Marine Compound Xyloketal B as a Potential Drug Development Target for Neuroprotection


Abstract
1. Introduction
2. The Anti-Oxidative and Anti-Apoptotic Effects of Xyloketal B in Endothelial Cells
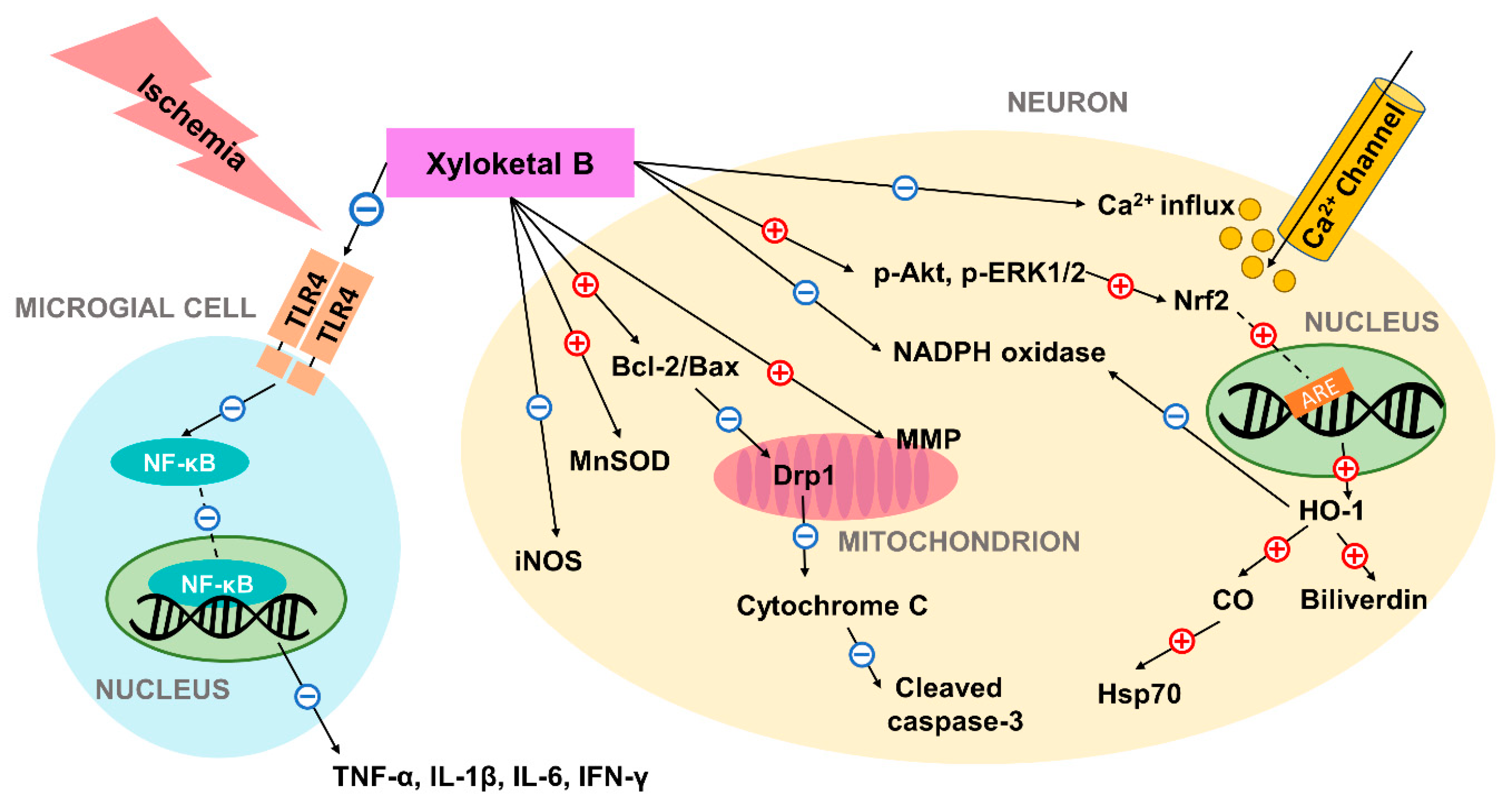
3. The ROS-Scavenging and Mitochondrial-Protective Abilities of Xyloketal B in Neurons
4. Xyloketal B Promotes Expression of HO-1 by Regulating the Upstream Signaling Pathway
5. Xyloketal B’s Neuroprotective Effect in Neonatal Hypoxic-Ischemic Brain Injury Model
6. The Neuroprotective Potential of Xyloketal B in Adult Ischemia Model
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Smajlovic, D. Strokes in Young Adults: Epidemiology and Prevention. Vasc. Health Risk Manag. 2015, 11, 157–164. [Google Scholar] [CrossRef]
- Rodgers, H. Stroke. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 110, pp. 427–433. [Google Scholar]
- Chaturvedi, M.; Kaczmarek, L. MMP-9 Inhibition: A Therapeutic Strategy in Ischemic Stroke. Mol. Neurobiol. 2014, 49, 563–573. [Google Scholar] [CrossRef]
- Gutierrez, J.; Esenwa, C. Secondary Stroke Prevention: Challenges and Solutions. Vasc. Health Risk Manag. 2015, 437–450. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M. Animal Models of Ischemic Stroke and Their Application in Clinical Research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef]
- Ding, Q.; Liao, S.-J.; Yu, J. Axon Guidance Factor Netrin-1 and Its Receptors Regulate Angiogenesis after Cerebral Ischemia. Neurosci. Bull. 2014, 30, 683–691. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Lipton, S.A. Failures and Successes of NMDA Receptor Antagonists: Molecular Basis for the Use of Open-Channel Blockers like Memantine in the Treatment of Acute and Chronic Neurologic Insults. NeuroRx J. Am. Soc. Exp. Neurother. 2004, 1, 101–110. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K.; Salter, M.W. NMDA Receptors in Clinical Neurology: Excitatory Times Ahead. Lancet Neurol. 2008, 7, 742–755. [Google Scholar] [CrossRef]
- Li, K.; Chung-Davidson, Y.-W.; Bussy, U.; Li, W. Recent Advances and Applications of Experimental Technologies in Marine Natural Product Research. Mar. Drugs 2015, 13, 2694–2713. [Google Scholar] [CrossRef]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New Drugs from Marine Organisms in Alzheimer’s Disease. Mar. Drugs 2016, 14, 5. [Google Scholar] [CrossRef]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S.; Vrijmoed, L.L.P.; Jones, E.B.G.; Krohn, K.; Steingröver, K.; et al. Five Unique Compounds: Xyloketals from Mangrove Fungus Xylaria Sp. from the South China Sea Coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Wilson, P.D. Synthesis of Xyloketal A, B, C, D, and G Analogues. J. Org. Chem. 2006, 71, 1620–1625. [Google Scholar] [CrossRef]
