2. Results
Compound
1 was obtained as an amorphous solid. Its molecular formula was established as C
16H
14O
7Cl
2 based on the HRESIMS and NMR data, containing two Cl-atoms and implying nine indices of hydrogen deficiency. The
1H NMR data of
1 (
Table 1) displayed signals of two aromatic protons [
δH 6.51 (1H, d,
J = 2.2 Hz); 6.62 (1H, d,
J = 2.2 Hz)], one olefin proton [6.68 (1H, s)], two methylenes [
δH 3.33 (2H, d,
J = 6.0 Hz); 3.04 (1H, dd,
J = 9.5, 14.7 Hz); 2.45 (1H, dd,
J = 6.9, 14.7 Hz)], two methines [
δH 4.75 (1H, dd,
J = 6.9, 9.5 Hz); 6.52 (1H, s)], and one methoxy group at
δH 3.92 (1H, s). The
13C NMR data of
1 (
Table 2) exhibited 16 carbon resonances assignable to one methyl, two methylenes, two sp
3 and three sp
2 methines, six quaternary carbons and two carbonyl carbons. These spectroscopic features suggested that
1 belong to the isocoumarin class [
8,
9,
10]. Analysis of the
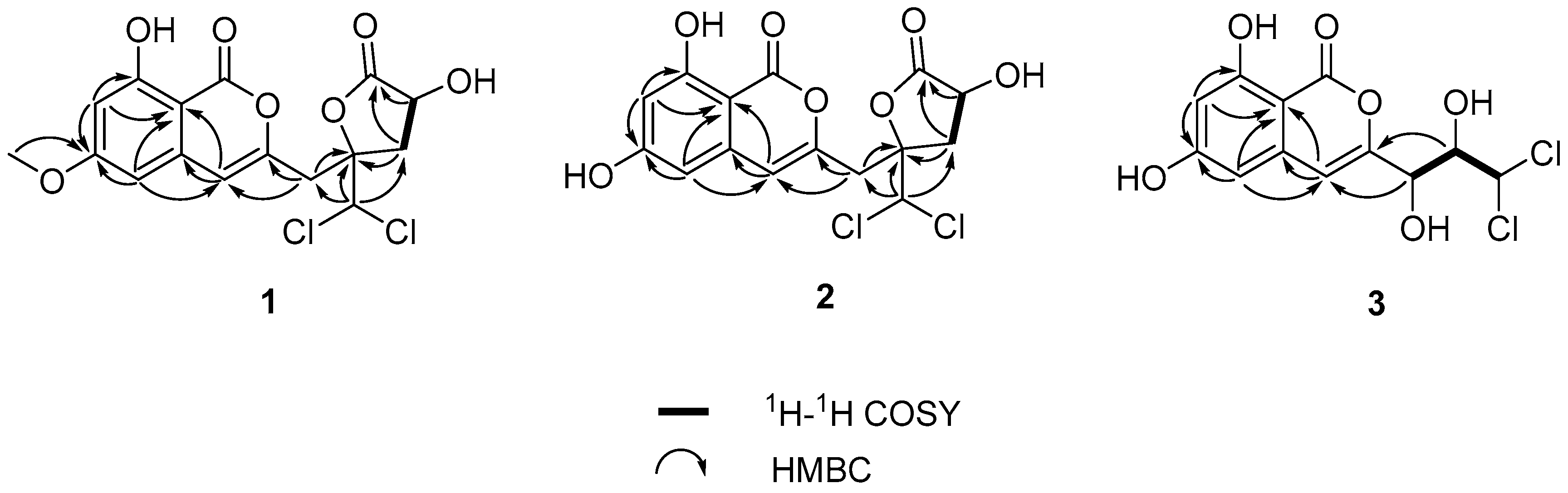
1H-
1H COSY spectrum (
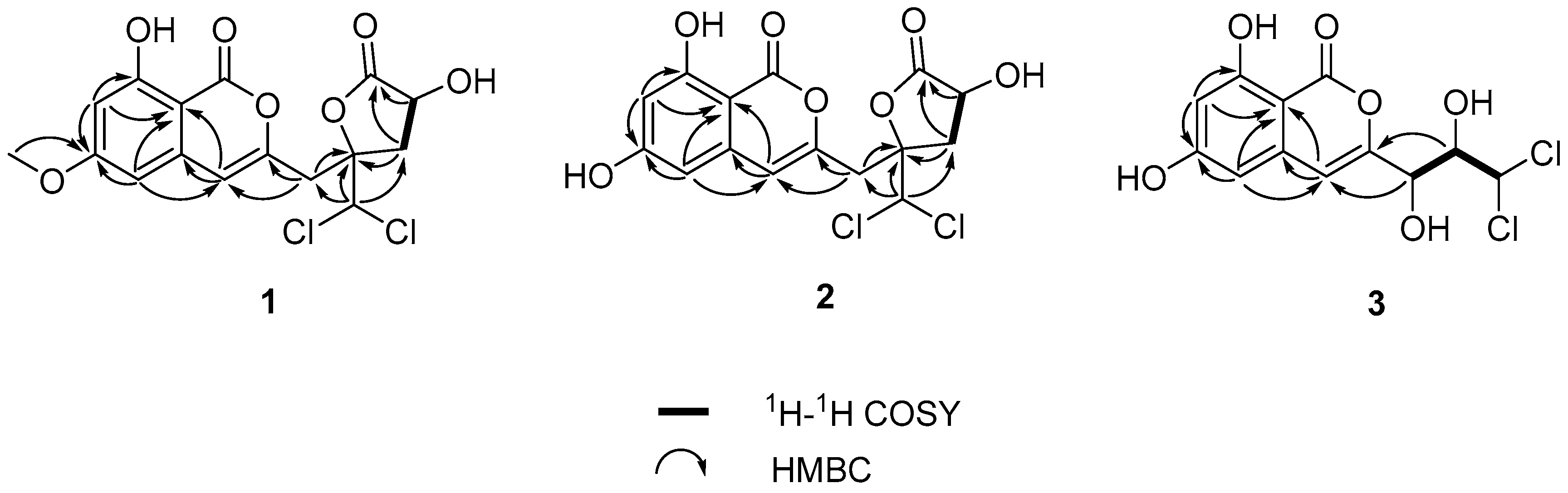
