Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells

 ,
,  , ,
, ,  ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
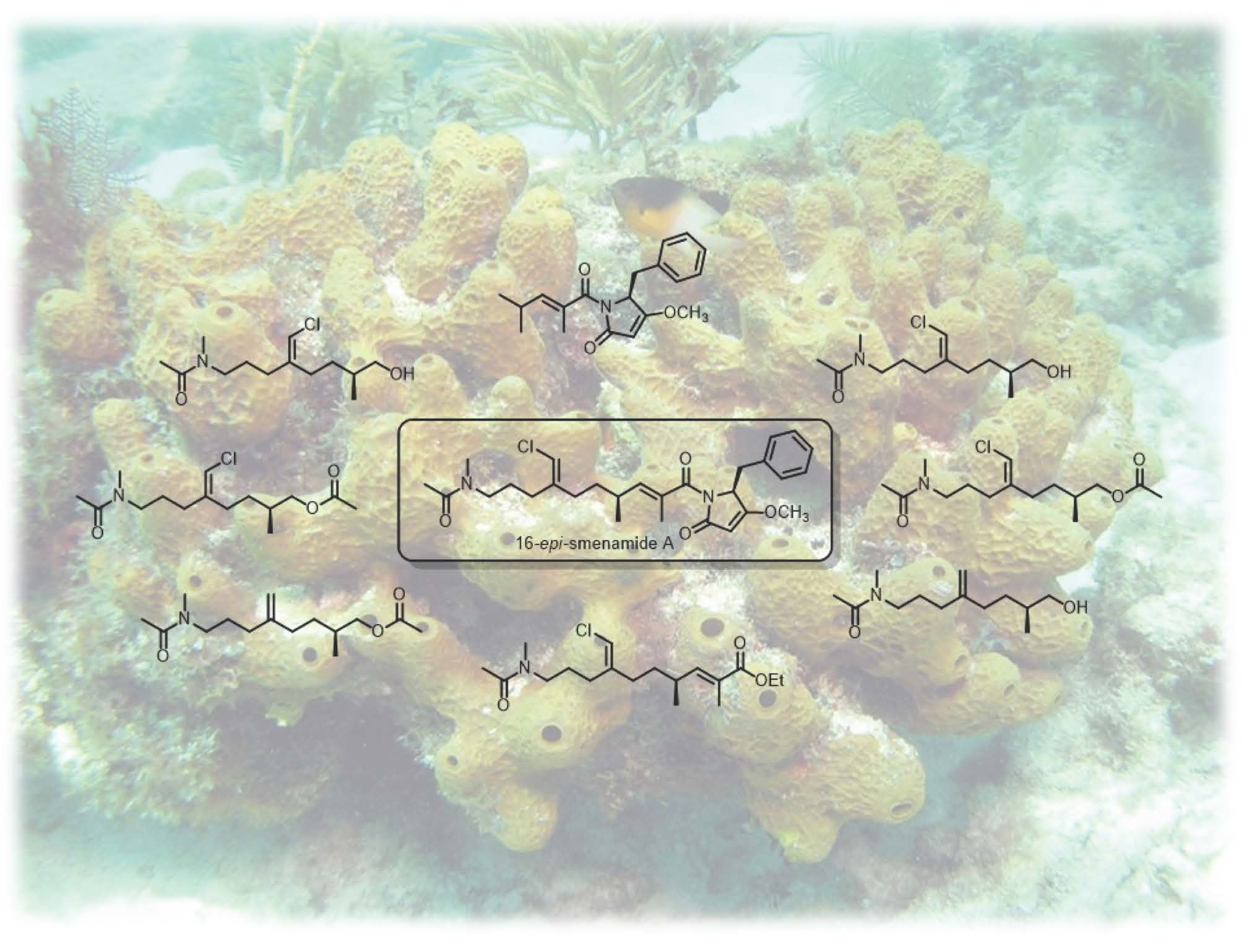
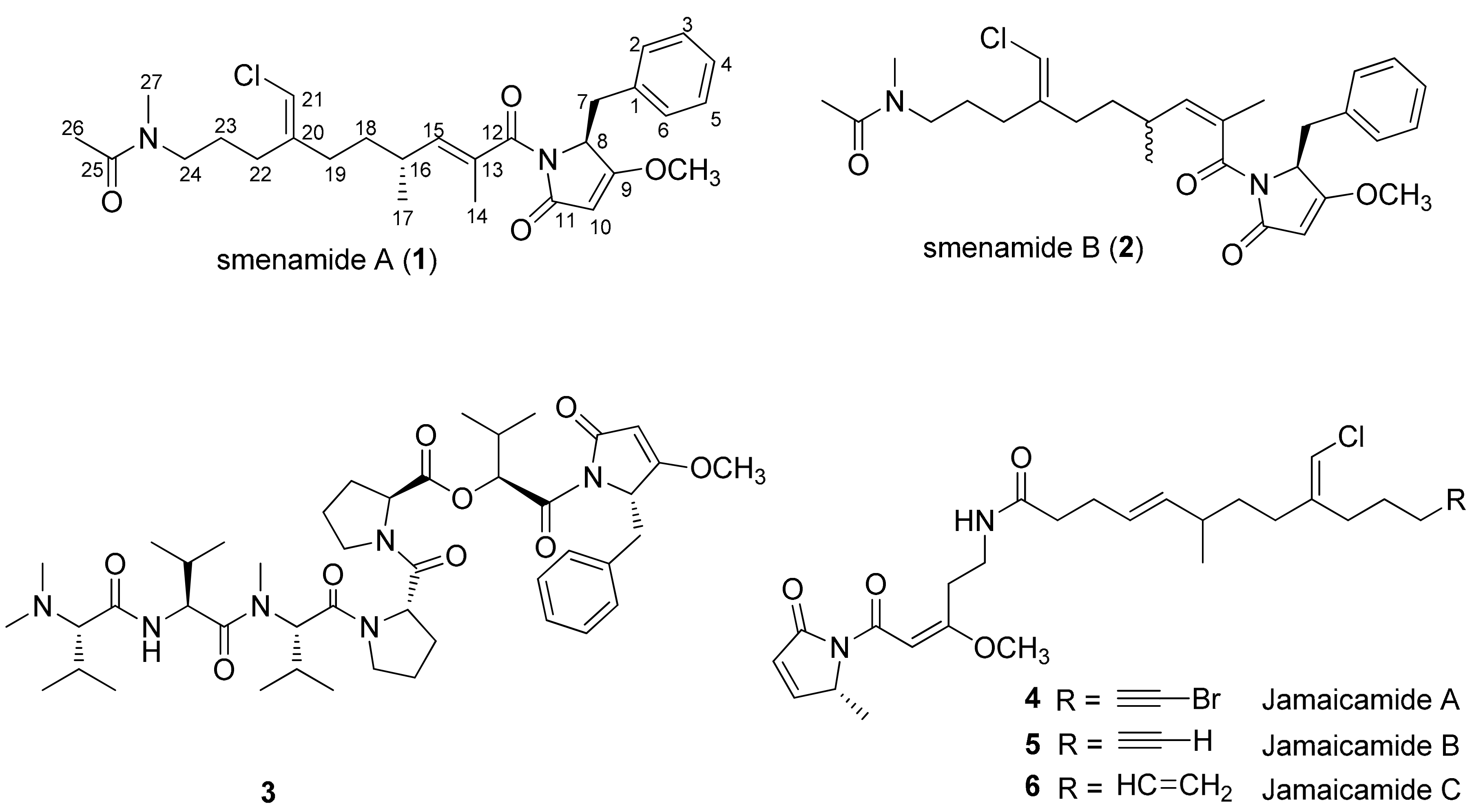
1. Introduction
2. Results and Discussion
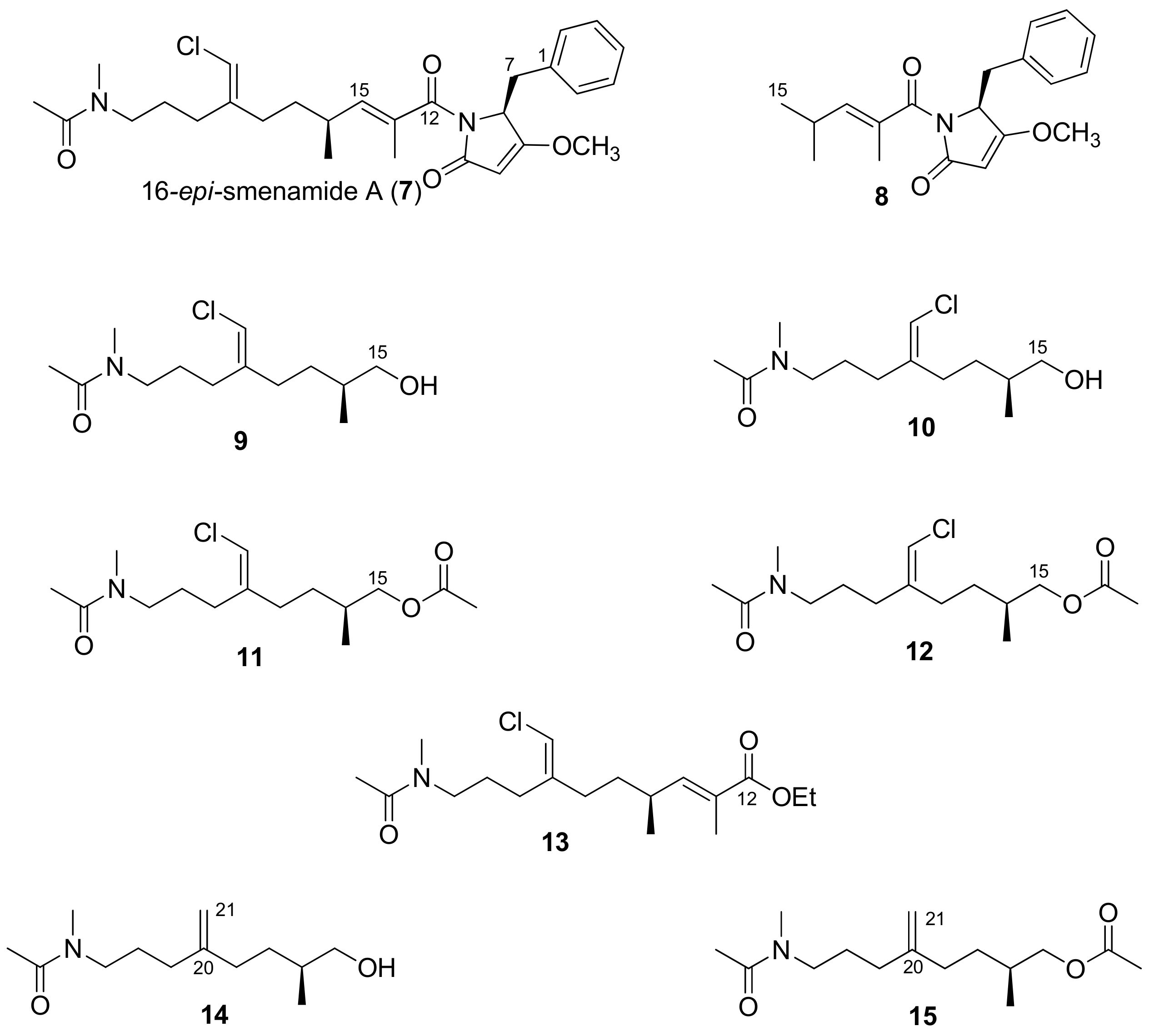
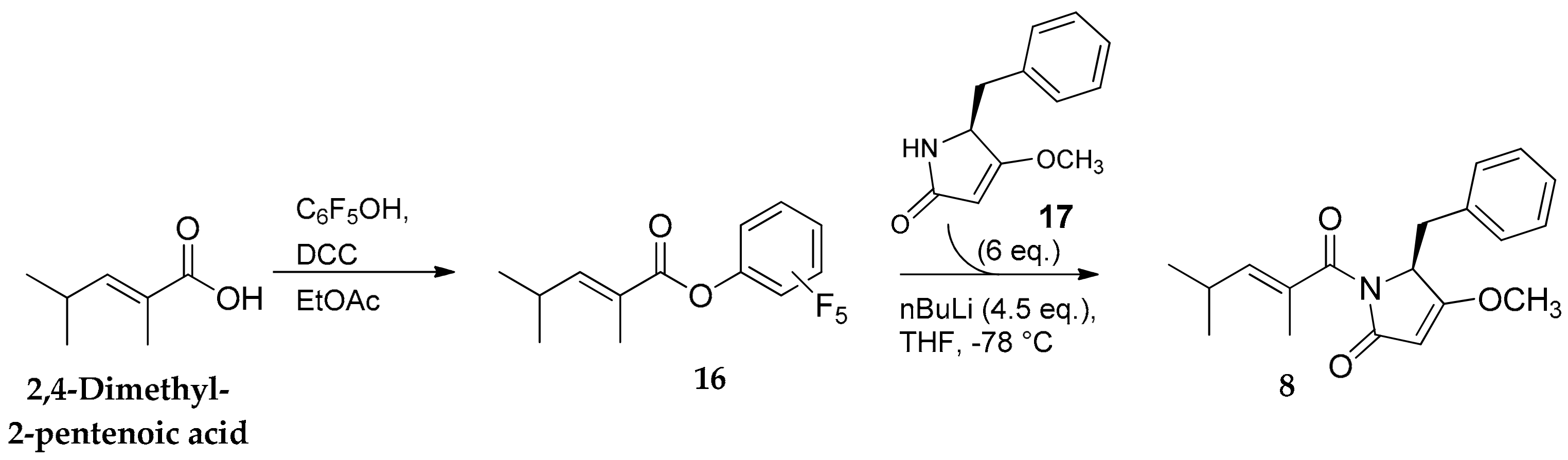
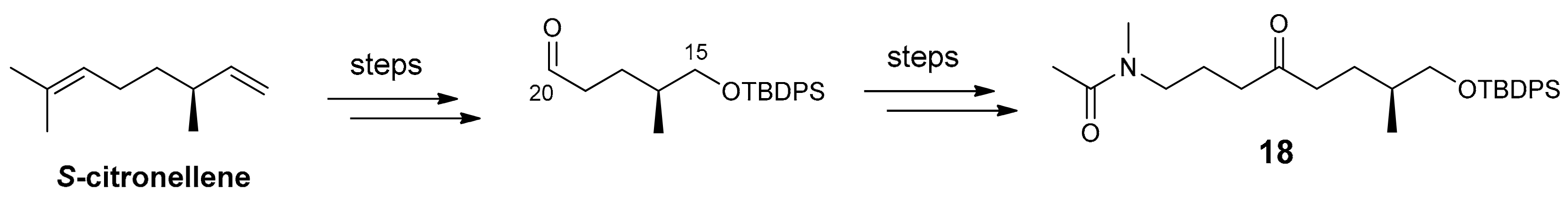
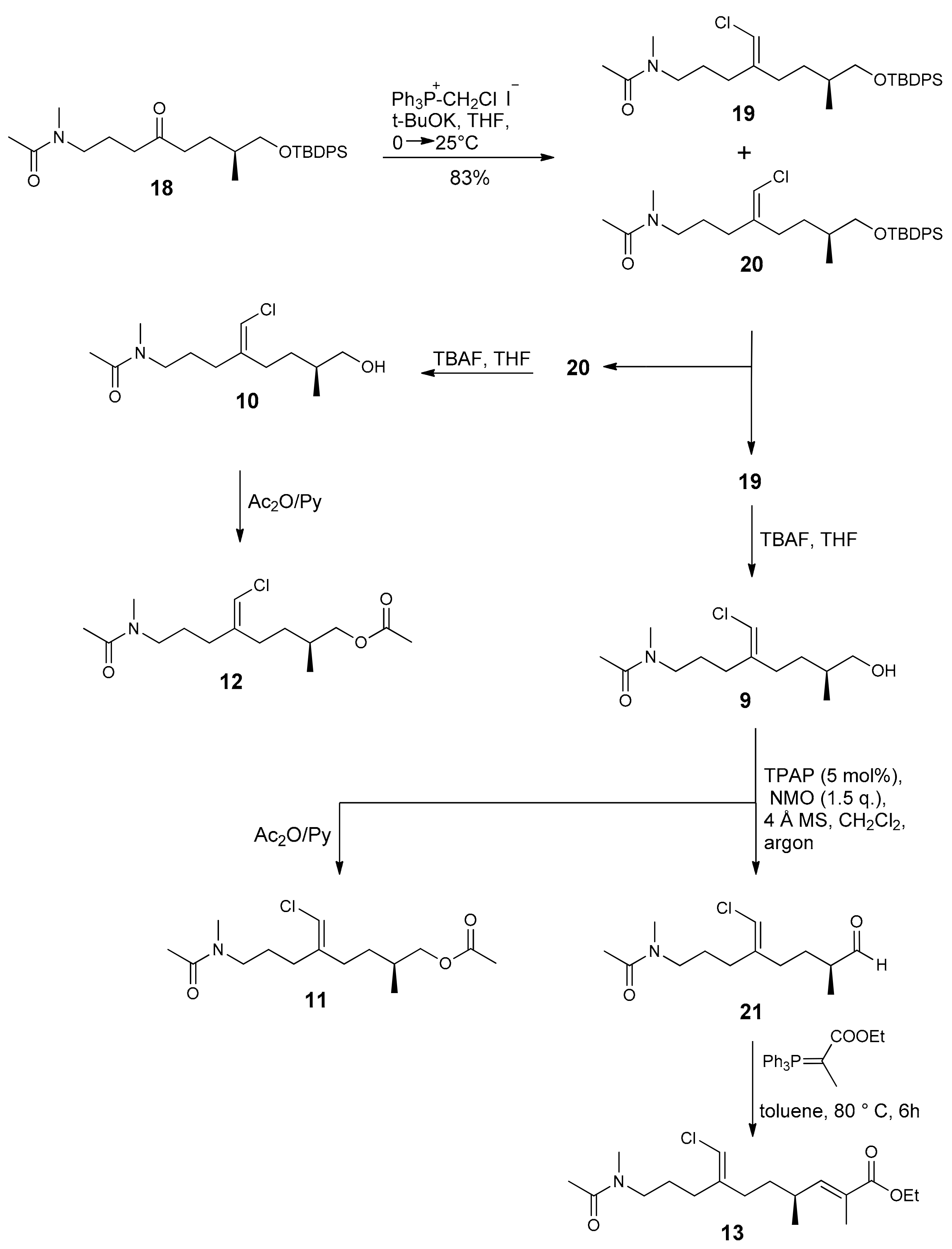
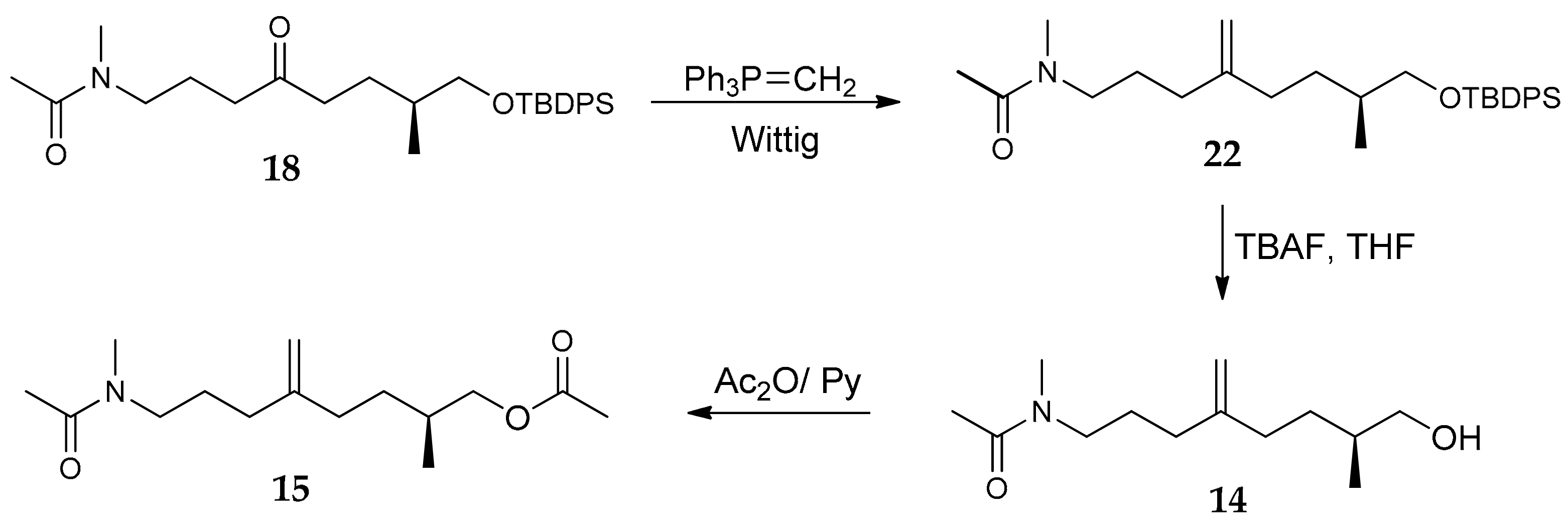
2.1. Compounds 7–15
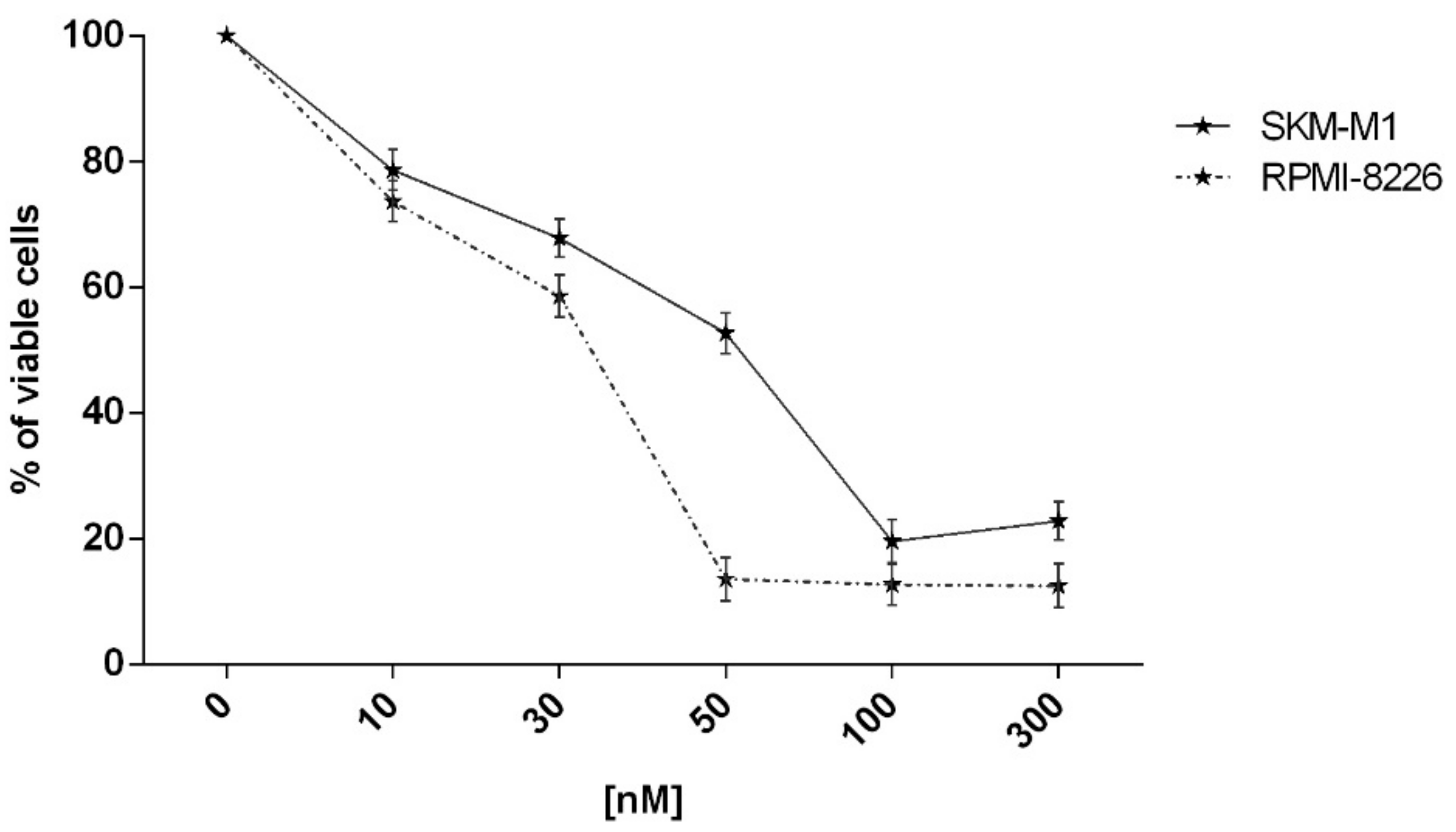
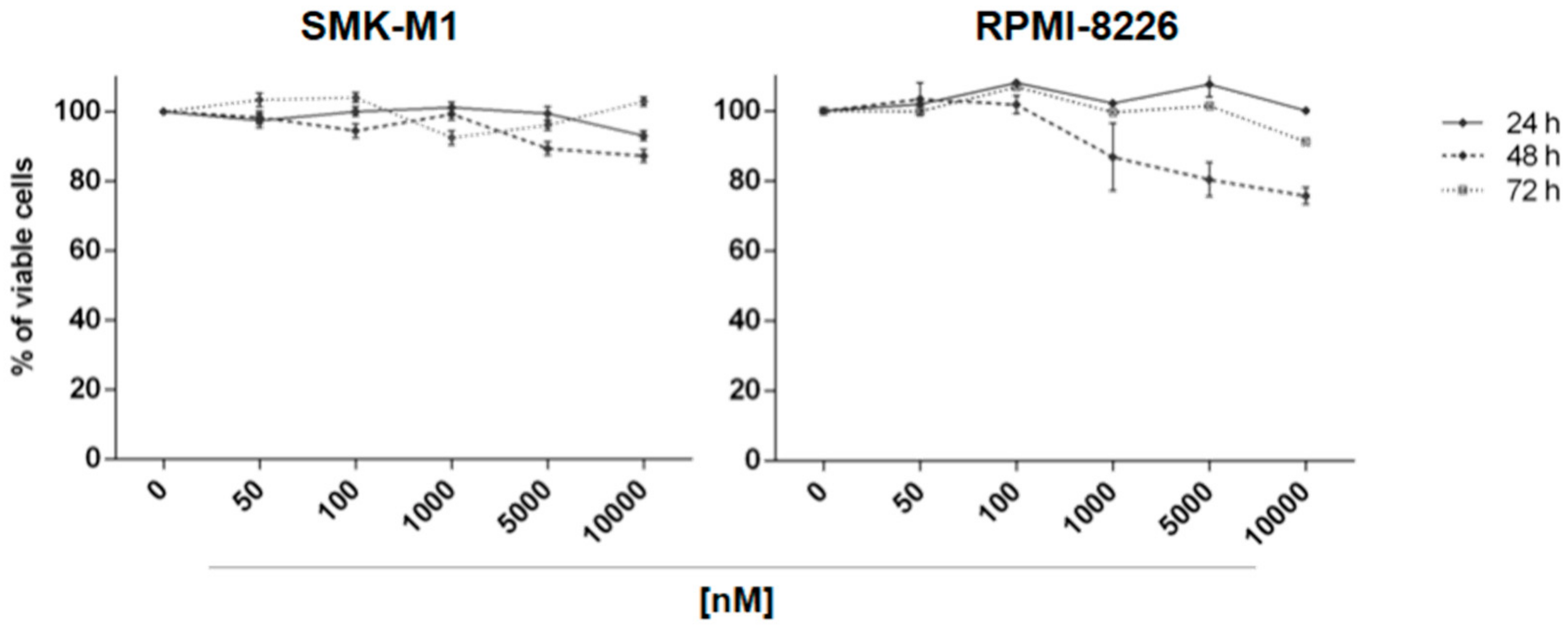
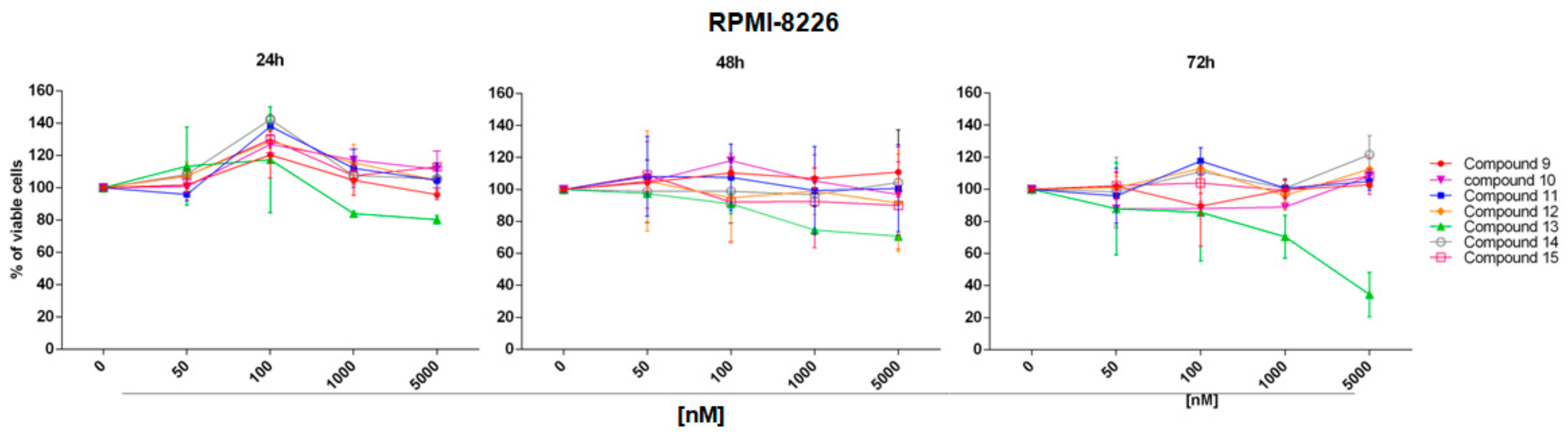
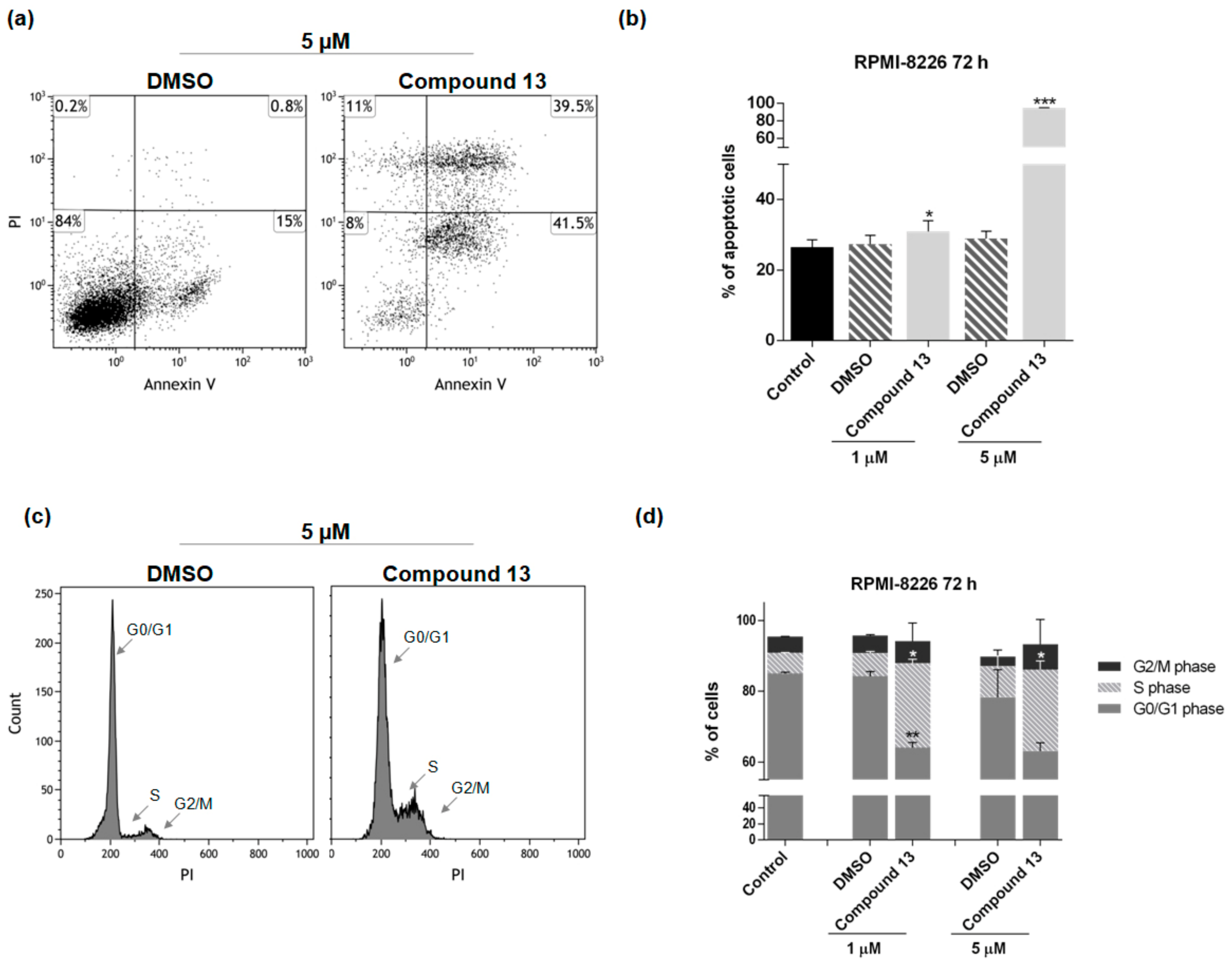
2.2. In Vitro Evaluation of Activity on Multiple Myeloma Cell Lines
3. Experimental Section
3.1. General Experimental Procedures









3.2. Biological Activity
3.2.1. Cell Lines and Chemical