- Chen, W.-L.; Qian, Y.; Meng, W.-F.; Pang, J.-Y.; Lin, Y.-C.; Guan, Y.-Y.; Chen, S.-P.; Liu, J.; Pei, Z.; Wang, G.-L. A Novel Marine Compound Xyloketal B Protects against Oxidized LDL-Induced Cell Injury in Vitro. Biochem. Pharmacol. 2009, 78, 941–950. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.-Y.; Lin, Y.-C.; Liu, J.; Huang, R.; Wang, G.-L.; Pei, Z.; et al. Marine Compound Xyloketal B Protects PC12 Cells against OGD-Induced Cell Damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef]
- Pan, N. Xyloketal B Alleviates Cerebral Infarction and Neurologic Deficits in a Mouse Stroke Model by Suppressing the ROS/TLR4/NF-ΚB Inflammatory Signaling Pathway. Acta Pharmacol. Sin. 2017, 338, 1236–1247. [Google Scholar] [CrossRef]
- Xiao, A.-J.; Chen, W.; Xu, B.; Liu, R.; Turlova, E.; Barszczyk, A.; Sun, C.; Liu, L.; Deurloo, M.; Wang, G.-L.; et al. Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury. Mar. Drugs 2014, 13, 29–47. [Google Scholar] [CrossRef]
- Li, Z.-X.; Chen, J.-W.; Yuan, F.; Huang, Y.-Y.; Zhao, L.-Y.; Li, J.; Su, H.-X.; Liu, J.; Pang, J.-Y.; Lin, Y.-C.; et al. Xyloketal B Exhibits Its Antioxidant Activity through Induction of HO-1 in Vascular Endothelial Cells and Zebrafish. Mar. Drugs 2013, 11, 504–522. [Google Scholar] [CrossRef]
- Esplugues, J.V. NO as a Signalling Molecule in the Nervous System. Br. J. Pharmacol. 2002, 135, 1079–1095. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H Oxidases: Specific Features, Expression, and Regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef]
- Rueckschloss, U.; Galle, J.; Holtz, J.; Zerkowski, H.-R.; Morawietz, H. Induction of NAD(P)H Oxidase by Oxidized Low-Density Lipoprotein in Human Endothelial Cells: Antioxidative Potential of Hydroxymethylglutaryl Coenzyme A Reductase Inhibitor Therapy. Circulation 2001, 104, 1767–1772. [Google Scholar] [CrossRef]
- Liu, S.; Luo, R.; Xiang, Q.; Xu, X.; Qiu, L.; Pang, J. Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury. Mar. Drugs 2015, 13, 948–973. [Google Scholar] [CrossRef]
- Ouyang, Y.-B.; Giffard, R.G. MicroRNAs Affect BCL-2 Family Proteins in the Setting of Cerebral Ischemia. Neurochem. Int. 2014, 77, 2–8. [Google Scholar] [CrossRef]
- Karsan, A.; Yee, E.; Poirier, G.G.; Zhou, P.; Craig, R.; Harlan, J.M. Fibroblast Growth Factor-2 Inhibits Endothelial Cell Apoptosis by Bcl-2-Dependent and Independent Mechanisms. Am. J. Pathol. 1997, 151, 1775–1784. [Google Scholar]