Figure 2) suggested the presence of one independent spin system H
2-11/H-12. Together with the HMBC cross-peaks (
Figure 2) of H-12/C-13; H-11/C-10, C-13; H-9/C-3, C-10; H-14/C-9, C-10, and C-11 indicated the side chain of isocaproicacid moiety location at C-3. The chemical shift of H-14 (
δH 6.52) indicated the dichloro substitution at C-14 [
8]. Apart from a carbonyl group and isocoumarin group, the remaining one indices of hydrogen deficiency was proved to be
α-hydroxyl-
γ-lactone ring. The chemical shift of quaternary carbon
δC 86.0 (C-10) confirmed the ring bridging C-10 and C-13. Moreover, the methoxy group was placed to C-6 based on the HMBC correlation of its proton to C-6. Thus, the constitution of
1 was established (
Figure 1).
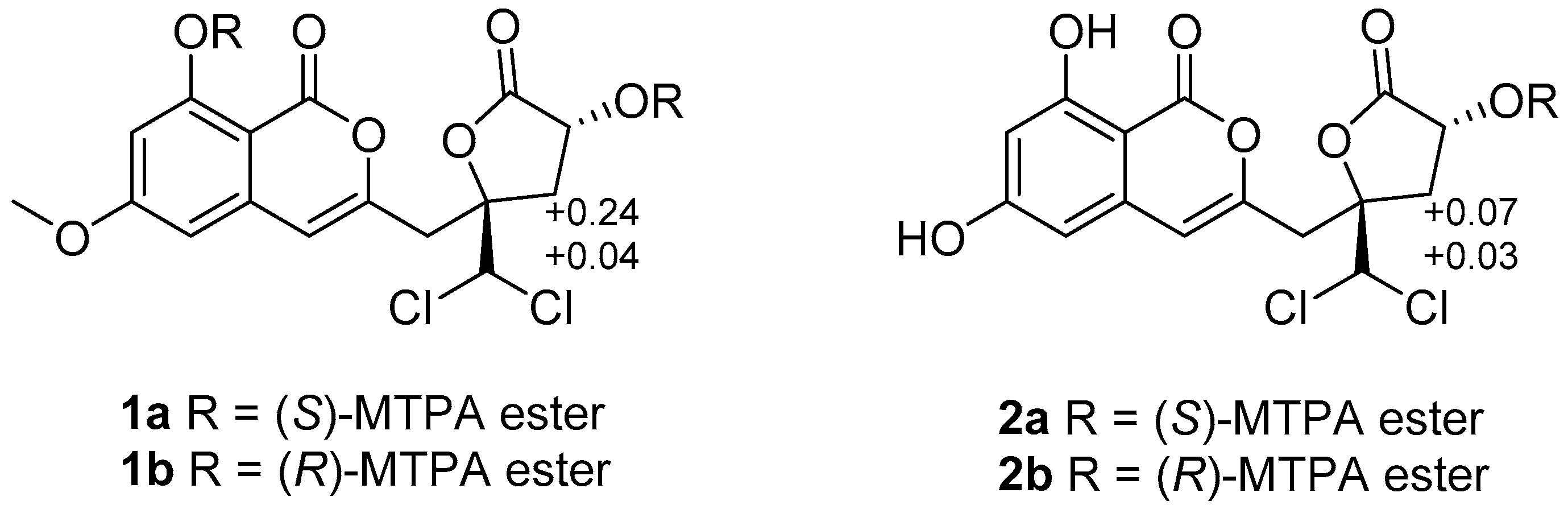
The relative configuration of
1 was determined by NOESY data (
Figure 3). The cross-peak of H-12 and H-14 indicated the
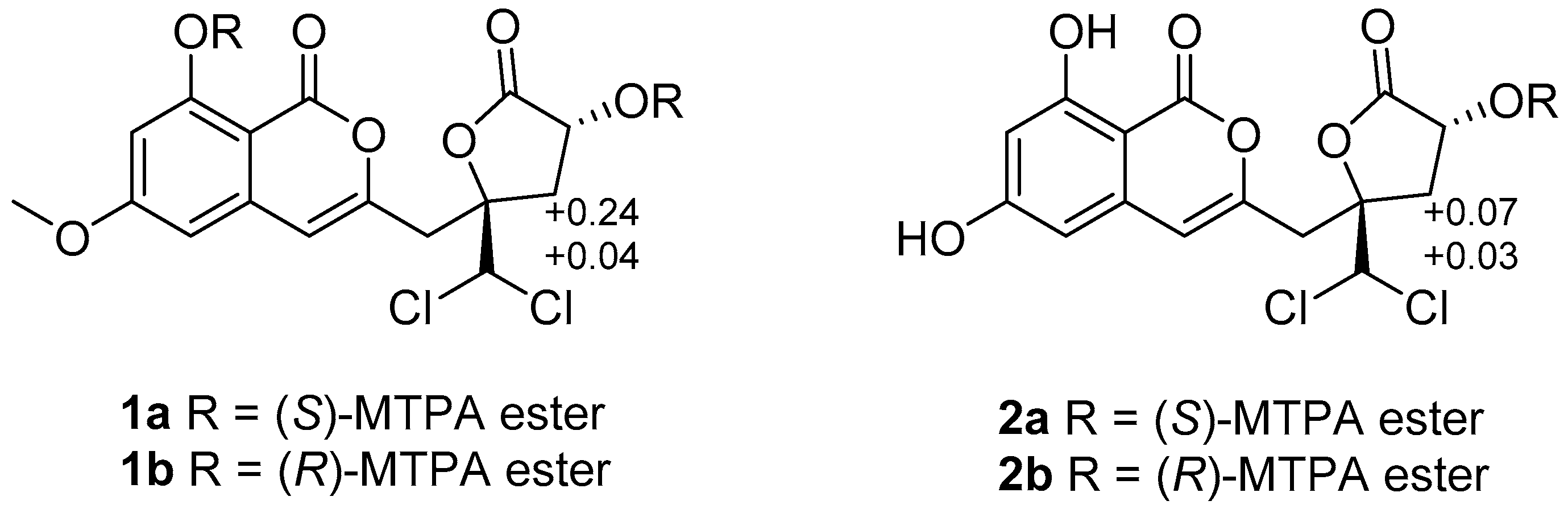