3.2.2. Cell Viability
3.2.3. Functional Tests
3.2.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Glycolipids from sponges. Part 17.1 Clathrosides and isoclathrosides, unique glycolipids from the Caribbean sponge Agelas clathrodes. J. Nat. Prod. 2006, 69, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; D’Esposito, M.; Fattorusso, E.; Mangoni, A.; Basilico, N.; Parapini, S.; Taramelli, D. Damicoside from Axinella damicornis: The Influence of a Glycosylated Galactose 4-OH Group on the Immunostimulatory Activity of α-Galactoglycosphingolipids. J. Med. Chem. 2005, 48, 7411–7417. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Perinu, C.; Teta, R.; Panza, E.; Ianaro, A. Tedarenes A and B: Structural and stereochemical analysis of two new strained cyclic diarylheptanoids from the marine sponge Tedania ignis. J. Org. Chem. 2012, 77, 6377–6383. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, I.; Caivano, A.; La Rocca, F.; Trino, S.; De Luca, L.; D’Alessio, F.; Schenone, S.; Falco, G.; Botta, M.; Del Vecchio, L.; et al. A pyrazolo[3,4-d]pyrimidine compound reduces cell viability and induces apoptosis in different hematological malignancies. Front. Pharmacol. 2016, 7, 416. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides A and B, chlorinated peptide/polyketide hybrids containing a dolapyrrolidinone unit from the Caribbean sponge Smenospongia aurea. Evaluation of their role as leads in antitumor drug research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Friedman, S.J.; Pettit, G.R.; Hamel, E. Dolastatin 15, a potent antimitotic depsipeptide derived from Dolabella auricularia: Interaction with tubulin and effects on cellular microtubules. Biochem. Pharmacol. 