plasma. Rapid Commun. Mass Spectrom. 2005, 19, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Del Bufalo, A.; Fini, M. Deep sea as a source of novel-anticancer drugs: Update on discovery and preclinical/clinical evaluaton in a systems medicine perspective. EXCLI J. 2015, 14, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Tanaka, K.; Nicholson, B.; Deyanat-Yazdi, G.; Potts, B.; Yoshida, T.; Oda, A.; Kitagawa, T.; Orikasa, S.; Kiso, Y.; et al. Synthesis and structure-activity relationship study of antimicrotubule agents phenylahistin derivatives with a didehydropiperazine-2,5-dione structure. J. Med. Chem. 2012, 55, 1056–1071. [Google Scholar] [CrossRef] [PubMed]
- Bourhill, T.; Narendran, A.; Johnston, R.N. Enzastaurin: A lesson in drug development. Crit. Rev. Oncol. Hematol. 2017, 112, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hexner, E.; Roboz, G.; Hoffman, R.; Luger, S.; Mascarenhas, J.; Carroll, M.; Clementi, R.; Bensen-Kennedy, D.; Moliterno, A. Open-label study of oral CEP-701 (lestaurtinib) in patients with polycythaemia vera or essential thrombocythaemia with JAK2-V617F mutation. Br. J. Haematol. 2014, 164, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.L.; Aleksic, A.; McGuffey, L.; Dauser, R.; Nuchtern, J.; Bernacky, B.; Blaney, S.M. Plasma and cerebrospinal fluid pharmacokinetics of rebeccamycin (NSC 655649) in nonhuman primates. Cancer Chemother. Pharmacol. 2004, 54, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Lopez-Lazaro, L.; Beijnen, J.H.; Schellens, J.H.M. Trabectedin (Yondelis, formerly ET-743), a mass balance study in patients with advanced cancer. Investig. New Drugs 2005, 23, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Omura, S. Chemical biology of natural indolocarbazole products: 30 Years since the discovery of staurosporine. J. Antibiot. 2009, 62, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Anizon, F.; Moreau, P.; Sancelme, M.; Laine, W.; Bailly, C.; Prudhomme, M. Rebeccamycin analogues bearing amine substituents or other groups on the sugar moiety. Bioorg. Med. Chem. 2003, 11, 3709–3722. [Google Scholar] [CrossRef]
- Marshall, J.L.; Kindler, H.; Deeken, J.; Bhargava, P.; Vogelzang, N.J.; Rizvi, N.; Luhtala, T.; Boylan, S.; Dordal, M.; Robertson, P.; et al. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Investig. New Drugs 2005, 23, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Kotliarova, S.; Walling, J.; Musib, L.; Thornton, D.; Albert, P.S.; Fine, H.A. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin. Cancer Res. 2009, 15, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kotliarova, S.; Butman, J.A.; Albert, P.S.; Kim, L.; Musib, L.; Thornton, D.; Fine, H.A. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol. 2010, 12, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations; Li, W., Zhang, J., Tse, F.L.S., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; ISBN 9781118671276. [Google Scholar]
