Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
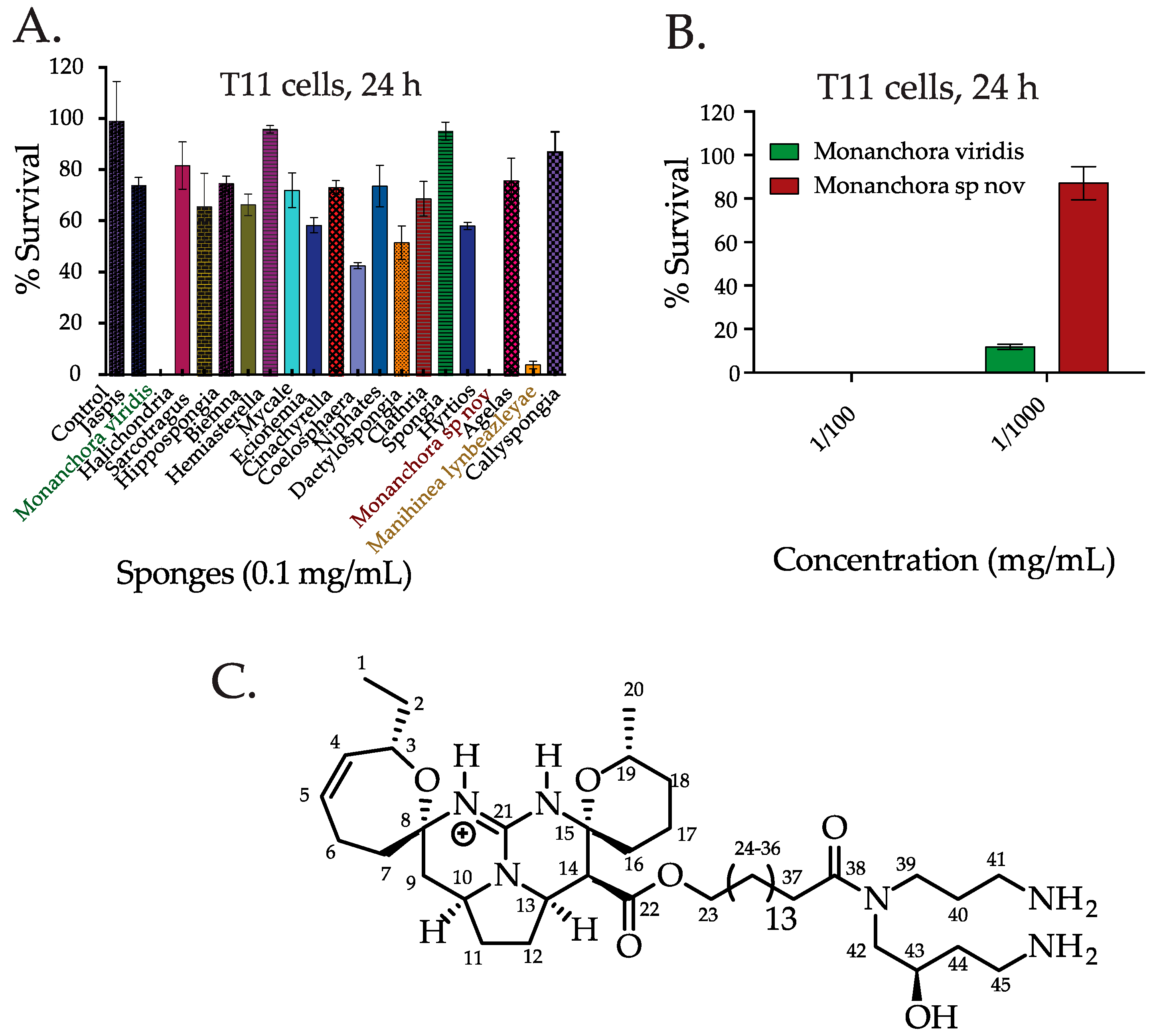
2.1. Screening the Extracts from Western Australian (WA) Marine Sponge in TNBC Cells and Bioassay Guided Isolation of C800
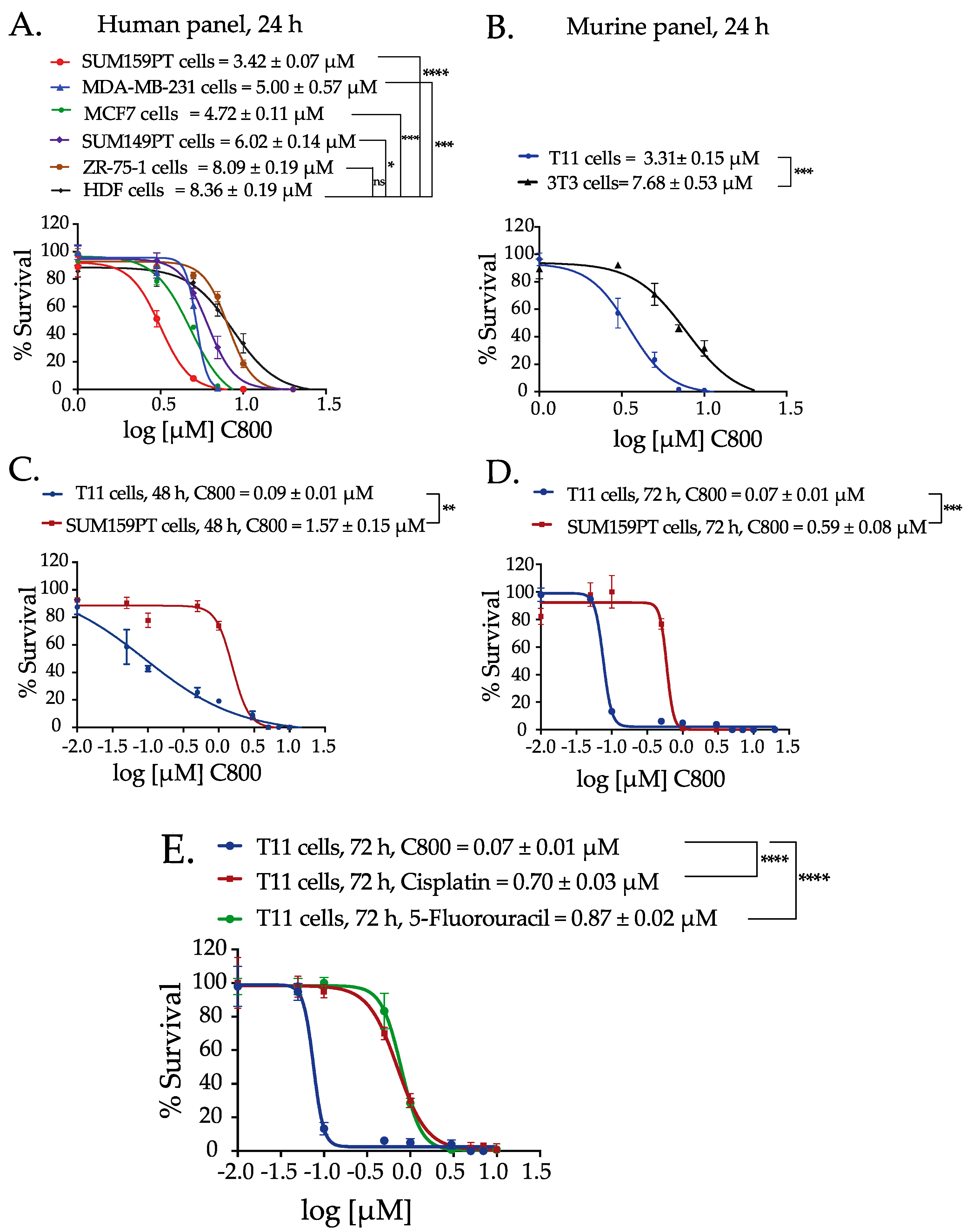
2.2. C800 Decreases Cell Viability in a Panel of Breast Cancer Cells
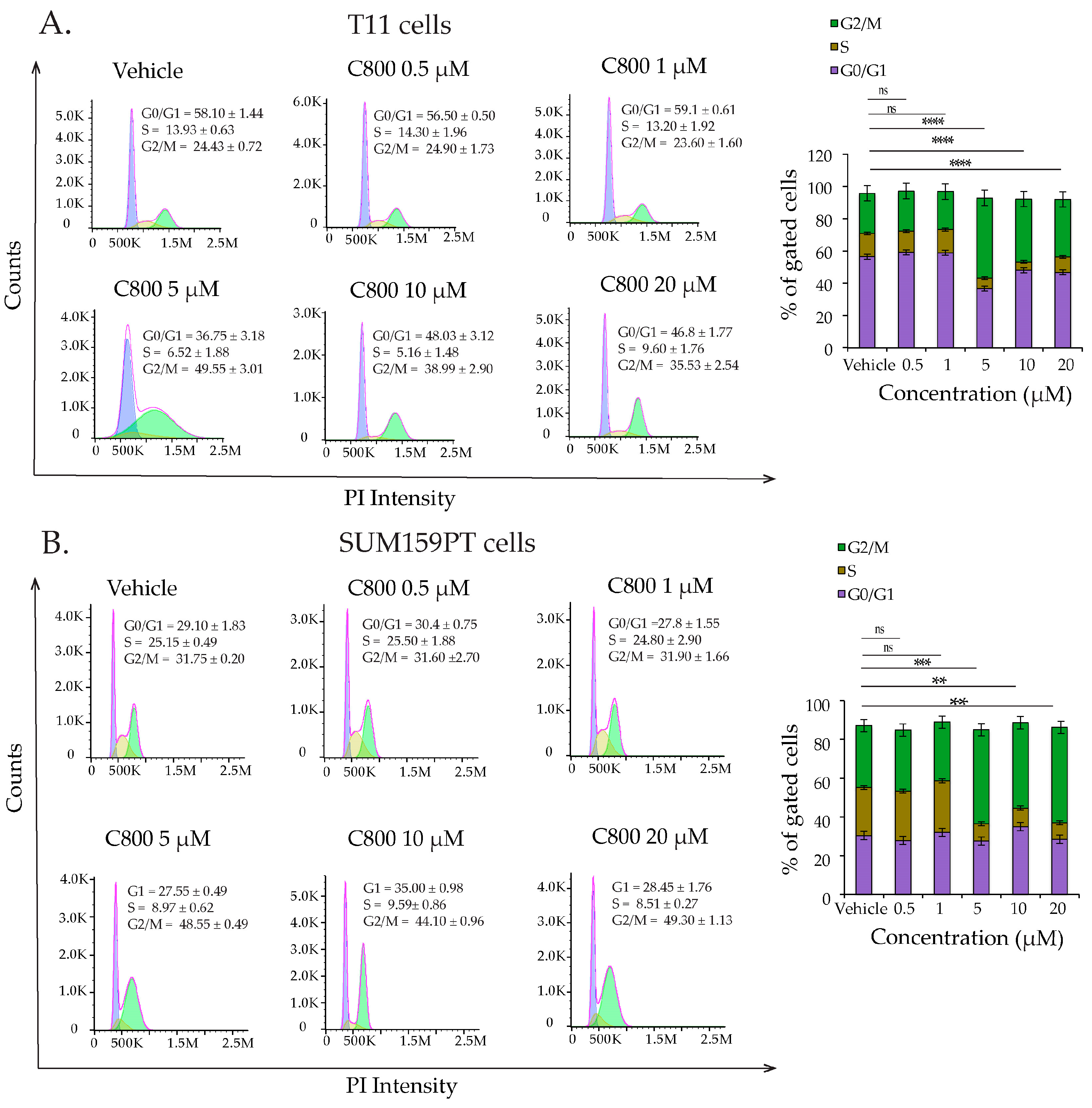
2.3. C800 Induces Cell Cycle Arrest in TNBC
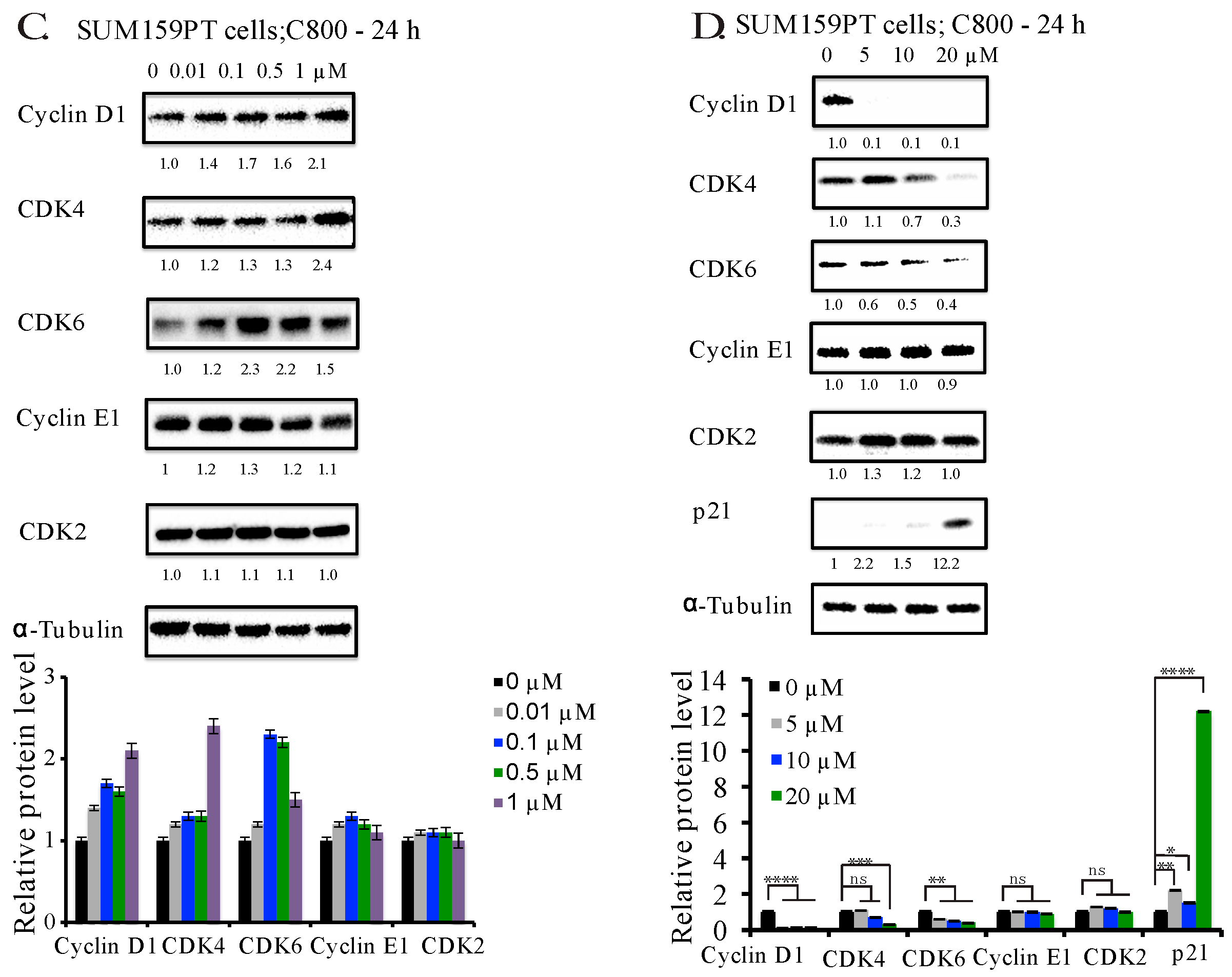
2.4. C800 Down-Regulates Cell Cycle Related Proteins in TNBC Cells
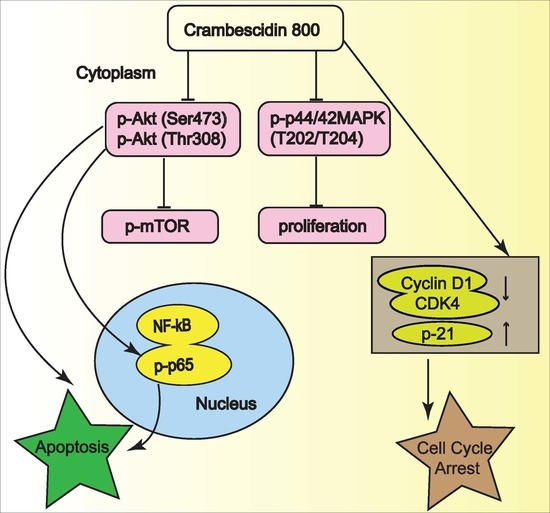
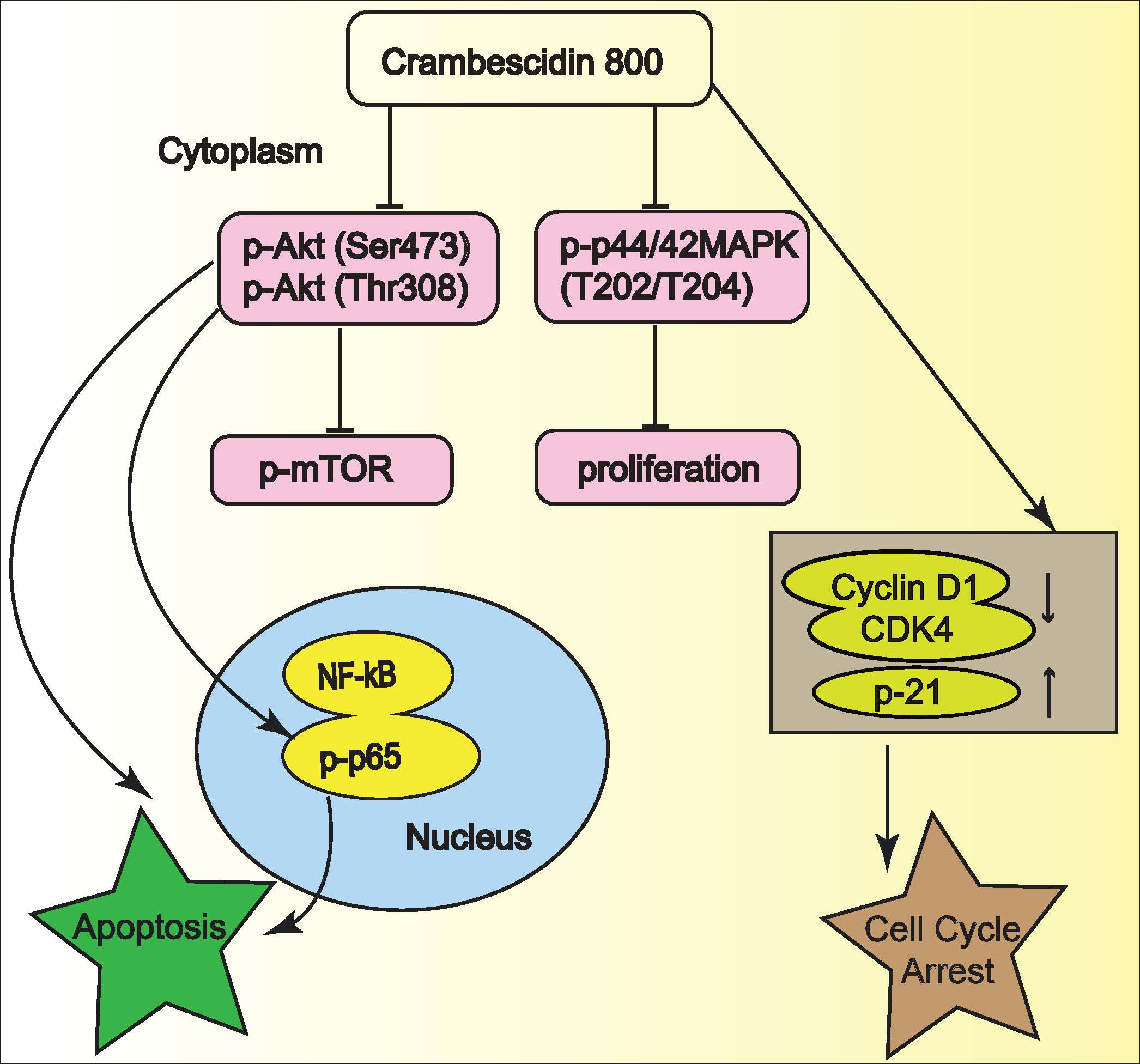
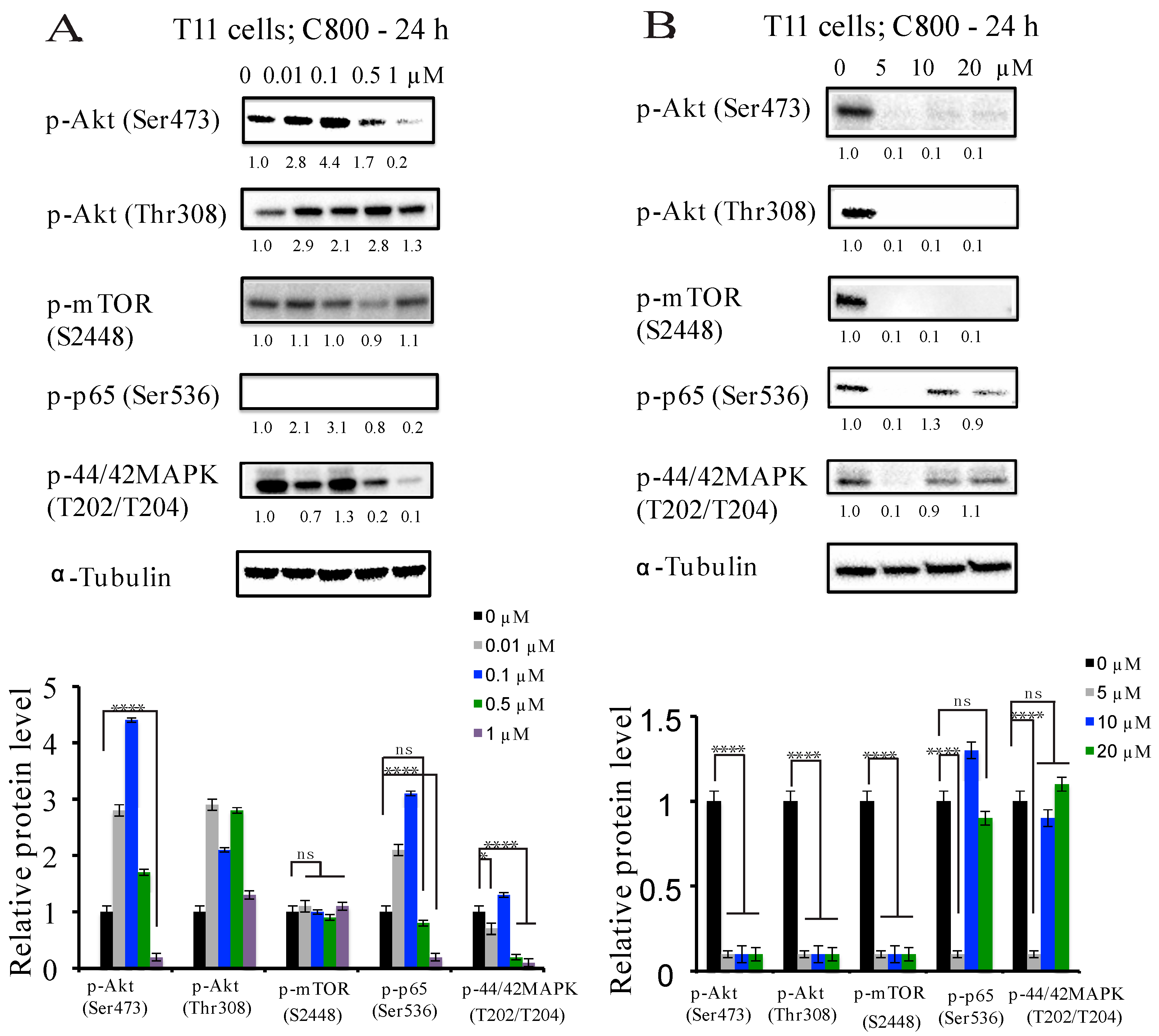
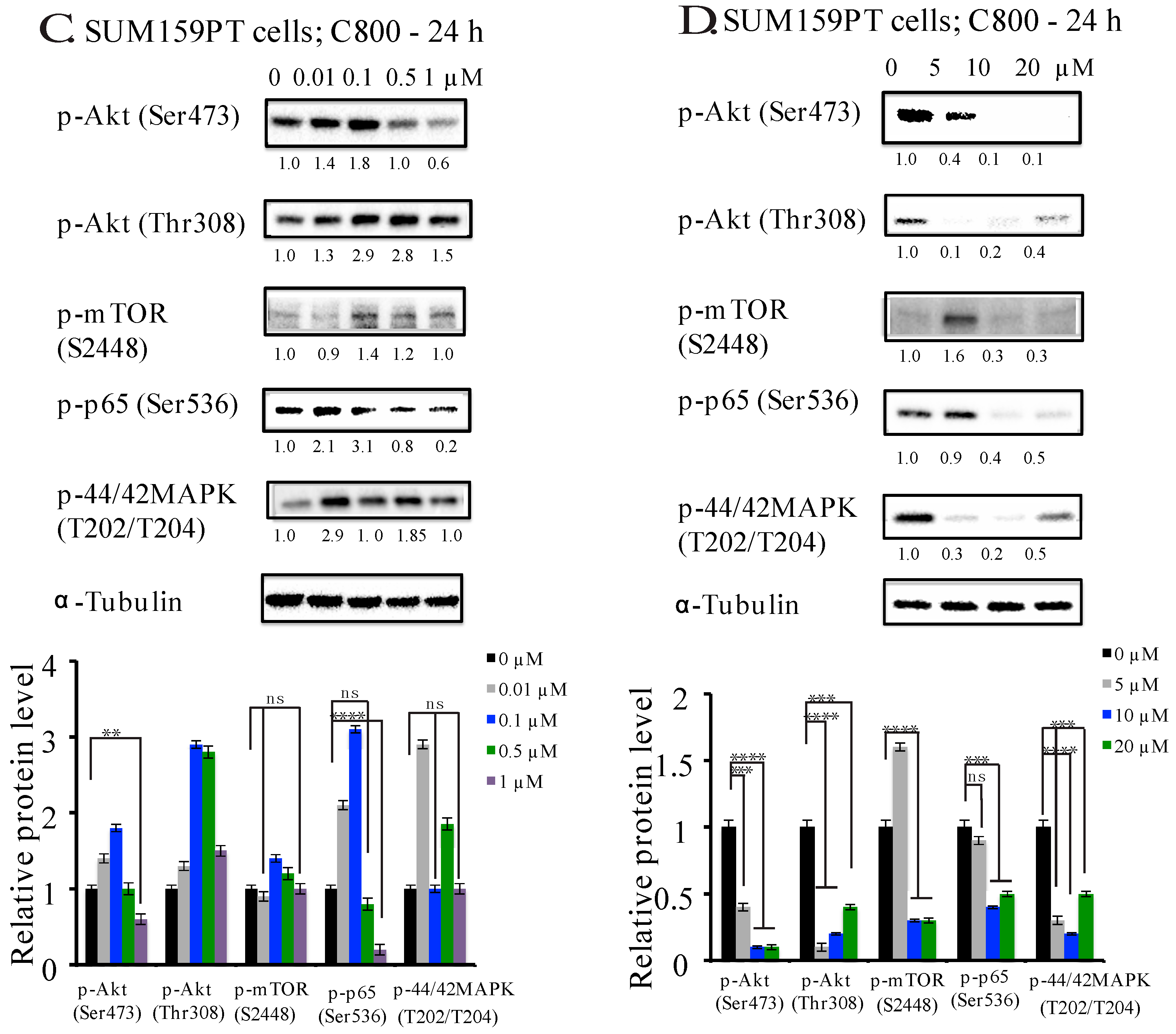
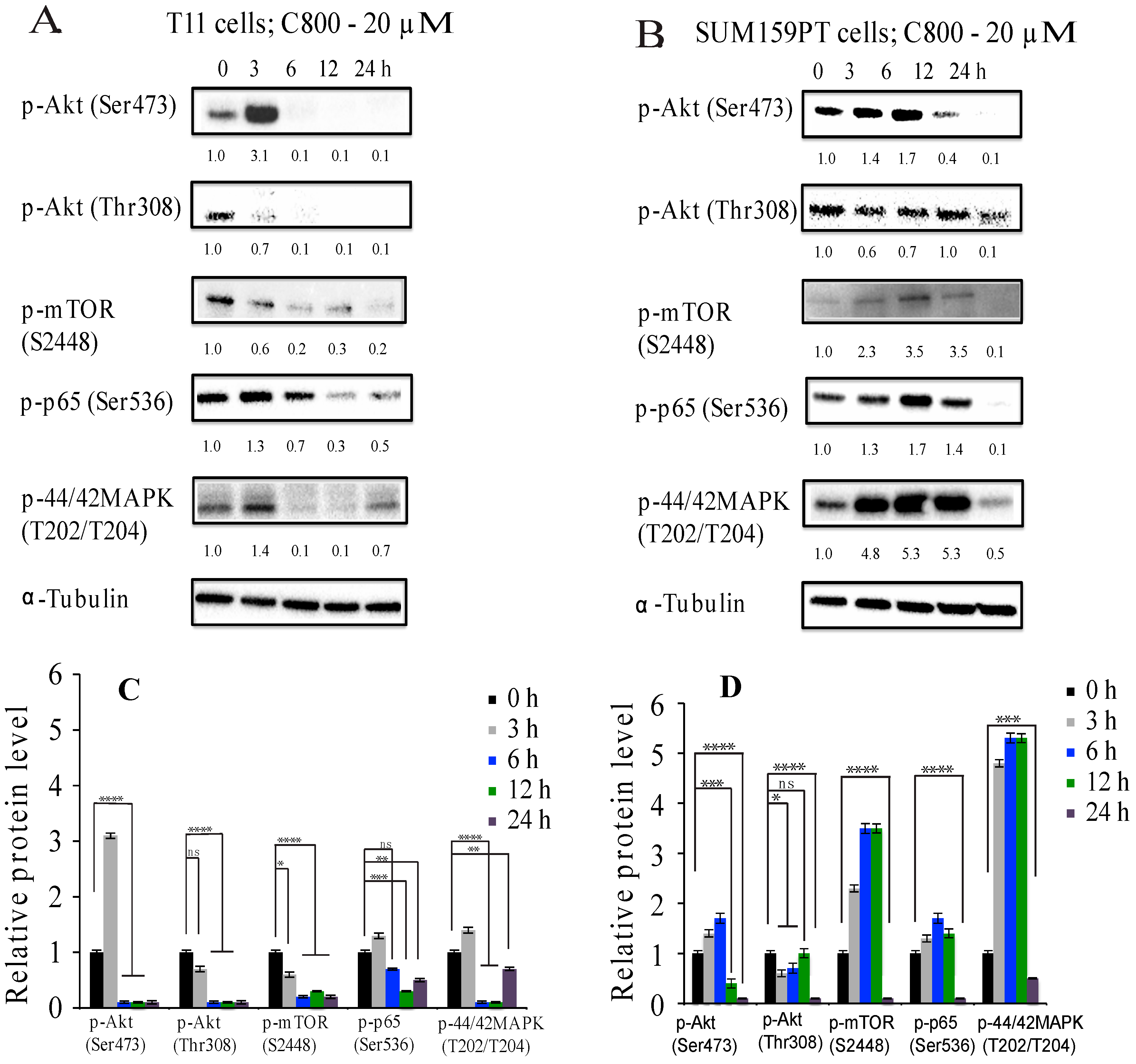
2.5. C800 Downregulates Phosphorylation of AKT/mTOR, MAPK, and NF-κB Signaling Pathways in TNBC Cells
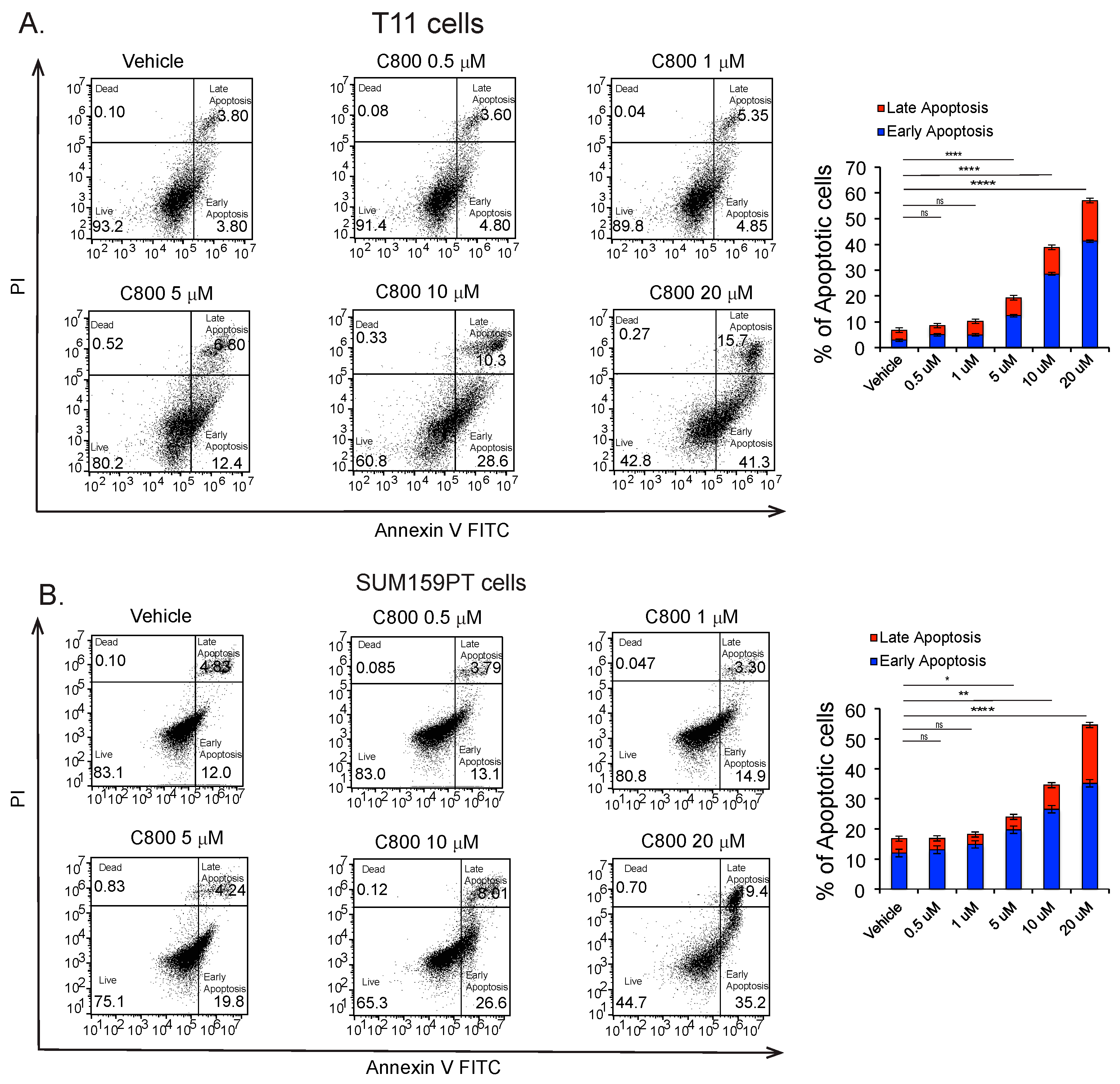
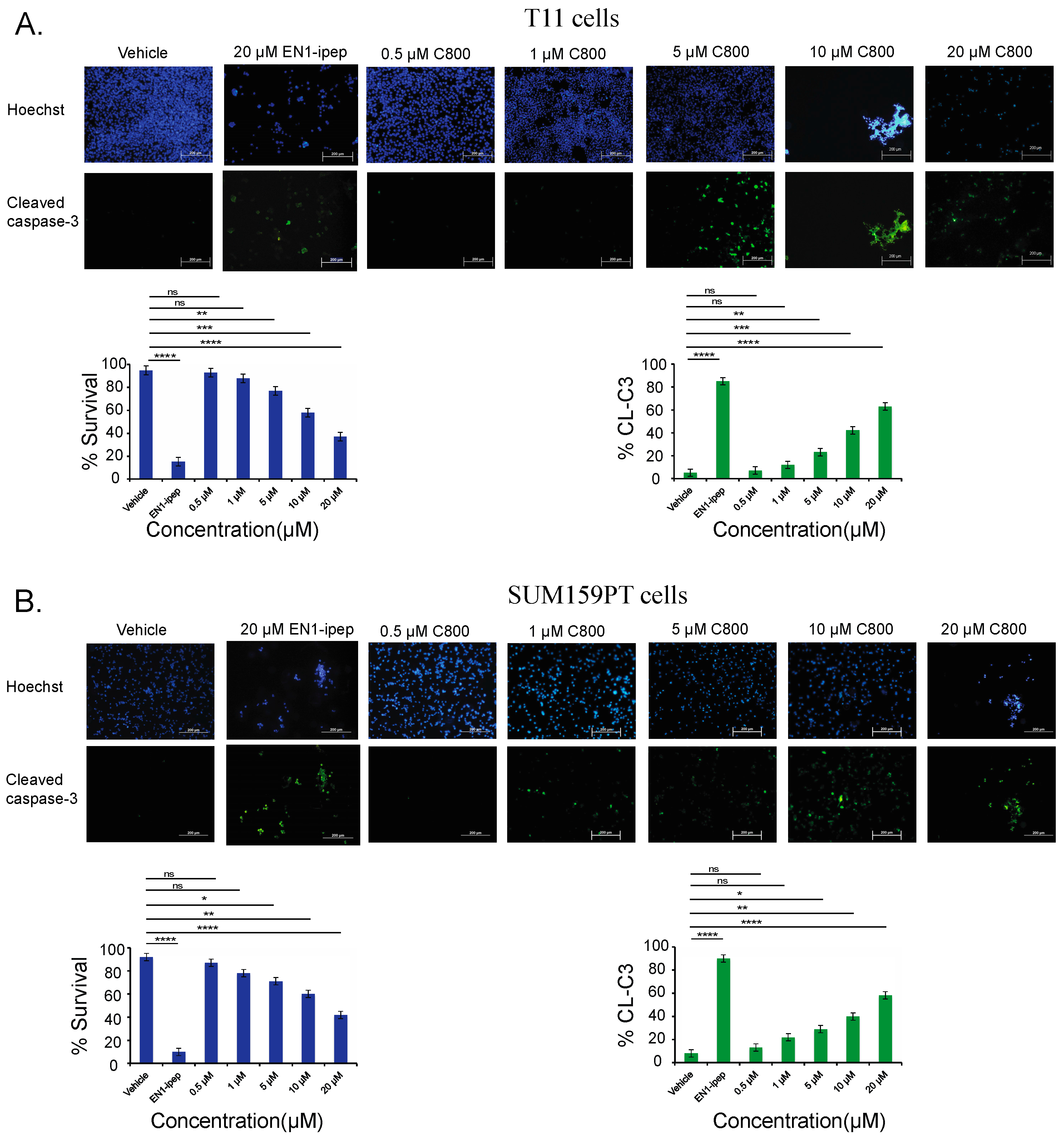
2.6. C800 Induces Apoptotic Cell Death in TNBC Cells
3. Materials and Methods
3.1. Experimental Procedures
3.2. Reagents
3.3. Details of Collection of Sponge Materials and Identification
3.4. Purification and Isolation of Crambescidin 800 (C800)
3.5. Cell Culture
3.6. Cell Viability Assay
3.7. Cell Cycle Analysis
3.8. Western Blot Analysis