1992, 43, 2637–2645. [Google Scholar] [CrossRef]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscule. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Caso, A.; Mangoni, A.; Piccialli, G.; Costantino, V.; Piccialli, V. Studies toward the Synthesis of Smenamide A, an Antiproliferative Metabolite from Smenospongia aurea: Total Synthesis of ent-Smenamide A and 16-epi-Smenamide A. ACS Omega 2017, 2, 1477–1488. [Google Scholar] [CrossRef]
- La Rocca, F.; Airoldi, I.; Di Carlo, E.; Marotta, P.; Falco, G.; Simeon, V.; Laurenzana, I.; Trino, S.; De Luca, L.; Todoerti, K.; et al. EphA3 targeting reduces in vitro adhesion and invasion and in vivo growth and angiogenesis of multiple myeloma cells. Cell. Oncol. 2017, 40, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Caivano, A.; La Rocca, F.; Laurenzana, I.; Annese, T.; Tamma, R.; Famigliari, U.; Simeon, V.; Trino, S.; De Luca, L.; Villani, O.; et al. Epha3 acts as proangiogenic factor in multiple myeloma. Oncotarget 2017, 8, 34298–34309. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Bourguet-Kondracki, M.-L.; Mai, L.H.; Longeon, A.; Teta, R.; Meijer, L.; Van Soest, R.; Mangoni, A.; Costantino, V. Chloromethylhalicyclamine B, a Marine-Derived Protein Kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016, 79, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Della Sala, G.; Teta, R.; Caso, A.; Bourguet-Kondracki, M.-L.; Pawlik, J.R.; Mangoni, A.; Costantino, V. Chlorinated thiazole containing polyketide-peptides from the Caribbean sponge Smenospongia conulosa: Structure elucidation on microgram scale. EJOC 2016, 16, 2871–2875. [Google Scholar] [CrossRef]