syn relationship between H-12 and H-14. The absolute configuration of C-12 was further confirmed by the modified Mosher ester method [
11]. The (
S)- and (
R)-MTPA esters of
1 (
1a and
1b) were prepared using (
R)- and (
S)-MTPA chloride, respectively. The differences in the
1H NMR chemical shifts of
1a and
1b were summarized to determine the absolute configuration of this position, which was clearly established as 12
R. Taking the data discussed above into account, the absolute configuration of
1 was assigned as 10
R,12
R (
Figure 4). Thus, compound
1 was determined as dichlorodiaportintone (
Figure 1).
Compound
2 was isolated as a white amorphous powder, having the molecular formula C
15H
12O
7Cl
2 based on the HREIMS at
m/
z 372.9885 [M − H]
−. The NMR data (
Table 1 and
Table 2) resembled those of
1, except for the disappearence of a methoxy group (
δC 56.3,
δH 3.92). The NOESY correlation (
Figure 3) from H-12 to H-14 proved that compounds
2 and
1 shared the same relative configuration. The absolute configuration at C-12 was also determined as
R by the modified Mosher’s method. Thus, the absolute configuration of
2 was determined as 10
R,12
R (
Figure 4). Therefore, the compound
2 was assigned as desmethyldichlorodiaportintone (
Figure 1).
Compound
3 was obtained as a white powder. The molecular formula was determined as C
12H
10O
6Cl
2 by HRESIMS. The
1H NMR spectrum (
Figure S18) of
3 showed signals for a 1,2,3,5-tetrasubstituted aromatic unit [
δH 6.44 (1H, d,
J = 2.1 Hz) and
δH 6.51 (1H, d,
J = 2.1 Hz)], one methine signal at
δH 6.43 (1H, d,
J = 1.6 Hz), two oxygenated methine signals at
δH 4.38 (1H, dd,
J = 1.6, 8.8 Hz) and
δH 4.28 (1H, d,
J = 8.8 Hz). The
13C NMR spectrum (
Figure S19) exhibited 13 carbon signals, indicating a carbonyl carbon, three methines, eight aromatic carbons. The
1H and
13C NMR spectra (
Table 1 and
Table 2) of
3 were similar to those of dichlorodiaportinol A (
4), except for the absence of the methoxy group (
δC 56.3,
δH 3.90) at C-6 in
3. The structure of
3 was also confirmed using
1H-
