Abstract
Ten new triterpenoid compounds with structure diversity of the C-17 side-chain, including nine tirucallanes, named xylocarpols A–E (1–5) and agallochols A–D (6–9), and an apotirucallane, named 25-dehydroxy protoxylogranatin B (10), were isolated from the mangrove plants Xylocarpus granatum, Xylocarpus moluccensis, and Excoecaria agallocha. The structures of these compounds were established by HR-ESIMS and extensive one-dimensional (1D) and two-dimensional (2D) NMR investigations. The absolute configurations of 1 and 2 were unequivocally determined by single-crystal X-ray diffraction analyses, conducted with Cu Kα radiation; whereas those of 4, 6–8 were assigned by a modified Mosher’s method and the comparison of experimental electronic circular dichroism (ECD) spectra. Most notably, 5, 6, 7, and 9 displayed potent activation effects on farnesoid–X–receptor (FXR) at the concentration of 10.0 μM; 10 exhibited very significant agonistic effects on pregnane–X–receptor (PXR) at the concentration of 10.0 nM.
1. Introduction
Cholestasis, a clinical syndrome of hepatobiliary diseases, is usually caused by accumulation of bile acids in the liver and systemic circulation [1]. Long-term cholestasis can lead to primary biliary cirrhosis, primary sclerosing cholangitis, and hepatic failure. In clinical practice, abnormal metabolism of bile acids is deemed to be a crucial risk factor that induces cholestasis and cholestatic liver injury [1,2]. Farnesoid–X–receptor (FXR) and pregnane–X–receptor (PXR) are two members of the nuclear receptor family. Due to the regulation capability of a suite of genes involved in the metabolism, transport, and elimination of bile acids, FXR and PXR are considered to be the key target proteins for the treatment of cholestasis and liver injury [1,3,4,5].
Xylocarpus granatum and Xylocarpus moluccensis, true-mangrove species of the family Meliaceae, have been used in folk medicine, particularly in South and Southeast Asian countries, for the treatment of cholera, diarrhea, malaria, and other fever diseases [6]. Mangroves of the genus Xylocarpus are mainly distributed in East Africa, India, Bangladesh, Southeast Asia, Southern China, and Northern Australia [7,8]. Previous chemical investigation of X. granatum and X. moluccensis, collected from different locations, led to the isolation and identification of various limonoids, protolimonoids, alkaloids, and flavanones. To the best of our knowledge, limonoids are the main secondary metabolites of these mangroves [7,8,9].
Excoecaria agallocha, a semi-mangrove species of the family Euphorbiaceae, has been used in Asia as a traditional medicine for the treatment of epilepsy, haematuria, leprosy, and toothache [10,11]. It is mainly distributed along the sea coasts of China, India, Philippines, and Oceania. The poisonous milky latex, exuded from the bark of this plant, can cause blistering of the skin and temporary blindness [12,13,14,15,16]. Previous chemical investigation of this plant resulted in the isolation and identification of diterpenoid, triterpenoid, tanninoid, and flavonoid natural compounds [10,17,18,19,20,21]. To date, only 18 triterpenoid compounds, including types of cycloartane, friedelane, lupane, oleanane, and taraxerane, have been reported from this plant [17].
As part of our ongoing investigation of bioactive natural products from mangrove plants, 10 new triterpenoid compounds, including nine tirucallanes (1–9) and one apotirucallane (10) (Figure 1), were isolated from mangrove plants X. granatum, X. moluccensis, and E. agallocha. We reported the isolation and structure elucidation of these compounds, and their agonistic effects on human FXR and PXR.
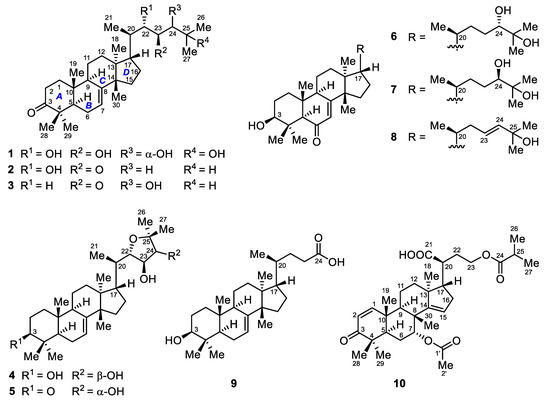
Figure 1.