- Zhao, L.-Y.; Li, J.; Yuan, F.; Li, M.; Zhang, Q.; Pang, J.-Y.; Zhang, B.; Sun, F.-Y.; Sun, H.-S.; Li, Q.; et al. Xyloketal B Attenuates Atherosclerotic Plaque Formation and Endothelial Dysfunction in Apolipoprotein E Deficient Mice. Mar. Drugs 2015, 13, 2306–2326. [Google Scholar] [CrossRef]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In Vitro Oxygen-Glucose Deprivation to Study Ischemic Cell Death. In Neuronal Cell Death; Lossi, L., Merighi, A., Eds.; Springer: New York, NY, USA, 2015; Volume 1254, pp. 197–210. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein Carbonyl Groups as Biomarkers of Oxidative Stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Pae, H.-O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.-H.; Choi, Y.K.; Lee, B.-S.; Kim, S.R.; Chung, H.-T. Heme Oxygenase in the Regulation of Vascular Biology: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Conradt, B. New Role of the BCL2 Family of Proteins in the Regulation of Mitochondrial Dynamics. Curr. Opin. Cell Biol. 2010, 22, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; Jacobson, M.R. Defects in Mitochondrial Fission Protein Dynamin-Related Protein 1 Are Linked to Apoptotic Resistance and Autophagy in a Lung Cancer Model. PLoS ONE 2012, 7, e45319. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yao, X.; Liu, Z.; Zhang, H.; Li, W.; Li, Z.; Wang, G.-L.; Pang, J.; Lin, Y.; Xu, Z.; et al. Protective Effects of Xyloketal B against MPP+-Induced Neurotoxicity in Caenorhabditis Elegans and PC12 Cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-S.; Huang, P.-H.; Wang, C.-H.; Lin, F.-Y.; Tsai, H.-Y.; Wu, T.-C.; Lin, S.-J.; Chen, J.-W. Nrf-2 Mediated Heme Oxygenase-1 Expression, an Antioxidant-Independent Mechanism, Contributes to Anti-Atherogenesis and Vascular Protective Effects of Ginkgo Biloba Extract. Atherosclerosis 2011, 214, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Dusting, G.J.; Mori, T.A.; Taylor, C.J.; Croft, K.D.; Jiang, F. Induction of Heme Oxygenase-1 In Vivo Suppresses NADPH Oxidase Derived Oxidative Stress. Hypertension 2007, 50, 636–642. [Google Scholar] [CrossRef]
- Zhou, J.-B.; Zheng, Y.-L.; Zeng, Y.-X.; Wang, J.-W.; Pei, Z.; Pang, J.-Y. Marine Derived Xyloketal Derivatives Exhibit Anti-Stress and Anti-Ageing Effects through HSF Pathway in Caenorhabditis Elegans. Eur. J. Med. Chem. 2018, 148, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, Q.; Wu, S.; Yang, L.; Ren, P.; Yang, Y.; Gao, J.; Li, L. Neonatal Hypoxic Ischemic Encephalopathy-Related Biomarkers in Serum and Cerebrospinal Fluid. Clin. Chim. Acta 2015, 450, 282–297. [Google Scholar] [CrossRef]