1H COSY and HMBC spectra (
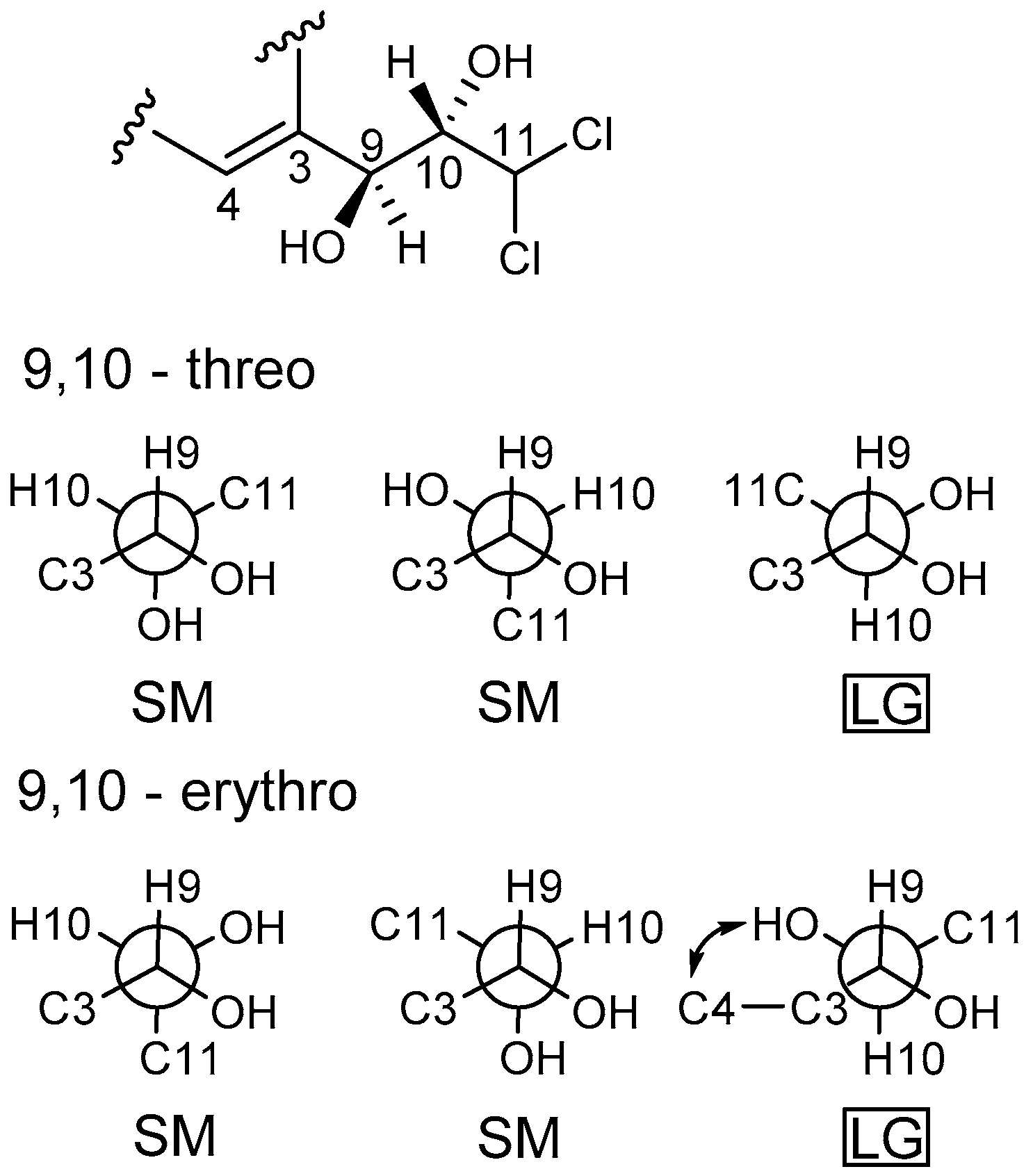
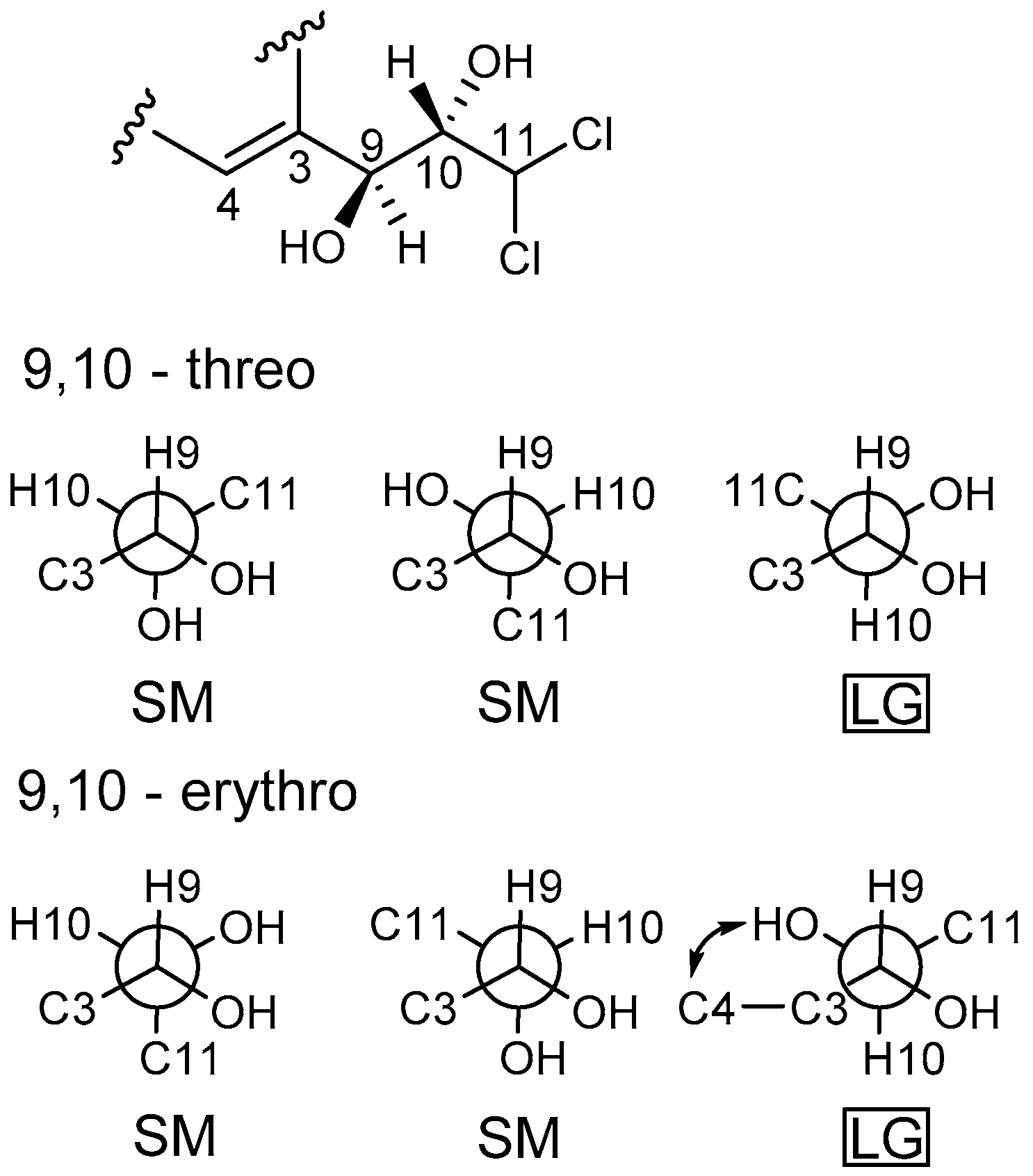
Figure 2). The configuration of two stereocenters (C-9 and C-10) were determined by coupling constants and NOESY experiments (
Figure 3). Protons H-9 and H-10 displayed a large coupling constant (
3JH-9,H-10 = 8.8 Hz), indicating them to be in an anti configuration. This allowed for only two of the six possible relative configuration for C-9 and C-10 could be satisfied. As well as the NOE correlation of H-4/OH-10 in DMSO, the relative configuration of C-9 and C-10 was unambiguously determined as 9
R* and 10
S* (
Figure 5) and named desmethyldichlorodiaportinol (
Figure 1).
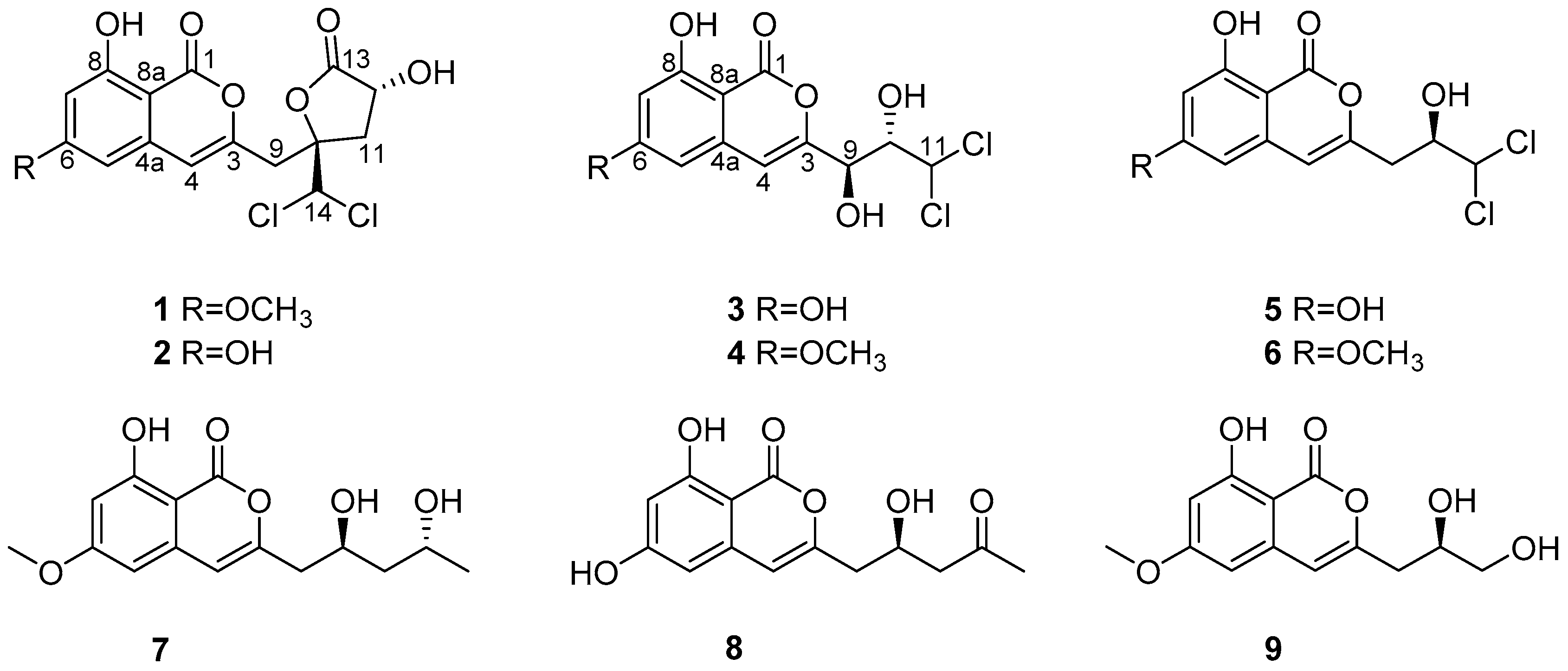
The other known compounds were identified as dichlorodiaportinol (
4) [
9], desmethyldichlorodiaportin (
5) [
8], dichlorodiaportin (
6) [
10], mucorisocoumarin B (
7) [
12], citroisocoumarin (
8) [
13], and diaportinol (
9) [
11] by comparison with NMR data in the literature.