- Faivre, S.; Chièze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef] [PubMed]
- Van Andel, L.; Fudio, S.; Rosing, H.; Munt, S.; Miguel-Lillo, B.; González, I.; Tibben, M.M.; de Vries, N.; de Vries Schultink, A.H.M.; Schellens, J.H.M.; et al. Pharmacokinetics and excretion of 14C–Plitidepsin in patients with advanced cancer. Investig. New Drugs 2017, 35, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Dubbelman, A.-C.; Rosing, H.; Jansen, R.S.; Mergui-Roelvink, M.; Huitema, A.D.R.; Koetz, B.; Lymboura, M.; Reyderman, L.; Lopez-Anaya, A.; Schellens, J.H.M.; et al. Mass balance study of [14C]eribulin in patients with advanced solid tumors. Drug Metab. Dispos. 2012, 40, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, D.A.; Holland, J.C.; Athanaselis, S. Testing for drugs of abuse in saliva and sweat. J. Chromatogr. B Biomed. Appl. 1998, 713, 111–135. [Google Scholar] [CrossRef]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics. Concepts and Applications, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1995; ISBN 978-0-683-07404-8. [Google Scholar]
- Fuse, E.; Tanii, H.; Takai, K.; Asanome, K.; Kurata, N.; Kobayashi, H.; Kuwabara, T.; Kobayashi, S.; Sugiyama, Y. Altered pharmacokinetics of a novel anticancer drug, UCN-01, caused by specific high affinity binding to alpa1-acid glycoprotein in humans. Cancer Res. 1999, 59, 1054–1060. [Google Scholar] [PubMed]
- Nijenhuis, C.M.; Schellens, J.H.M.; Beijnen, J.H. Regulatory aspects of human radiolabeled mass balance studies in oncology: Concise review. Drug Metab. Rev. 2016, 48, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Raju, K.S.R.; Taneja, I.; Singh, S.P. Wahajuddin Utility of noninvasive biomatrices in pharmacokinetic studies. Biomed. Chromatogr. 2013, 27, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Rizk, M.L.; Zou, L.; Savic, R.M.; Dooley, K.E. Importance of Drug Pharmacokinetics at the Site of Action. Clin. Transl. Sci. 2017, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Bauer, K.S.; Wu, S.; Smith, R.; Bisacia, S.; Dancey, J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin. Cancer Res. 2007, 13, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Dubbelman, A.-C.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Bioanalytical aspects of clinical mass balance studies in oncology. Bioanalysis 2011, 3, 2637–2655. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Aubry, A.-F. Best Practices in Biological Sample Preparation for LC-MS Bioanalysis. In Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations; Li, W., Zhang, J., Tse, F.L.S., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; pp. 165–184. [Google Scholar]