Structures of compounds 1–10.
2. Results and Discussion
Compound 1 was obtained as colorless crystals. Its molecular formula C30H50O5 with six indices of hydrogen deficiency was established by the negative HR-ESIMS quasi molecular ion peak at m/z 525.3356 ([M + Cl]−, calculated for 525.3352) (Supplementary Materials S6 and S7). According to the 1H and 13C NMR spectroscopic data of 1 (Table 1 and Table 2) (Supplementary Materials S8‒S13), two elements of unsaturation were due to a carbon-carbon double bond and a carbonyl group. Thus, the molecule was tetracyclic. The 13C NMR spectroscopic data and the DEPT135 experiment of 1 (Supplementary Materials S14 and S15) revealed the presence of eight methyl groups, seven methylene groups, eight methine groups (including three oxygenated ones at δC 75.8, 70.9, and 80.1 ppm, respectively, and an olefinic one at δC 118.0 ppm), and seven quaternary carbons (including an oxygenated one at δC 74.1, an olefinic one at δC 145.7, and a ketone carbon at δC 217.0 ppm, respectively).

Table 1.
1H (400 MHz) NMR spectroscopic data for compounds 1–5 in CDCl3 (δ in ppm, J in Hz).
Table 2.
13C (100 MHz) NMR spectroscopic data for compounds 1‒5 in CDCl3 (δ in ppm).
1H‒1H COSY correlations (Supplementary Materials S16‒S18) between H-22/H-23 and H-23/H-24 and HMBC correlations from H-24 to C-25, C-26, and C-27 confirmed the connection of the fragment of C-22(OH)–C-23(OH)–C-24(OH)–C-25(OH)–(Me)2 (Figure 2a). The NMR spectroscopic data of 1 resembled those of toonaciliatavarin E [22], except for different orientations of three chiral centers on the C-17 side-chain, videlicet C-22, C-23, and C-24.
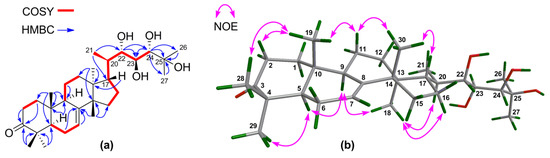
Figure 2.
(a) Key 1H‒1H COSY and HMBC correlations for 1; (b) Selected NOE interactions for 1.
The relative configuration of the tetracyclic tirucallane core (rings-A, B, C, and D) of 1 was corroborated by diagnostic NOE interactions (Supplementary Materials S28‒S32). Those between H-5/H-9, H-5/H3-29, H-9/H3-18, and H3-18/H-20 revealed the α-oriented H-5, H-9, H3-18, H-20, and H3-29. Similarly, NOE interactions between H3-19/H-2β, H3-19/H3-28, H3-19/H-11β, H3-30/H-11β, and H3-30/H-17 assigned the β-oriented H-17, H3-19, H3-28, and H3-30 (Figure 2b).
In order to establish the absolute configuration of the whole molecule of 1, particularly the absolute configuration of three chiral centers on the C-17 side-chain, a single-crystal X-ray diffraction analysis, conducted with Cu Kα radiation (Flack parameter of 0.01(6)) (Figure 3), was employed. Thus, the absolute configuration of 1, named xylocarpol A, was unequivocally assigned as (5R,9R,10R,13S,14S,17S,20R,22S,23S,24S)-22,23,24,25-tetrahydroxytirucalla-7-ene-3-one.
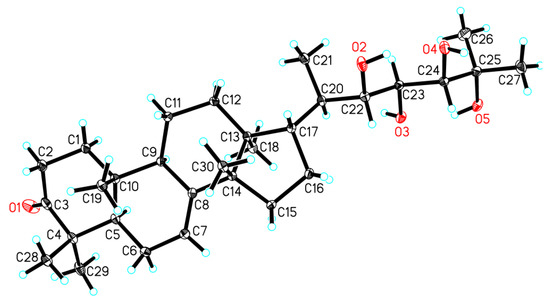
Figure 3.
Perspective drawing of the single-crystal X-ray structure of 1.