The anti-inflammatory activities of all compounds were evaluated against nitric oxide (NO) production in the lipopolysaccharide (LPS)-stimulated mouse macrophage RAW 264.7. The results suggest that the compound
2 showed potent inhibitory activities with IC
50 value of 15.8 μM, and compounds
1,
5, and
6 exhibited weak inhibitory activity in comparison with the indomethacin (the positive control, IC
50 = 37.5 μM). Other compounds showed no inhibitory effect (IC
50 > 100 μM) (
Table 3). All compounds showed no cytotoxic effect at the tested concentration. Compounds
2 and
5 which have a hydroxyl group at C-6 showed batter than compounds
1 and
6 with a methoxy group at C-6. The structure-activity relationships of these dichloroisocoumarins indicated that a hydroxyl group was more significance than a methoxy group on anti-inflammatory activity. Isocoumarins were previously reported to have radical scavenging and antioxidant [
14], anti-HIV [
15], antimicrobial [
16], anti-
γ-secretase [
17], antitumor [
18], immunomodulatory [
19], antifungal [
20], toxicity to zebrafish embryos [
13], and
α-glucosidase inhibitory activities [
21]. This is the first report of anti-inflammatory activity of dichloroisocoumarins.
The antibacterial activities of the isolated compounds
1–
9 against two Gram-positive bacteria (
Staphylococcus aureus and
Bacillus subtilis) and three Gram-negative bacteria (
Escherichia coli,
Klebsiella pneumoniae, and
Acinetobacter calcoaceticus) were tested (
Table 4). Compounds
5 and
6 showed antibacterial activities with the MIC values between 25 and 50 μg·mL
–1 against
S. aureus,
B. subtilis,
E. coli,
K. pneumoniae, and
A. calcoaceticus. Compound
1 exhibited antibacterial activities with the MIC values at 50 μg·mL
−1 against
S. aureus,
E. coli and
K. pneumoniae. Other compounds did not exhibit obvious activity at 50 μg·mL
−1.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on an MCP 300 (Anton Paar, Shanghai, China) polarimeter at 25 °C. UV spectra were recorded in MeOH using a PERSEE TU-1900 spectrophotometer (Persee, Beijing, China). IR spectra were recorded on a Nicolet Nexus 670 spectrophotometer (Nicolet, Madison, WI, USA) in KBr discs. NMR spectra were carried out on Bruker Avance 400 spectrometer (1H 400 MHz, 13C 100 MHz) (Bruker Bio Spin Corporation, Bellerica, MA, USA) and Bruker Avance 500 spectrometer (1H 500 MHz, 13C 125 MHz) (Bruker Bio Spin Corporation, Bellerica, MA, USA). ESIMS spectra were measured on a Finnigan LCQ-DECA mass spectrometer (Finnigan, Beijing, China), and HRESIMS spectra were obtained on a Thermo Fisher Scientic Q-TOF mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Column chromatography (CC) was conducted using silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China) and Sephadex LH-20 (Amersham Pharmacia, Piscataway, NJ, USA). Thin-layer chromatography (TLC) was performed on silica gel plates (Qingdao Huang Hai Chemical Group Co., G60, F-254, Qingdao, China).
3.2. Fungal Material and Fermentation
The fungus CYSK-4 used in this study was isolated from healthy branch of the marine semimangrove
Pluchea indica, which was collected in July 2015 from Shankou Mangrove Nature Reserve in Guangxi Province, China. It was obtained using the standard protocol for isolation [
21]. Initially, the plant tissue was washed with sterile water and surface-sterilized in a 200 mL beaker with 75% ethanol for 1 min. This was followed by dipping the sample into 5% sodium hypochlorite for 1 min, then the plant parts were rinsed with sterile water, cut into 3 mm sections, and plated on PDA with penicillin (100 units per mL) and streptomycin (0.08 mg·mL
−1). The plates were incubated at 25 ± 1 °C. The endophytic fungal strains were isolated by routine microbiological methods. The fungal isolates were numbered and stored at 4 °C in triplicate on PDA slants. Fungal identification was carried out using a molecular biological protocol by DNA amplification and sequencing of the ITS region [
22]. The sequence data obtained from the fungal strain have been deposited at Gen Bank with accession no. MG571637. A BLAST search result showed that the sequence was the most similar (99%) to the sequence of
Ascomycota sp. (compared to KT240142.1 EF060747.1). A voucher strain was deposited in the Guangdong Microbial Culture Center under patent depository number GDMCC 60100. The fungus
Ascomycota sp. CYSK-4 was cultured on autoclaved rice solid-substrate medium (60 × 500 mL Erlenmeyer flasks, each containing 50 g rice and 50 mL 3‰ of saline water) for 30 days at room temperature.