- Stokvis, E.; Rosing, H.; Beijnen, J.H. Liquid chromatography-mass spectrometry for the quantitative bioanalysis of anticancer drugs. Mass Spectrom. Rev. 2005, 24, 887–917. [Google Scholar] [CrossRef] [PubMed]
- Danaceau, J.P.; Haynes, K.; Chambers, E.E. A Comprehensive Comparison of Solid Phase Extraction (SPE) vs. Solid Liquid Extraction (SLE) vs. Liquid Liquid Extraction (LLE) Sample Prep Techniques in Bioanalysis and Forensic Toxicology Analyses; Waters Corporation: Milford, MA, USA, 2017. [Google Scholar]
- Pucci, V.; Di Palma, S.; Alfieri, A.; Bonelli, F.; Monteagudo, E. A novel strategy for reducing phospholipids-based matrix effect in LC-ESI-MS bioanalysis by means of HybridSPE. J. Pharm. Biomed. Anal. 2009, 50, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Nováková, L.; Vlčková, H. A review of current trends and advances in modern bio-analytical methods: Chromatography and sample preparation. Anal. Chim. Acta 2009, 656, 8–35. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Tran, P.; Gu, H.; Tedesco, V.; Zhang, J.; Lin, W.; Gatlik, E.; Klein, K. Midostaurin, a novel protein kinase inhibitor for the treatment of acute nyelogenous leukemia: Insights from human absorption, metabolism, and excretion studies of a BDDCS II Drugs. Drug Metab. Dispos. 2017, 412, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopez-Martin, J.A.; Pollard, P.; Dumez, H.; Del Muro, X.G.; et al. Phase II randomized study of plitidepsin (aplidin), alone or in association with L-carnitine, in patients with unresectable advanced renal cell carcinoma. Mar. Drugs 2009, 7, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; Lee-Scott, M.; Adamson, P.C.; Blaney, S.M. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: A report from the Children’s Oncology Group. Eur. J. Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.A.; Arbuck, S.G.; Messmann, R.; Headlee, D.; Bauer, K.S.; Lush, R.M.; Murgo, A.; Figg, W.D.; Lahuse, T.; Jaken, S.; et al. Phase I trial of 72-hour continuous infusion UCN-01 in patients with refractory neoplasms. J. Clin. Oncol. 2001, 19, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, F.; Thormann, M.; Feliz, M.; Caba, J.M.; Lloyd-Williams, P.; Giralt, E. Conformational analysis of dehydrodidemnin B (aplidine) by NMR spectroscopy and molecular mechanics/dynamics calculations. J. Org. Chem. 2001, 66, 4580–4584. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.; Anthoney, A.; Metges, J.P.; Alsina, M.; Gonçalves, A.; Brown, J.; Montagut, C.; Gunzer, K.; Laus, G.; Iglesias Dios, J.L.; et al. Phase Ib/II study of elisidepsin in metastatic or advanced gastroesophageal cancer (IMAGE trial). Cancer Chemother. Pharmacol. 2016, 77, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.R.; Sarantopoulos, J.; Lee, L.; Reyderman, L.; He, Y.; Olivo, M.; Goel, S. Pharmacokinetics of eribulin mesylate in cancer patients with normal and impaired renal function. Cancer Chemother. Pharmacol. 