- Ley, S.V.; Norman, J.; Griffith, W.P.; Marsden, S.P. Tetrapropylammoniumperruthenate, Pr4N+RuO4−, TPAP: A catalytic oxidant for organic synthesis. Synthesis 1994, 639–666. [Google Scholar] [CrossRef]
- Piccialli, V. Ruthenium tetroxide and perruthenate chemistry. Recent advances and related transformations mediated by other transition metal oxospecies. Molecules 2014, 19, 6534–6582. [Google Scholar] [CrossRef] [PubMed]
- Zerk, T.J.; Moore, P.W.; Harbort, J.S.; Chow, S.; Byrne, L.; Koutsantonis, G.A.; Harmer, J.R.; Martínez, M.; Williams, C.M.; Bernhardt, P.V. Elucidating the mechanism of the Ley–Griffith (TPAP) alcohol oxidation. Chem. Sci. 2017, 8, 8435–8442. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, I.; Caivano, A.; Trino, S.; De Luca, L.; La Rocca, F.; Simeon, V.; Tintori, C.; D’Alessio, F.; Teramo, A.; Zambello, R.; et al. A Pyrazolo[3,4-d]pyrimidine compound inhibits Fyn phosphorylation and induces apoptosis in natural killer cell leukemia. Oncotarget 2016, 7, 65171–65184. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Teta, R.; Miceli, R.; Ceccarelli, L.S.; Della Sala, G.; Camerlingo, R.; Irollo, E.; Mangoni, A.; Pirozzi, G.; Costantino, V. Isolation and assessment of the in vitro anti-tumor activity of smenothiazole A and B, chlorinated thiazole-containing peptide/polyketides from the Caribbean sponge, Smenospongiaaurea. Mar. Drugs 2015, 13, 444–459. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caso, A.; Laurenzana, I.; Lamorte, D.; Trino, S.; Esposito, G.; Piccialli, V.; Costantino, V. Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells. Mar. Drugs 2018, 16, 206. https://doi.org/10.3390/md16060206
Caso A, Laurenzana I, Lamorte D, Trino S, Esposito G, Piccialli V, Costantino V. Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells. Marine Drugs. 2018; 16(6):206. https://doi.org/10.3390/md16060206
Chicago/Turabian StyleCaso, Alessia, Ilaria Laurenzana, Daniela Lamorte, Stefania Trino, Germana Esposito, Vincenzo Piccialli, and Valeria Costantino. 2018. "Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells" Marine Drugs 16, no. 6: 206. https://doi.org/10.3390/md16060206
APA StyleCaso, A., Laurenzana, I., Lamorte, D., Trino, S., Esposito, G., Piccialli, V., & Costantino, V. (2018). Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells. Marine Drugs, 16(6), 206. https://doi.org/10.3390/md16060206