3.3. Extraction and Isolation
Following incubation. The mycelia and solid rice medium were extracted three times with EtOAc. The extract was evaporated under reduced pressure to yield 60 g of residue. The residue was subjected to a silica gel column chromatography, eluting with a gradient of petroleum ether/EtOAc from 1:0 to 0:1, to obtain 36 fractions. Fraction 8 (120 mg) was subjected to silica gel CC (CH2Cl2/MeOH v/v, 98:2) to yield compounds 1 (8.2 mg), 3 (3.8 mg), and 7 (2.5 mg). Fraction 12 (90 mg) was applied to Sephadex LH-20 CC (CH2Cl2/MeOH v/v, 1:1) to give compounds 2 (5.1 mg) and 5 (3.4 mg). Fraction 16 (68 mg) was purified by Sephadex LH-20 CC (CH2Cl2/MeOH v/v, 1:1) and silica gel CC (CH2Cl2/MeOH v/v, 88:12) afford 4 (4.6 mg) and 8 (2.2 mg). Fraction 18 (62 mg) was chromatographed on Sephadex LH-20 CC (100% MeOH) to obtain compound 6 (1.8 mg). Fraction 20 was separated by silica gel CC (CH2Cl2/MeOH v/v, 86:14) to give subfraction fraction 20.2, which was purified by Sephadex LH-20 CC (CH2Cl2/MeOH v/v, 1:1) to yield 9 (2 mg).
3.3.1. Dichlorodiaportintone (1)
Amorphous solid; [
α = +11.9 (
c 0.12, acetone); UV (MeOH)
λmax (log
ε): 332 (3.78), 280 (3.87), 245 (4.61) nm; IR (KBr)
νmax: 3319, 1800, 1681, 1621, 1386, 1238, 1209, 1167, 856, 795, 712 cm
−1; HRESIMS
m/z 387.0041 [M − H]
– (calcd. for C
16H
13O
7Cl
2, 387.0048);
1H NMR (500 MHz, acetone-
d6) data, see
Table 1;
13C NMR (125 MHz, acetone-
d6) data, see
Table 2.
3.3.2. Desmethyldichlorodiaportintone (2)
White amorphous powder; [
α = +6.9 (
с 0.06, acetone); UV (MeOH)
λmax (log
ε): 331 (3.85), 262 (4.20), 246 (4.44) nm; IR (KBr)
νmax: 3232, 1781, 1679, 1630, 1455, 1372, 1248, 1166, 1071, 781 cm
−1; HRESIMS
m/z 372.9885 [M − H]
− (calcd. for C
15H
11O
7Cl
2, 372.9887);
1H NMR (500 MHz, acetone-
d6) data, see
Table 1;
13C NMR (125 MHz, acetone-
d6) data, see
Table 2.
3.3.3. Desmethyldichlorodiaportinol (3)
White powder; [
α = +18.3 (
с 0.1, acetone); UV (MeOH) λ
max (log
ε): 329 (3.62), 268 (3.65), 245 (3.26) nm; IR (KBr)
νmax: 3543, 3461, 3232, 1683, 1631, 1497, 1399, 1293, 1242, 1188, 1071, 1038, 837, 703 cm
−1; HRESIMS
m/z 318.9782 [M − H]
− (calcd. for C
12H
9O
6Cl
2, 318.9782);
1H NMR (500 MHz, acetone-
d6) data, see
Table 1;
13C NMR (125 MHz, acetone-
d6) data, see
Table 2.
3.3.4. Preparation of the (S)- and (R)-MTPA Esters 1a and 1b
Compound 1 (1 mg) was treated with (R)-MTPACl (10 μL) and pyridine (0.5 mL). The mixture was reacted at room temperature for 24 h. The solution was extracted with 5 mL of CH2Cl2, and the organic phase was concentrated under reduced pressure. The residue was purified by preparative TLC (CH2Cl2) to yield the (S)-MTPA ester 1a (0.8 mg). In a similar way, (R)-MTPA ester 1b (0.5 mg) was obtained from compound 1 (1 mg) reacted with (S)-MTPACl (10 μL).
(S)-MTPA ester 1a: 1H NMR (acetone-d6, 400 MHz) δH: 6.74 (1H, s, H-4), 6.65 (1H, s, H-14), 7.12 (1H, d, J = 2.2 Hz, H-5), 6.72 (1H, d, J = 2.2 Hz, H-7), 5.95 (1H, dd, J = 8.0, 10.3 Hz, H-12), 3.98 (3H, s, 6-OCH3), 3.76 (3H, s, OCH3-MTPA), 3.52 (3H, s, OCH3-MTPA), 3.43 (2H, d, J = 4.6 Hz, H-9), 3.33 (1H, dd, J = 7.8, 15.0 Hz, H-11a), 2.85 (1H, dd, J = 10.4, 15.0 Hz, H-11b). ESIMS m/z 821.0 [M + 1]+.
(R)-MTPA ester 1b: 1H NMR (acetone-d6, 400 MHz) δH: 6.65 (1H, s, H-4), 6.63 (1H, s, H-14), 6.57 (1H, d, J = 2.2 Hz, H-5), 6.52 (1H, d, J = 2.2 Hz, H-7), 6.03 (1H, dd, J = 8.0, 10.3 Hz, H-12), 3.92 (3H, s, 6-OCH3), 3.58 (3H, s, OCH3-MTPA), 3.52 (3H, s, OCH3-MTPA), 3.38 (2H, d, J = 4.6 Hz, H-9), 3.29 (1H, dd, J = 7.8, 15.0 Hz, H-11a), 2.61 (1H, dd, J = 10.4, 15.0 Hz, H-11b). ESIMS m/z 821.0 [M + 1]+.
3.3.5. Preparation of the (S)- and (R)-MTPA Esters 2a and 2b
Following the same way as described for compound 1, R- and S- MTPA ester derivatives 2a and 2b were prepared from 2.