2015, 76, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Gutierrez, M.E.; Gardner, E.R.; Figg, W.D.; Melillo, G.; Dancey, J.; Sausville, E.A.; Conley, B.A.; Murgo, A.J.; Doroshow, J.H. A phase I trial of UCN-01 and prednisone in patients with refractory solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2010, 65, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Perl, A.; Luger, S.; Baer, M.R.; Norsworthy, K.J.; Bauer, K.S.; Tidwell, M.; Fleckinger, S.; Carroll, M.; Sausville, E.A. Phase I study of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Investig. New Drugs 2013, 31, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Hotte, S.J.; Oza, A.; Winquist, E.W.; Moore, M.; Chen, E.X.; Brown, S.; Pond, G.R.; Dancey, J.E.; Hirte, H.W. Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: A Princess Margaret Hospital Phase II Consortium study. Ann. Oncol. 2006, 17, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Stokvis, E.; Rosing, H.; Beijnen, J.H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: Necessity or not? Rapid Commun. Mass Spectrom. 2005, 19, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Stokvis, E.; Rosing, H.; López-Lázaro, L.; Schellens, J.H.M.; Beijnen, J.H. Switching from an analogous to a stable isotopically labeled internal standard for the LC-MS/MS quantitation of the novel anticancer drug Kahalalide F significantly improves assay performance. Biomed. Chromatogr. 2004, 18, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Hillebrand, M.J.X.; Bosch, T.M.; Lopez-Lazaro, L.; Schellens, J.H.M.; Beijnen, J.H. Metabolism of trabectedin (ET-743, Yondelis) in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 825–837. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Synonym (s) | Natural Product or Derivative | Origin | Administration | Dose | Indication | Status |
|---|---|---|---|---|---|---|---|
| AGS-67E | Derivative | Cyanobacterium Caldora penicillata [15] | intravenous infusion (i.v.) | Escalating doses | Lymphoid malignancies [18] | Phase I | |
| Brentuximab vedotin (Adcetris®) | SGN-35/cAC10-vcMMAE | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 1.8 mg/kg [19] | Hodgkin lymphoma, Systemic anaplastic large cell lymphoma [19] | Approved |
| Cytarabine (Cytosar-U®/DepoCyte®) | Ara-C/Cytosine arabinoside/1-ß-D-Arabinofuranosylcytosine | Derivative | Sponge Cryptotethya crypta [12] | i.v./intrathecal | 75–200 mg/m2 [20] | Acute myelogenous leukemia, Chronic myelogenous leukemia, Acute lymphoblastic leukemia non-Hodgkin’s lymphoma [20] | Approved |
| Denintuzumab mafodotin | SGN-CD19A/SGN-19A | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 3 mg/kg | Diffuse large B-cell lymphoma [21] | Phase II |