Compound 2 was isolated as colorless crystals. Its molecular formula C30H48O3 (seven degrees of unsaturation) was established by the positive HR-ESIMS quasi molecular ion peak at m/z 457.3670 ([M + H]+, calculated for 457.3676) (Supplementary Material S33). The NMR spectroscopic data of 2 (Table 1 and Table 2) (Supplementary Materials S34‒S39) were closely related to those of aphagranin D [23], the difference being the absence of the 24-OH and 25-OH groups in 2. This deduction were corroborated by the upshifted C-24 (δC 82.1 CH in aphagranin D; whereas δC 46.8 CH2 in 2) and C-25 (δC 79.0 qC in aphagranin D; whereas δC 24.5 CH in 2), and the proton spin-spin system, i.e., H2-24‒H-25(H3-26)‒H3-27, observed in the 1H‒1H COSY spectrum of 2 (Supplementary Materials S42‒S44).
NOE interactions (Supplementary Materials S53‒S58) between H-5/H-9, H-5/H3-29, H-9/H3-18, and H3-18/H-20 assigned the α-oriented H-5, H-9, H3-18, and H3-29; whereas those between H3-19/H-2β, H3-19/H3-28, H3-30/H-12β, H3-30/H-17 concluded the β-oriented H-17, H3-19, H3-28, and H3-30. Finally, single-crystal X-ray diffraction analysis, conducted with Cu Kα radiation (Flack parameter of 0.08(13)) (Figure 4), unambiguously established the absolute configuration of the whole molecule of 2. Therefore, the absolute configuration of 2, named xylocarpol B, was unambiguously determined to be (5R,9R,10R,13S,14S,17S,20R,22S)-22-hydroxytirucalla-7-ene-3,23-dione.
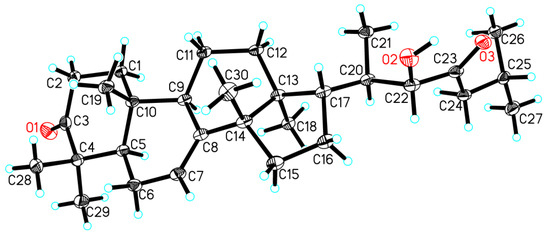
Figure 4.
Perspective drawing of the single-crystal X-ray structure of 2.
Compound 3 provided the molecular formula C30H48O3 as deduced from the positive HR-ESIMS quasi molecular ion peak at m/z 457.3654 ([M + H]+, calculated for 457.3676) (Supplementary Material S59). The NMR spectroscopic data of 3 (Table 1 and Table 2) (Supplementary Materials S60‒S64) resembled those of 2, except for the different location of the only hydroxy group on the C-17 side-chain. A 24-OH group was observed in 3 instead of the 22-OH function in 2. HMBC correlations (Supplementary Materials S72‒S77) from the oxygenated H-24 (δH 4.07, s) to C-23, C-25, C-26, and C-27 confirmed the presence of the 24-OH group. NOE interactions (Supplementary Materials S78‒S82) between H-5/H-9, H-5/H3-29, H-9/H3-18, and H3-18/H-20, and those between H3-19/H-2β, H3-19/H3-28, H3-30/H-11β, and H3-30/H-17 established the same tetracyclic tirucallane core (rings-A, B, C, and D) in 3 as that of 2. However, due to the limited amount of 3, the chirality of C-24 could not be determined. Thus, the structure of 3, named xylocarpol C, was assigned as 24-hydroxytirucalla-7-ene-3,23-dione.