(S)-MTPA ester 2a: 1H NMR (CHCl3, 400 MHz) δH: 6.96 (1H, s, H-4), 6.95 (1H, s, H-14), 7.01 (1H, s, H-5), 6.69 (1H, s, H-7), 6.35 (1H, d, J = 2.31 Hz, H-12), 3.53 (3H, s, OCH3-MTPA), 3.46 (2H, m, H-9), 2.98 (1H, dd, J = 10.6, 14.9 Hz, H-11a), 2.55 (1H, dd, J = 7.7, 14.9 Hz, H-11b). ESIMS m/z 591.0 [M + 1]+.
(R)-MTPA ester 2b: 1H NMR (CHCl3, 400 MHz) δH: 6.91 (1H, s, H-4), 6.93 (1H, s, H-14), 7.0 (1H, s, H-5), 6.62 (1H, s, H-7), 6.33 (1H, d, J = 2.31 Hz, H-12), 3.53 (3H, s, OCH3-MTPA), 3.40 (2H, m, H-9), 2.91 (1H, dd, J = 10.6, 14.9 Hz, H-11a), 2.52 (1H, dd, J = 7.7, 14.9 Hz, H-11b). ESIMS m/z 591.0 [M + 1]+.
3.4. Nitric Oxide Production Assay
Murine macrophage RAW 264.7 cells purchased from the Shanghai Institutes for Biological Sciences in DMEM (high glucose) medium supplemented with 10% (
v/
v) fetal bovine serum, 100 μg·mL
‒1 penicillin and streptomycin, and 10 mM HEPES at 37 °C in a 5% CO
2 atmosphere [
23]. Cells were pretreated with different samples dissolved in serum-free culture medium containing 0.5% DMSO (10, 5, 2.5, 1.25, and 0.625 μM) for 4 h, followed by stimulation with 1 μg·mL
–1 LPS for 24 h. Fifty μL of cell culture medium was mixed with 100 μL of Griess reagent I and II and incubated at room temperature for 10 min with horizontal shaking, after which the absorbance at 540 nm was measured in a microplate reader. Indomethacin was used as a positive control and was purchased from Sigma-Aldrich Co. (CAS number: 53-86-1, EINECS number: 200-186-5; Buchs, Switzerland). Wells with DMSO were used as a negative control (final DMSO concentration was 0.1%). The NO production inhibition rate was calculated by the flowing formula:
IC50 was defined as the concentration of compound that inhibited 50% NO production relative to the LPS group and was calculated using SPSS 16.0 software. All assays were performed in triplicate.
3.5. Antimicrobial Activity
Antimicrobial activities were evaluated by the conventional broth dilution assay [
24]. Two Gram-positive—
S. aureus (ATCC 12228) and
B. subtilis (ATCC 6633)—and three Gram-negative—
E. coli (ATCC 25922),
K. pneumoniae (ATCC 13883), and
A. calcoaceticus (ATCC 23055)—were used. Overnight cultures of five bacterial strains were made up in 0.9% saline to an inoculum density of 5 × 10
5 cfu by comparison with a MacFarland standard. All compounds were dissolved in DMSO and diluted by Mueller Hinton broth to a starting concentration of 2 mg·mL
–1. Ninety-five μL of MHB and 5 μL of test compounds or the antibiotic were dispensed into wells as well as the 100 μL bacterial suspension. After incubation at 37 °C for 24 h, the inhibitory effect was evaluated by optical density measurement. The MIC was determined as the concentration which the growth was inhibited 80% of bacterial. One hundred μL bacterial suspension were added to the solutions in 96-well to achieve a final volume of 200 μL and final sample concentrations from 50 to 0.125 μg·mL
–1. The blank well was also incubated with only medium under the same conditions. OD measurement was record at 595 nm. All experiments were performed in triplicate and with ciprofloxacin and gentamicin as the positive control.
4. Conclusions
In conclusion, nine secondary metabolites including three new dichloroisocumarins—dichlorodiaportintone (1), desmethyldichlorodiaportintone (2), and desmethyldichloro-diaportinol (3)—and six known compounds—dichlorodiaportinol (4), desmethyldichlorodiaportin (5), dichlorodiaportin (6), mucorisocoumarin B (7), citroisocoumarin (8), and diaportinol (9)—were isolated from the marine mangrove-derived endophytic fungus Ascomycota sp. CYSK-4. Their structures were clarified by analysis of NMR data. Compounds 1–2 and 5–6 showed anti-inflammatory activities by inhibiting the production of NO in LPS-induced RAW 264.7 cells with IC50 values of 41.5, 15.8, 33.6, and 67.2 μM, respectively. Compounds 1, 5, and 6 showed inhibitory activity against S. aureus, B. subtilis, E. coli, K. pneumoniae and A. calcoaceticus with MIC values of 25–50 μg·mL–1. The results above proved that the dichloroisocumarins have the potential to be used as natural anti-inflammatory agents and antibiotics through appropriate structural modification.


 and
and 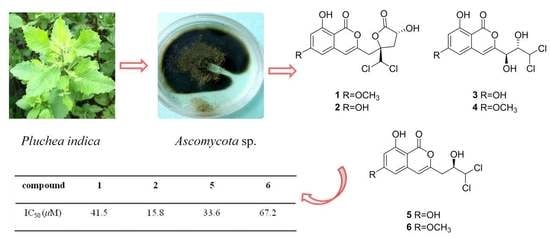


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}