| Depatuxizumab mafodotin | ABT-414 | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 1.25 mg/kg | Glioblastoma/Pediatric brain tumors [22] | Phase II |
| Enfortumab vedotin | ASG-22ME/ASG-22CE | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | Urothelial cancer [23] | Phase II | |
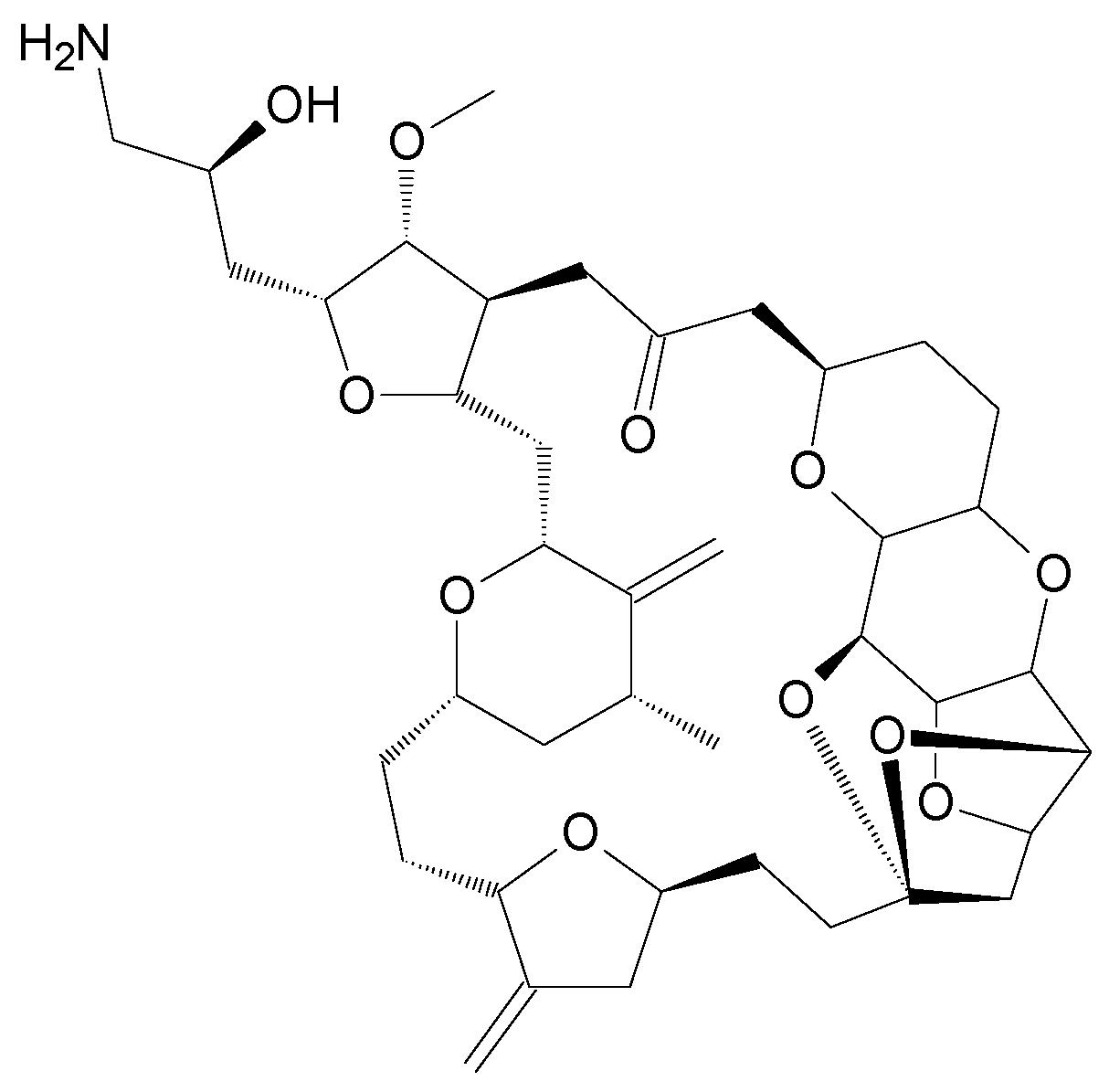
| Eribulin mesylate (Halaven®) | E7389 | Derivative | Sponge Halichodria okadai [12] | i.v. | 1.23 mg/m² [24] | Breast cancer, Liposarcoma [24] | Approved |
| Glembatumumab vedotin | CDX-011/CR011-vcMMAE | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 1.9, 2.2 mg/kg | Breast cancer, Metastatic melanoma [25] | Phase II |
| GSK2857916 | J6M0-mcMMAF | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | Escalating doses | Multiple myeloma [26] | Phase I |
| Ladiratuzumab vedotin | SGN-LIV1A | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | Escalating doses | Human epidermal growth factor receptor 2 (HER2)-negative breast cancer, triple negative breast cancer | Phase I |
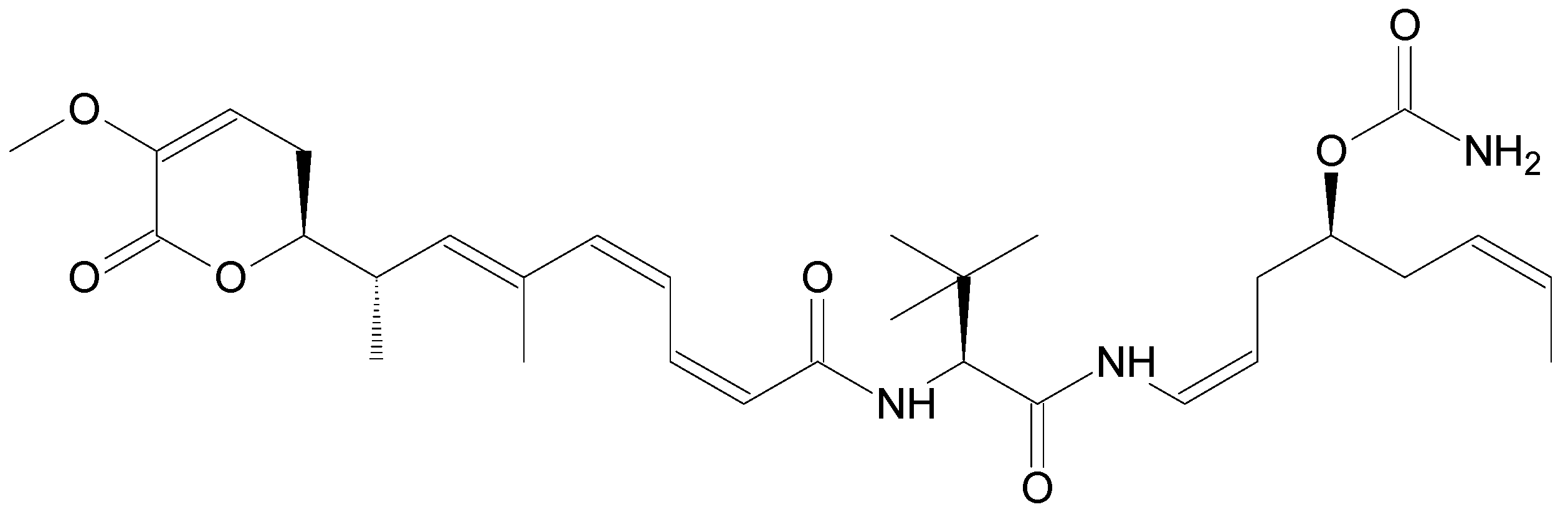
| Lurbinectedin (Zepsyre®) | PM01183 | Trabectedin analogue | Tunicate Ecteinascidia turbinata [12] | i.v. | 3.2 mg/mL | Platinum-resistant ovarian cancer [27] | Phase III |
| Marizomib | Salinosporamide A/NPI-0052 | Natural product | Marine actinomycete Salinispora tropica [12] | i.v. | 0.5 mg/m2 | Multiple myeloma, glioblastoma [28] | Phase II |
| Midostaurin (Rydapt®) | PKC412/CGP41251/N-benzoylstaurosporine | Staurosporine analogue | Bacterium Streptomyces staurosporeus [29] | oral | 50 mg twice daily [30] | FLT3+ Acute myeloid leukemia [30] | Approved |
| Pinatuzumab vedotin | DCDT-2980S/RG7593 | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 1.8, 2.4 mg/kg | Follicular lymphoma, Diffuse large B-cell lymphoma | Phase I |
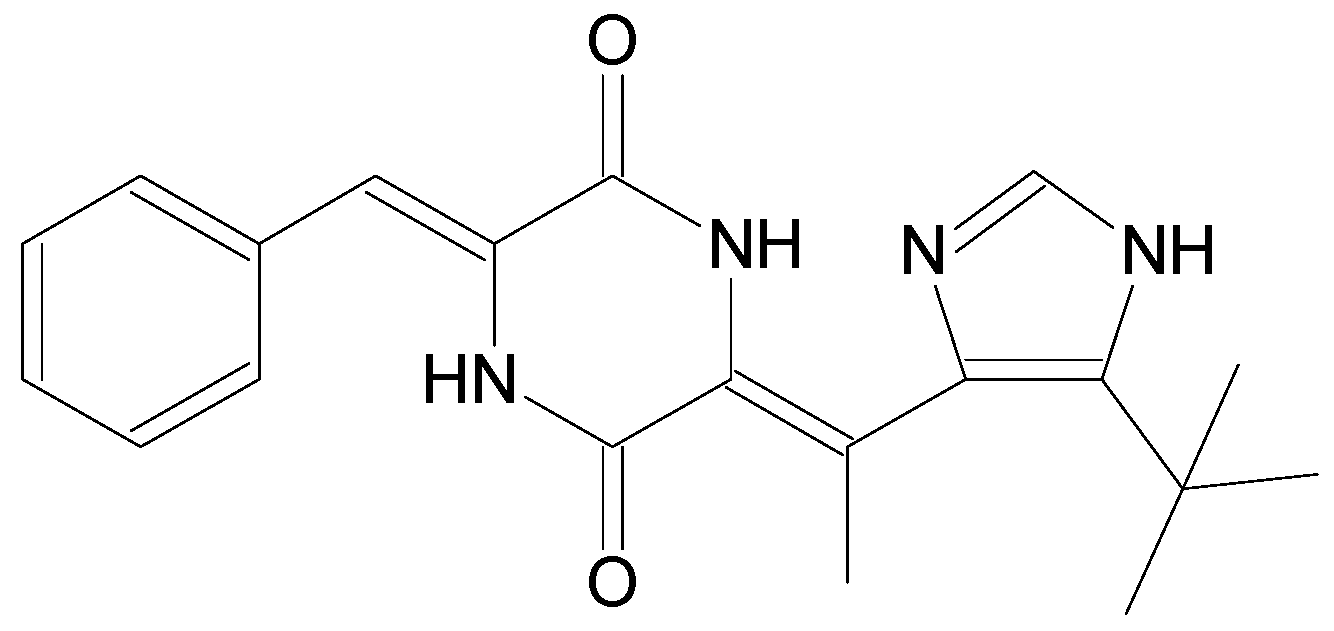
| Plinabulin | NPI2358 | Derivative | Marine fungus Aspergillus sp. [12] | i.v. | 30 mg/m2 | Non-small cell lung cancer [31] | Phase III |
| Plitidepsin (Aplidin®) | Dehydrodidemnin B | Natural product | Tunicate Aplidium albicans [12] | i.v. | 5 mg/m2 | Multiple Myeloma, Lymphoma [27] | Phase III |
| PM060184 | PM0184/Plocabulin | Natural product | Sponge Lithoplocamia lithistoides [12] | i.v. | Starting dose 4 mg/m2 | Breast cancer, Solid tumors [27] | Phase I |
| Polatuzumab vedotin | DCDS-4501A | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | 1.8, 2.4 mg/kg | Non-Hodgkin’s lymphoma, B-cell lymphoma [32] | Phase II |
| Tisotumab vedotin | HuMax-TF-ADC/HuMab-TF-011-vcMMAE/TF-011-vcMMAE | Derivative | Cyanobacterium Caldora penicillata [15] | i.v. | Escalating doses | Solid tumors [33] | Phase I |
| Trabectedin (Yondelis®) | ET-743 | Natural product | Tunicate Ecteinascidia turbinate [12] | i.v. | 1.1/1.5 mg/m2 [34] | Soft tissue sarcoma [34] | Approved |