Compound 4 gave the molecular formula C30H50O4 as obtained from the negative HR-ESIMS quasi molecular ion peak at m/z 509.3402 ([M + Cl]−, calculated for 509.3403) (Supplementary Materials S83 and S84). The 1H and 13C NMR spectroscopic data of 4 (Table 1 and Table 2) (Supplementary Materials S85‒S90) were closely related to those of odoratone [24], except for the replacement of the C-3 ketone group in odoratone by a hydroxy group in 4. The above deduction was corroborated by the upshifted C-3 (δC 216.0 qC in odoratone; whereas δC 79.3 CH in 4) and 1H‒1H COSY correlations between H2-2/H-3 (Figure 5a) (Supplementary Materials S95‒S97). The relative configuration of 4 was established by NOE interactions (Supplementary Materials S107‒S110). Those between H-3/H-5, H-5/H-9, H-5/H3-29, H-9/H3-18, and H3-18/H-20 assigned the α-oriented H-3, H-5, H-9, H3-18, H-20, and H3-29; NOE interactions between H3-19/H3-28, H3-30/H-11β, H3-19/H-11β, and H3-30/H-17 concluded the β-oriented H-17, H3-19, H3-28, and H3-30 (Figure 5b). The above results concluded the same relative configuration for the tetracyclic tirucallane core (rings-A, B, C, and D) in 4 as that of 1–3, except for the additional chiral C-3 in 4. Moreover, NOE interactions between H-23/H3-21 and H-24/H3-26 assigned the α-oriented H-23, H-24, and H3-26; those between H-22/H3-27 concluded the β-oriented H-22 and H3-27 (Figure 5b). Thus, the relative configuration of the C-17 side-chain of 4 was determined.
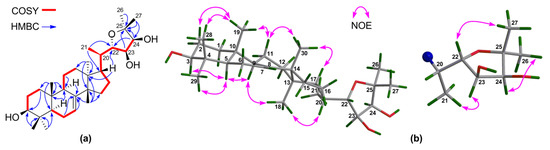
Figure 5.
(a) Key 1H‒1H COSY and HMBC correlations for 4; (b) Selected NOE interactions for 4.
In order to establish the absolute configuration of 4, a modified Mosher’s α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) ester method was applied [25]. The (3,23,24)-tri(S)- and (3,23,24)-tri(R)-MTPA esters of 4, videlicet 4s and 4r (Supplementary Materials S280‒S289), were successfully prepared. Based on the MTPA ester rule of Δδ (δS − δR) values (Figure 6) [25], the absolute configurations of C-3, C-23, and C-24 were assigned as S, R, and S, respectively. Therefore, the absolute configuration of 4, named xylocarpol D, was unequivocally established as (3S,5R,9R,10R,13S,14S,17S,20R,22S,23R,24S)- 3,23,24-trihydroxy-22,25-epoxytirucalla-7-ene. The absolute configuration of the 2,2-dimethyltetrahydrofuran-3,4-diol moiety of odoratone [24] was first clarified as 22S,23R,24S.
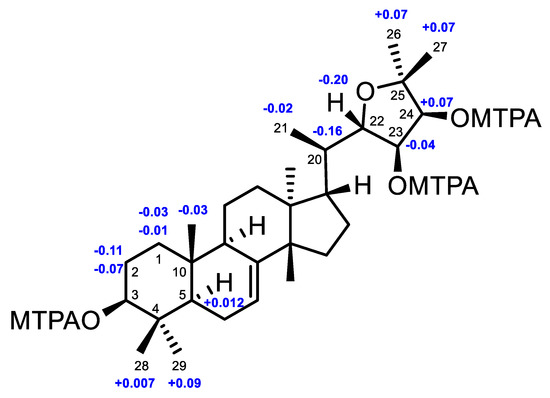
Figure 6.
Δδ values (Δδ [ppm] = [δS − δR]) that were obtained for the (3,23,24)-tri(S)- and (3,23,24)-tri(R)-MTPA esters of 4 (4s, 4r).
The molecular formula of 5 was determined to be C30H48O4 (seven degrees of unsaturation) by the negative HR-ESIMS quasi molecular ion peak at m/z 507.3245 ([M + Cl]−, calculated for 507.3247) (Supplementary Materials S111 and S112). Two elements of unsaturation were due to a carbon-carbon double bond and a ketone group; thus, the molecule was pentacyclic. The NMR spectroscopic data of 5 (Table 1 and Table 2) (Supplementary Materials S113‒S118) were similar to those of 4, the difference being the opposite orientation of the 24-OH group and the replacement of the 3-OH group in 4 by a ketone function in 5. Diagnostic NOE interactions (Supplementary Materials S134‒S137) between H-22/H3-27 and H-24/H3-27 assigned the β-oriented H-22 and H-24; whereas those between H-23/H3-26 concluded the α-oriented H-23 (Figure 7). HMBC correlations (Supplementary Materials S128‒S133) from H2-1, H2-2, H3-28, and H3-29 to the carbon (δC 217.2 qC) of a ketone group confirmed its location at C-3. Therefore, the structure of 5, named xylocarpol E, was assigned as depicted.