- Sun, H.-S. Role of TRPM7 in Cerebral Ischaemia and Hypoxia: TRPM7 in Cerebral Ischaemia. J. Physiol. 2017, 595, 3077–3083. [Google Scholar] [CrossRef]
- Griesmaier, E.; Stock, K.; Medek, K.; Stanika, R.I.; Obermair, G.J.; Posod, A.; Wegleiter, K.; Urbanek, M.; Kiechl-Kohlendorfer, U. Levetiracetam Increases Neonatal Hypoxic-Ischemic Brain Injury under Normothermic, but Not Hypothermic Conditions. Brain Res. 2014, 1556, 10–18. [Google Scholar] [CrossRef]
- Salakou, S.; Kardamakis, D.; Tsamandas, A.C.; Zolota, V.; Apostolakis, E.; Tzelepi, V.; Papathanasopoulos, P.; Bonikos, D.S.; Papapetropoulos, T.; Petsas, T.; et al. Increased Bax/Bcl-2 Ratio Up-Regulates Caspase-3 and Increases Apoptosis in the Thymus of Patients with Myasthenia Gravis. In Vivo 2007, 21, 123–132. [Google Scholar]
- Perlman, H.; Zhang, X.; Chen, M.W.; Walsh, K.; Buttyan, R. An Elevated Bax/Bcl-2 Ratio Corresponds with the Onset of Prostate Epithelial Cell Apoptosis. Cell Death Differ. 1999, 6, 48–54. [Google Scholar] [CrossRef]
- Bederson, J.B.; Pitts, L.H.; Tsuji, M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H. Rat Middle Cerebral Artery Occlusion: Evaluation of the Model and Development of a Neurologic Examination. Stroke 1986, 17, 472–476. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Oxidative Stress and Its Role in the Pathogenesis of Ischaemic Stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St. Clair, D.; Batinic-Haberle, I. Manganese Superoxide Dismutase, MnSOD and Its Mimics. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 794–814. [Google Scholar] [CrossRef]
- Jin, R.; Yang, G.; Li, G. Inflammatory Mechanisms in Ischemic Stroke: Role of Inflammatory Cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef]
- Gesuete, R.; Kohama, S.G.; Stenzel-Poore, M.P. Toll-Like Receptors and Ischemic Brain Injury. J. Neuropathol. Exp. Neurol. 2014, 73, 378–386. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Huang, X.; Wang, G.; Lv, X.; Meng, W.; Chen, W.; Pang, J.; Lin, Y.; Sun, H.; et al. Xyloketal B Exerts Antihypertensive Effect in Renovascular Hypertensive Rats via the NO-SGC-CGMP Pathway and Calcium Signaling. Acta Pharmacol. Sin. 2018, 39, 875–884. [Google Scholar] [CrossRef]


{kind=link}
| In Vitro/In Vivo | Cells/Animals | Model | Dose/Concentration | Major Findings |
|---|---|---|---|---|
| In vitro | HUVECs | OxLDL induced oxidative injury | 0.3 to 40 μM | Cytoprotective effect; Decreased ROS generation; Increased NO generation [17] |
| In vitro | PC12 | OGD model of ischemic stroke | 12.5 to 800 μM | ROS-scavenging and mitochondrial-protective abilities [18] |