| Name | Synonym (s) | Natural Product or Derivative | Origin | Discontinued/Inactive | Reason for Discontinuation |
|---|---|---|---|---|---|
| ASG-15ME | AGS15E | Derivative | Cyanobacterium Caldora penicillata [15] | Discontinued | Unspecified [35] |
| Becatecarin | XL-119/NSC 655649/BMY 27557/BMS 181176 | Rebeccamycin analogue | Marine actinomycete Saccharothrix aerocolonigenes [36] | Discontinued | Not superior to existing therapies [37] |
| CEP-2563 | KT-8391 | Staurosporine derivative | NAT | ||
| Didemnin B | Natural product | Tunicate Trididemnin cyanophorum [38] | Discontinued | Toxicity [39] | |
| Edotecarin | J-107088/PF-804950/PHA-782615/ED-749 | Derivative | NAT | ||
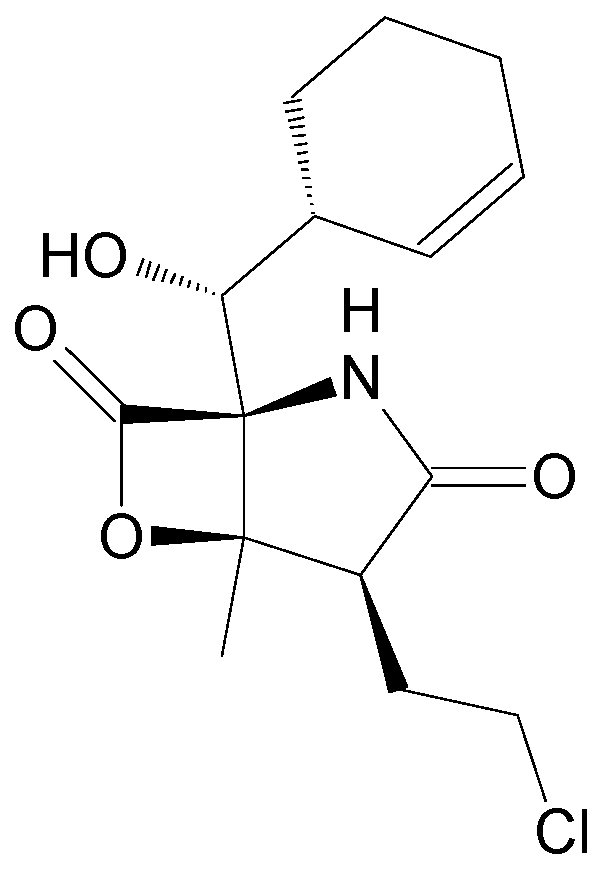
| Elisidepsin (Irvalec®) | PM02734 | Structural analogue | Mollusk Elysia rufescens [40] | Discontinued | Strategic [41] |
| Enzastaurin | LY317615 | Staurosporine derivative | Bacterium Streptomyces staurosporeus | Discontinued | Lack of efficacy [42] |
| Indusatumab vedotin | MLN-0264/TAK-0264 | Derivative | Cyanobacterium Caldora penicillata [15] | Discontinued | Lack of efficacy [43] |
| Lestaurtinib | CEP-701 | Staurosporine derivative | Bacterium Streptomyces staurosporeus | Discontinued | Strategic [41] |
| Lifastuzumab vedotin | DNIB0600A/Anti-NaPi2B ADC/RG7599 | Derivative | Cyanobacterium Caldora penicillata [15] | Discontinued | Lack of efficacy [44] |
| Pipecolidepsin A | Natural product | Sponge Homophymia lamellose [13] | NAT | ||
| Stellatolide A | Natural product | Sponge Ecionemia acervus [45] | NAT | ||
| UCN-01 | 7-hydroxystaurosporine | Staurosporine analogue | Bacterium Streptomyces staurosporeus [29] | NAT | |
| Vandortuzumab vedotin | DSTP-3086 S/RG-7450/thio-antiSTEAP1-MC-vc-PAB-MMAE | Derivative | Cyanobacterium Caldora penicillata [15] | NAT | |
| Zalypsis | PM00104/PM-10450 | Derivative | Sponge Netropsia sp. [15] | NAT |
| Compound | Matrix | Sample Pre-Treatment | Stationary Phase | Mobile Phase | Detection | Internal Standard | Linear Range | LOD | Metabolites | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Elisidepsin | Plasma (Dog) | LLE (ethyl acetate) | YMC Pro C18, S-5 (50 × 2.0 mm, 120 Å) | A: 5 mM ammonium acetate, 0.1% FA in H2O B: 0.1% FA in MeOH | MS/MS | 2H8-PM02734 (SIL) | 0.05–100 ng/mL | No | [62] | |
| Eribulin mesylate | Plasma Urine (Human) | LLE (ethyl acetate/MeOH/EtOH) | Polaris® C18 (30 × 2.0 mm, 3 μm) | A: 0.1% FA in H2O-ACN (87:13, v/v) B: 0.1% FA in THF-ACN (30:70, v/v) | MS/MS | ER-076349 (Structural analogue) | 0.2–100 ng/mL | No | [63] | |
| Eribulin mesylate | Plasma Whole blood Urine Feces (Human) | P/WB/U: LLE (ethyl acetate/MeOH/EtOH) F: dilution (ACN) | Polaris® C18-A (30 × 2.0 mm, 3 μm) | A: 0.1% FA in H2O B: 0.1% FA in ACN | MS/MS | ER-076349 (Structural analogue) | 0.2–100 ng/mL (P); 0.5–100 ng/mL (WB/U); 100–25,000 ng/mL (F) | No | [64] | |