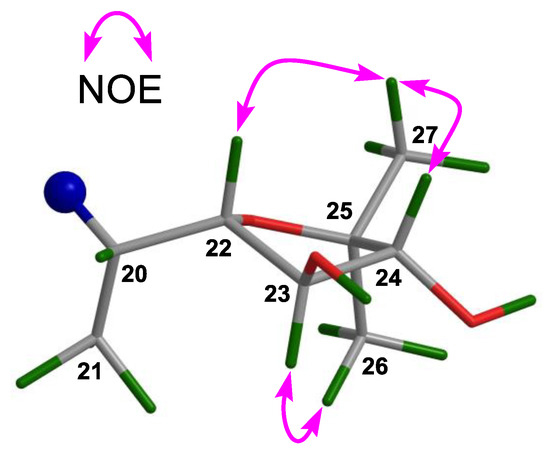
Figure 7.
Diagnostic NOE interactions for the 2,2-dimethyltetrahydrofuran-3,4-diol moiety of 5.
The molecular formula of 6 was concluded to be C30H50O4 by the positive HR-ESIMS quasi molecular ion peak at m/z 475.3790 ([M + H]+, calculated for 475.3782) (Supplementary Materials S138 and S139). The 1H and 13C NMR spectroscopic data of 6 (Table 3 and Table 4) (Supplementary Materials S140‒S146) resembled those of brumollisol B [26], except for the absence of the 23-OH group, being corroborated by the upshifted C-23 (δC 69.5 CH in brumollisol B; whereas δC 28.4 CH2 in 6), 1H‒1H COSY correlations (Supplementary Materials S149‒S152) between H2-23/H-24, and HMBC correlations (Supplementary Materials S157‒S164) from H3-26 and H3-27 to C-24 (Figure 8a).
Table 3.
1H (400 MHz) NMR spectroscopic data for compounds 6‒10 in CDCl3 (δ in ppm, J in Hz).
Table 4.
13C (100 MHz) NMR spectroscopic data for compounds 6‒10 in CDCl3 (δ in ppm).
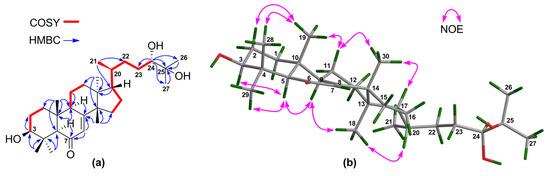
Figure 8.
(a) Key 1H‒1H COSY and HMBC correlations for 6; (b) Selected NOE interactions for 6.
The relative configuration for the tetracyclic tirucallane core (rings-A, B, C, and D) of 6 was established by NOE interactions (Supplementary Materials S165‒S168). Those between H-3/H-5, H-5/H-9, H-5/H3-29, H-9/H3-18, and H3-18/H-20 assigned the α-oriented H-3, H-5, H-9, H3-18, H-20, and H3-29; NOE interactions between H3-19/H3-28, H3-30/H-11β, H3-19/H-11β, and H3-30/H-17 concluded the β-oriented H-17, H3-19, H3-28, and H3-30 (Figure 8b).
The absolute configuration of 6 was established by the application of the modified Mosher’s MTPA ester method [25]. The (3,24)-di(S)- and (3,24)-di(R)-MTPA esters of 6 were prepared (Supplementary Materials S290‒S297). Based on the MTPA ester rule of Δδ (δS − δR) values (Figure 9) [25], the absolute configurations of C-3 and C-24 were assigned as S and S, respectively. Hence, the absolute configuration of 6, named agallochol A, was unequivocally assigned as (3S,5R,9R,10R,13S,14S,17S,20S,24S)- 3,24,25-trihydroxytirucalla-7-ene-6-one.
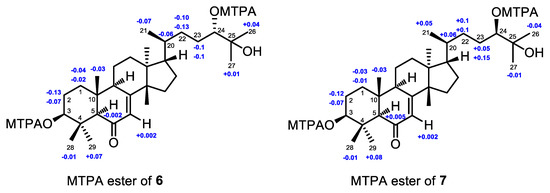
Figure 9.