| Both | PC12 and C. elegans | MPP+-induced neurotoxicity | 25 to 250 μM | Reduction of ROS generation and restoration of anti-oxidant glutathione level [36] |
| Both | HUVECs and zebrafish | AngII-Induced HUVEC apoptosis and PMA-induced respiratory burst of zebrafish embryos | 0.2 to 80 μM | Increase in HO-1 expression through PI3K/Akt signaling pathway [21] |
| Both | Mouse primary cortical cells and CD1 mice | OGD in cortical cells and neonatal hypoxic-ischemic brain injury | 10 to 100 μM in vitro; 5 mg/kg body weight in vivo | Neuro-protection in neonatal ischemic brain injury [20] |
| In vitro | HUVECs | H2O2-induced HUVEC injury | 20 and 25 μM | Xyloketal B and its two derivatives inhibited H2O2-induced HUVEC injury [26] |
| Both | HUVECs and apolipoprotein E-deficient mice | High-fat diet-induced atherosclerosis | 10 to 80 μM in vitro; 7, 14, and 28 mg/kg/day in vivo | Reduction of aortic atherosclerotic lesion area and improved endothelia function via increasing NO generation [29] |
| In vivo | C57 mice | Transient middle cerebral artery occlusion (tMCAO) | 50 mg/kg body weight | Pretreatment reduced infarction volume dose-dependently [19] |
| In vivo | C. elegans | Heat stress | 100 μM | Xyl-B derivative increased the expression of Hsp70 by upregulating HSF1 activity [39] |
| In vivo | Sprague Dawley rats | Two-kidney, two-clip renovascular hypertensive model | 20 μM in aortic ring function; 20 mg/kg body weight | Reduced blood pressure and enhanced relaxation of aortic rings in 2K2C renovascular hypertensive rats [50] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, H.; Luo, Z.; Chen, W.; Feng, Z.-P.; Wang, G.-L.; Sun, H.-S. Marine Compound Xyloketal B as a Potential Drug Development Target for Neuroprotection. Mar. Drugs 2018, 16, 516. https://doi.org/10.3390/md16120516
Gong H, Luo Z, Chen W, Feng Z-P, Wang G-L, Sun H-S. Marine Compound Xyloketal B as a Potential Drug Development Target for Neuroprotection. Marine Drugs. 2018; 16(12):516. https://doi.org/10.3390/md16120516
Chicago/Turabian StyleGong, Haifan, Zhengwei Luo, Wenliang Chen, Zhong-Ping Feng, Guan-Lei Wang, and Hong-Shuo Sun. 2018. "Marine Compound Xyloketal B as a Potential Drug Development Target for Neuroprotection" Marine Drugs 16, no. 12: 516. https://doi.org/10.3390/md16120516
APA StyleGong, H., Luo, Z., Chen, W., Feng, Z.-P., Wang, G.-L., & Sun, H.-S. (2018). Marine Compound Xyloketal B as a Potential Drug Development Target for Neuroprotection. Marine Drugs, 16(12), 516. https://doi.org/10.3390/md16120516