| Lurbinectedin | Plasma (Cynomolgus monkey Dog Mice Mini-pig Rat) | SLE (TBME) | ACE C18 PFP (30 × 2.1 mm, 3 μm) | A: 0.1% FA in H2O B: 0.1% FA in ACN | MS/MS | PM040038 (SIL) | 0.1–100 ng/mL | 0.025 ng/mL | No | [65] |
| Midostaurin | Plasma (Human) | LLE (diisopropyl ether) | μBondapak RP-18 (300 × 3.9 mm, 10 μm) | ACN-0.001 M ammonium acetate in H2O, pH 4.0 (45:55, v/v) | FLD 286/386 nm | CGP 41 126 (Structural analogue) | 1–1000 ng/mL | 0.5 ng/mL | CGP 50 723; CGP 50 750; CGP 52 421 | [66] |
| Midostaurin | Plasma (Human) | LLE (diisopropyl ether) | RP LiChrospher C18 end-capped (125 × 4.0 mm, 5 μm) | A: ACN B: 445 μL TEA in 1 L phosphate buffer, pH 3.6 | FLD 286/386 nm | CGP 41 126 (Structural analogue) | 0.2–1000 ng/mL | 0.1 ng/mL | CGP 50 673; CGP 50 723; CGP 50 750; CGP 52 421 | [67] |
| Midostaurin | Plasma (Human) | LLE (diethyl ether) | Prodigy ODS-2 (150 × 3.2 mm, 5 μm) | 0.4 mL Titriplex III-solution in MeOH-ACN-0.05 M ammonium acetate in water (40:26:34, v/v/v) | FLD 286/386 nm | N-phenyl-1- naphthylamine | 10–10,000 ng/mL | 10 ng/mL | CGP 52421e1; CGP 52421e2; CGP 62221; CGP 62221e1; CGP 62221e2; | [68] |
| Midostaurin | Plasma (Human) | PP (MeOH) | SunFire bonded and end-capped C18 silica (150 × 2.1 mm, 3.5 μm) | A: 10 mM ammonium formate, 0.1% FA in H2O B: 0.1% FA in ACN | MS/MS | Midostaurin-d5 (SIL) | 75–2500 ng/mL | No | [69] | |
| Plitidepsin | Plasma Urine (Rat) | PP (0.1% FA in ACN) + LLE (chloroform) | RP C18 Hypersil-5 ODS (100 × 3.0 mm; 5 μm, 120 Å) | A: 0.5% FA in H2O B: 0.5% FA in ACN | MS/MS | Didemnin B (Structural analogue) | 5–100 ng/mL (P); 1.25–125 ng/mL (U) | 1 ng/mL (P); 0.5 ng/mL (U) | No | [70] |
| Plitidepsin | Plasma (Mice) | Derivatization + SPE | Symmetry C18 (100 × 4.6 mm, 3.5 μm) | ACN–water–TFA (47:52.9:0.1, v/v/v) | FLD 410/560 nm | None | 2–100 ng/mL | No | [71] | |
| Plitidepsin | Plasma (Human) | LLE (TBME) | Zorbax Bonus-RP (50 × 2.0 mm, 5 μm) | 0.1% FA in ACN-5 mM ammonium acetate, 0.1% FA in H2O (80:20, v/v) | MS/MS | Didemnin B (Structural analogue) | 0.05–50 ng/mL | No | [72] | |
| Plitidepsin | Plasma Whole Blood Urine (Human) | PP (0.1% FA in ACN) + LLE (chloroform) | Hypersil-5 ODS (100 x 3.0 mm, 5 μm, 120 Å) | A: 0.5% FA in ACN B: 0.5% FA in H2O | MS/MS | Didemnin B (Structural analogue) | 1–250 ng/mL | 0.25 ng/ml | No | [73] |
| Plitidepsin | Whole Blood (Human) | Derivatization + SPE | Symmetry C18 (100 × 4.6 mm, 3.5 μm) | ACN-0.1% TFA in H2O (50:50, v/v) | FLD 410/560 nm | None | 2–100 ng/mL | No | [74] | |
| Plitidepsin | Plasma Whole Blood Urine (Human) | LLE (TBME) | SunFire C18 (50 × 2.1 mm, 5 μm) | A: 5 mM ammonium acetate, 0.1% FA in H2O B: 0.1% FA in ACN | MS/MS | (PM130461) 13C5-15N-plitidepsin (SIL) | 0.1–100 ng/mL | No | [75] | |