3.9. Apoptosis Assay (Annexin-V-PT-Binding Assay)
3.10. Apoptosis Assay (Immunofluorescence)
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.P.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer—Deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Eroles, P.; Zaragoza, R.; Vina, J.R.; Lluch, A. Triple-negative breast cancer: Molecular features, pathogenesis, treatment and current lines of research. Cancer Treat. Rev. 2010, 36, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Suleria, H.A.; Osborne, S.; Masci, P.; Gobe, G. Marine-Based Nutraceuticals: An Innovative Trend in the Food and Supplement Industries. Mar. Drugs 2015, 13, 6336–6351. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Suleria, H.A.R.; Gobe, G.; Masci, P.; Osborne, S.A. Marine bioactive compounds and health promoting perspectives; innovation pathways for drug discovery. Trends Food Sci. Technol. 2016, 50, 44–55. [Google Scholar] [CrossRef]
- Mann, J. Natural products in cancer chemotherapy: Past, present and future. Nat. Rev. Cancer 2002, 2, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Cortes, J.; Vahdat, L.T.; Wanders, J.; Akerele, C.; Kaufman, P.A. Phase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic breast cancer. Clin. Breast Cancer 2010, 10, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor Polyether Macrolides from a Marine Sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.N.A.; Kennedy, J.A.; Quinn, R.J. Biodiversity ‘hotspots’, patterns of richness and endemism, and taxonomic affinities of tropical Australian sponges (Porifera). Biodivers. Conserv. 2002, 11, 851–885. [Google Scholar] [CrossRef]
- Fromont, J.; Vanderklift, M.A.; Kendrick, G.A. Marine sponges of the Dampier Archipelago, Western Australia: Patterns of species distributions, abundance and diversity. Biodivers. Conserv. 2006, 15, 3731–3750. [Google Scholar] [CrossRef]
- Schonberg, C.H.L.; Fromont, J. Sponge gardens of Ningaloo Reef (Carnarvon Shelf, Western Australia) are biodiversity hotspots. Hydrobiologia 2012, 687, 143–161. [Google Scholar] [CrossRef]
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 2002, 19, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.; Kossuga, M.H. Natural guanidine derivatives. Nat. Prod. Rep. 2005, 22, 516–550. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.; Trindade-Silva, A.E.; Santos, M.F. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2012, 29, 1382–1406. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Kelly, M.; Bowling, J.; Sims, J.; Waters, A.; Hamann, M. Advancement into the Arctic region for bioactive sponge secondary metabolites. Mar. Drugs 2011, 9, 2423–2437. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.S.; Stonik, V.A. Monanchocidins B–E: Polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchomycalins A and B, unusual guanidine alkaloids from the sponge Monanchora pulchra. Tetrahedron Lett. 2012, 53, 4228–4231. [Google Scholar] [CrossRef]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Hauschild, J.; Amann, K.; Tabakmakher, K.M.; Venz, S.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; Fedorov, S.N.; et al. Marine alkaloid Monanchocidin a overcomes drug resistance by induction of autophagy and lysosomal membrane permeabilization. Oncotarget 2015, 6, 17328–17341. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Whittaker, N.F.; Bewley, C.A. Crambescidin 826 and dehydrocrambine A: New polycyclic guanidine alkaloids from the marine sponge Monanchora sp that inhibit HIV-1 fusion. J. Nat. Prod. 2003, 66, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Korolkova, Y.; Makarieva, T.; Tabakmakher, K.; Shubina, L.; Kudryashova, E.; Andreev, Y.; Mosharova, I.; Lee, H.S.; Lee, Y.J.; Kozlov, S. Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors. Mar. Drugs 2017, 15, E87. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, A.; Ho, D.; Wang, E.; Evans, C.W.; Ormonde, C.F.; Rashwan, R.; Singh, R.; Iyer, K.S.; Blancafort, P. Sensitizing basal-like breast cancer to chemotherapy using nanoparticles conjugated with interference peptide. Nanoscale 2016, 8, 9343–9353. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Usary, J.E.; Darr, D.B.; Dillon, P.M.; Pfefferle, A.D.; Whittle, M.C.; Duncan, J.S.; Johnson, S.M.; Combest, A.J.; Jin, J.; et al. Combined PI3K/mTOR and MEK Inhibition Provides Broad Antitumor Activity in Faithful Murine Cancer Models. Clin. Cancer Res. 2012, 18, 5290–5303. [Google Scholar] [CrossRef] [PubMed]
- Jareserijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins—New Antiviral and Cytotoxic Compounds from the Sponge Crambe-Crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Gallimore, W.A.; Kelly, M.; Scheuer, P.J. Alkaloids from the sponge Monanchora unguifera. J. Nat. Prod. 2005, 68, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Bondu, S.; Genta-Jouve, G.; Leiros, M.; Vale, C.; Guigonis, J.M.; Botana, L.M.; Thomas, O.P. Additional bioactive guanidine alkaloids from the Mediterranean sponge Crambe crambe. Rsc Adv. 2012, 2, 2828–2835. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic Guanidine Alkaloids from the Marine Sponge Crambe-Crambe and Ca++ Channel Blocker Activity of Crambescidin-816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Sfecci, E.; Lacour, T.; Amade, P.; Mehiri, M. Polycyclic Guanidine Alkaloids from Poecilosclerida Marine Sponges. Mar. Drugs 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.; Daloze, D.; Braekman, J.C.; Hajdu, E.; Muricy, G.; Vansoest, R.W.M. Isolation of Crambescidin-800 from Monanchora-Arbuscula (Porifera). Biochem. Syst. Ecol. 1994, 22, 645–646. [Google Scholar] [CrossRef]
- Heys, L.; Moore, C.G.; Murphy, P.J. The guanidine metabolites of Ptilocaulis spiculifer and related compounds; isolation and synthesis. Chem. Soc. Rev. 2000, 29, 57–67. [Google Scholar] [CrossRef]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid differentiation in K562 chronic myelogenous cells induced by crambescidin 800, a pentacyclic guanidine alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; Lopez-Alonso, H.; Roel, M.; Vieytes, M.R.; Thomas, O.; Ternon, E.; Vega, F.V.; Botana, L.M. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharmacol. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, M.C.; Crosariol, M.; Loro, E.; Li, Z.; Pestell, R.G. Cyclins and cell cycle control in cancer and disease. Genes Cancer 2012, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J. A gene transcription signature of the Akt/mTOR pathway in clinical breast tumors. Oncogene 2007, 26, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]
- Gholami, S.; Chen, C.H.; Gao, S.; Lou, E.; Fujisawa, S.; Carson, J.; Nnoli, J.E.; Chou, T.C.; Bromberg, J.; Fong, Y. Role of MAPK in oncolytic herpes viral therapy in triple-negative breast cancer. Cancer Gene Ther. 2014, 21, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Umemura, S.; Yoshida, S.; Ohta, Y.; Naito, K.; Osamura, R.Y.; Tokuda, Y. Increased phosphorylation of Akt in triple-negative breast cancers. Cancer Sci. 2007, 98, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.A.; Lee, H.N.; Choo, G.S.; Kim, H.J.; Che, J.H.; Jung, J.Y. Ixeris dentata (Thunb. Ex Thunb.) Nakai Extract Inhibits Proliferation and Induces Apoptosis in Breast Cancer Cells through Akt/NF-kappaB Pathways. Int. J. Mol. Sci. 2017, 18, 275. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Pise-Masison, C.A.; Radonovich, M.F.; Park, H.U.; Brady, J.N. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 2005, 24, 6719–6728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; LAllemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Ma, Q.; Li, J.; Zhang, D.; Liu, Z.G.; Rustgi, A.K.; Huang, C. Cyclin D1 induction through IkappaB kinase beta/nuclear factor-kappaB pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res. 2005, 65, 9287–9293. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Li, J.; Ma, Q.; Huang, C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis 2006, 27, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Apoptosis in the Pathogenesis and Treatment of Disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 53. https://doi.org/10.3390/md16020053
Shrestha S, Sorolla A, Fromont J, Blancafort P, Flematti GR. Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Marine Drugs. 2018; 16(2):53. https://doi.org/10.3390/md16020053
Chicago/Turabian StyleShrestha, Sumi, Anabel Sorolla, Jane Fromont, Pilar Blancafort, and Gavin R. Flematti. 2018. "Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells" Marine Drugs 16, no. 2: 53. https://doi.org/10.3390/md16020053
APA StyleShrestha, S., Sorolla, A., Fromont, J., Blancafort, P., & Flematti, G. R. (2018). Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Marine Drugs, 16(2), 53. https://doi.org/10.3390/md16020053