Δδ values (Δδ [ppm] = [δS − δR]) that were obtained for the (3,24)-di(S)- and (3,24)-di(R)-MTPA esters of 6 (6s, 6r) and 7 (7s, 7r).
Compound 7 provided the same molecular formula (C30H50O4) as that of 6 based on the positive HR-ESIMS quasi molecular ion peak at m/z 475.3792 ([M + H]+, calculated for 475.3782) (Supplementary Materials S169 and S170). The 1H and 13C NMR spectroscopic data of 7 (Table 3 and Table 4) (Supplementary Materials S171‒S175) resembled those of 6, except for the slight difference of 1H and 13C chemical shifts of CH-24 (δH 3.36 (br s), δC 78.6 in 6; whereas δH 3.30 dd (J = 11.6, 1.6 Hz), δC 79.5 in 7), indicating that both compounds are a pair of C-24 epimers. The above deduction was further corroborated by 1H‒1H COSY correlations between H-24/H2-23 (Supplementary Materials S178‒S180) and HMBC interactions between H-24/C-23, H-24/C-25, H3-26/C-24, and H3-27/C-24 (Supplementary Materials S184‒S189).
In order to determine the absolute configurations of C-3 and C-24 of 7, (3,24)-di(S)- and (3,24)-di(R)-MTPA esters of 7 were prepared (Supplementary Materials S298‒S305). Based on the MTPA ester rule of Δδ (δS − δR) values (Figure 9) [25], the absolute configurations of C-3 and C-24 were assigned as S and R, respectively. Hence, the absolute configuration of 7, named agallochol B, was unambiguously concluded to be (3S,5R,9R,10R,13S,14S,17S,20S,24R)-3,24,25-trihydroxytirucalla-7-ene-6-one.
The molecular formula of 8 was established as C30H48O3 by the positive HR-ESIMS quasi molecular ion peak at m/z 457.3673 ([M + H]+, calculated for 457.3682) (Supplementary Material S194). The NMR spectroscopic data of 8 (Table 3 and Table 4) (Supplementary Materials S195‒S200) were similar to those of 7, except for the presence of a Δ23(24) double bond (δH 5.60 (dd, J = 15.6, 4.8 Hz, 1H), 5.62 (d, J = 15.6 Hz, 1H); δC 125.2, CH, 139.7, CH) and the absence of the 24-OH group. 1H‒1H COSY correlations between H-23/H2-22 and H-23/H-24 (Supplementary Materials S203‒S206), and HMBC correlations between H-23/C-22, H-23/C-24, H-23/C-25, H-24/C-22, H-24/C-23, and H-24/C-25 in 8 (Supplementary Materials S211‒S217) confirmed the above deduction. In addition, the coupling constant of 15.6 Hz between H-23 and H-24 assigned the E-geometry of the Δ23(24) double bond.
The relative configuration of the tetracyclic tirucallane core (rings-A, B, C, and D) of 8 was established as the same as that of 7 by NOE interactions (Supplementary Materials S218‒S222). Those between H-5/H-3, H-5/H-9, H-5/H3-29, H-9/H3-18, H3-18/H-20, H3-19/H3-28, H3-19/H3-30, and H3-30/H-17 assigned the same relative configuration of 8 as that of 7. Moreover, the absolute configuration of 8, particularly that of the tetracyclic tirucallane core (rings-A, B, C, and D), was established to be the same as that of 7, except for the deficiency of the chiral C-24, by the accurate fit of their experimental ECD spectra (Figure 10). Therefore, the structure of 8, named agallochol C, was determined to be (3S,5R,9R,10R,13S,14S,17S,20S)-3,25-dihydroxytirucalla-7,23-diene-6-one.
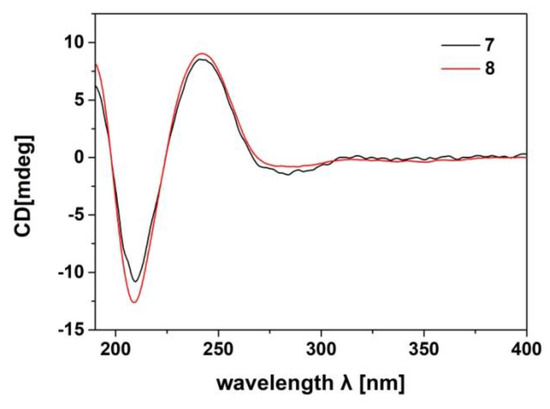
Figure 10.