| Trabectedin | Plasma (Human) | SPE | Zorbax SB-C18 column (75 × 4.6 mm, 3.5 μm) | ACN–25 mM phosphate buffer, pH 5.0 (70:30, v/v) | UV 210 nm | POB | 1–50 ng/mL | No | [76] | |
| Trabectedin | Plasma (Human) | SPE | Zorbax Rx-C18 (150 × 2.1 mm, 5 μm) | MeOH-5 mM ammonium acetate, 0.4% FA in H2O (75:25, v/v) | MS/MS | ET-729 (Structural analogue) | 0.01–2.5 ng/mL | No | [77] | |
| Trabectedin | Plasma (Human) | PP (MeOH) | Zorbax Rx-C18 (150 × 2.1 mm, 5 μm) | MeOH–H2O (85:15, v/v) | MS/MS | 2H3-ET-743 (SIL) | 0.05–2.5 ng/mL | No | [78] | |
| Trabectedin | Plasma (Human) | PP (HCl in MeOH) | Accucore XL C18 (50 × 2.1 mm, 4 μm) | A: 10 mM ammonium acetate in H2O, pH 6.8 B: MeOH | MS/MS | 2H3-ET-743 (SIL) | 0.025–1.0 ng/ml | No | [79] | |
| Trabectedin | Liver cells Tumor cells (Mice) | Lysis | Accucore XL C18 (50 × 2.1 mm, 4 μm) | A: 10 mM ammonium acetate in H2O, pH 6.8B: MeOH | MS/MS | 2H3-ET-743 (SIL) | 0.1–3 ng/mL (TC); 0.25–6 ng/mL (LC) | No | [80] | |
| UCN-01 | Plasma Urine (Human) | PP (ACN) | AM-312 ODS (150 × 6.0 mm S-5mm, 120 Å) | ACN–0.1% TEA in 0.05 M phosphate buffer, pH 7.3 (50:50, v/v). | FLD 310/410 nm | Staurosporine (Structural analogue) | 0.2–100 ng/mL (P); 1–400 ng/mL (U) | No | [81] | |
| UCN-01 | Plasma Saliva (Human) | PP (ACN) | Nova-Pak Phenyl (150 × 3.9 mm, 4 μm) | A: 0.05 M ammonium acetate in H2O, pH 4.15 B: ACN | UV (P) 295 nm FLD (Sal) 290/400 nm | Umbelliferone | 200–20,000 ng/mL (P); 4–200 ng/mL (S) | No | [82] | |
| UCN-01 | Plasma (Human) | PP (ACN) | Nova-Pak Phenyl (150 × 3.9 mm, 4 μm, 60 Å) | ACN–0.5 M ammonium acetate, 0.2% TEA in H2O (45:55, v/v) | FLD 310/410 nm | Staurosporine (Structural analogue) | 200–30,000 ng/mL | 0.1 μg/mL | No | [83] |
| Zalypsis | Plasma (Dog Human Mice Rat) | LLE (TBME) | Zorbax SB-C18 (50 × 2.1 mm, 5 μm, 80 Å) | A: 5 mM ammonium acetate, 0.1% FA in H2O B: 0.1% FA in MeOH | MS/MS | 13C2,2H3-PM00104 (SIL) | 0.01–5 ng/mL | No | [84] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Andel, L.; Rosing, H.; Schellens, J.H.; Beijnen, J.H. Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment. Mar. Drugs 2018, 16, 246. https://doi.org/10.3390/md16070246
Van Andel L, Rosing H, Schellens JH, Beijnen JH. Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment. Marine Drugs. 2018; 16(7):246. https://doi.org/10.3390/md16070246
Chicago/Turabian StyleVan Andel, Lotte, Hilde Rosing, Jan HM Schellens, and Jos H Beijnen. 2018. "Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment" Marine Drugs 16, no. 7: 246. https://doi.org/10.3390/md16070246
APA StyleVan Andel, L., Rosing, H., Schellens, J. H., & Beijnen, J. H. (2018). Review of Chromatographic Bioanalytical Assays for the Quantitative Determination of Marine-Derived Drugs for Cancer Treatment. Marine Drugs, 16(7), 246. https://doi.org/10.3390/md16070246