Comparison of the experimental ECD spectra of 7 and 8 (measured at concentrations of 100 and 167 µg/mL in acetonitrile, respectively).
Compound 9 had the molecular formula C27H44O3 as determined from its negative HR-ESIMS quasi molecular ion peak at m/z 451.2985 ([M + Cl]−, calculated for 451.2984) (Supplementary Materials S223 and S224). The NMR spectroscopic data of 9 (Table 3 and Table 4) (Supplementary Materials S225‒S231) resembled those of a trinortirucalla-7-ene, i.e., sikkimenoid F [27], except for the replacement of the C-24 aldehyde group in sikkimenoid F by a C-24 carboxyl group in 9. The above deduction was corroborated by the upshifted C-24 (δH 9.75 (br s), δC 203.1 CH in sikkimenoid F; whereas δC 178.5 qC in 9) and HMBC correlations from H2-22 and H2-23 to the carbonyl carbon (C-24) of this carboxyl group. The relative configuration of the tetracyclic tirucallane core (rings-A, B, C, and D) of 9 was established by NOE interactions (Supplementary Materials S247‒S250). Those between H-5/H-3, H-5/H-9, H-5/H3-29, H-9/H3-18, H3-18/H-20, H3-19/H3-28, H3-19/H3-30, and H3-30/H-17 assigned the same relative configuration of the tetracyclic tirucallane core of 9 as that of 8. Thus, the structure of 9, named agallochol D, was assigned as 3β-hydroxy-25,26,27-trinortirucalla-7-ene-24-oic acid.
Compound 10 provided the molecular formula C32H46O7 based on the positive HR-ESIMS quasi molecular ion peak at m/z 543.3303 ([M + H]+, calculated for 543.3316) (Supplementary Material S251), requiring ten degrees of unsaturation. According to the 1H and 13C NMR spectroscopic data of 10 (Table 3 and Table 4) (Supplementary Materials S252‒S258), six of the 10 elements of unsaturation were due to two carbon-carbon double bonds and four carbonyls. Thus, the molecule was tetracyclic. The DEPT135 experiment of 10 (Supplementary Materials S259‒S261) combined with its 13C NMR spectroscopic data revealed the presence of eight methyl groups, six methylene groups, nine methine groups (including three olefinic ones), and nine quaternary carbons (including four carbonyl carbons).
The above NMR characteristic features of 10 closely resembled those of an apotirucallane protolimonoid, i.e., protoxylogranatin B [28], except for the absence of the 25-OH group in 10. 1H–1H COSY correlations from the proton of a methine moiety (δH 2.56 m; δC 34.1) to H3-26 and H3-27 confirmed the presence of the CH-25 group (Figure 11a). The relative configuration of 10 was established on the basis of NOE interactions (Supplementary Materials S275‒S279). Those between H-5/H-9, H-5/H3-29, H-9/H3-18, H-20/H3-18 assigned the α-oriented H-5, H-9, H3-18, and H-20; whereas those between H3-19/H3-28, H3-19/H-6β, and H3-30/H-6β concluded the β-oriented H3-19 and H3-30 (Figure 11b). The NOE interaction between H-7/H3-30, but not between H-7/H-5 and H-7/H3-18, indicated the β-oriented H-7 and the corresponding α-oriented 7-acetoxy group (Figure 11b). Thus, the structure of 10, named 25-dehydroxy protoxylogranatin B, was established as depicted.
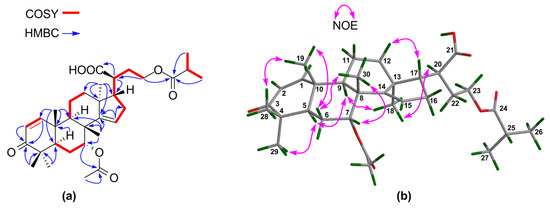
Figure 11.
(a) Key 1H‒1H COSY and HMBC correlations for 10; (b) Selected NOE interactions for 10.
In order to search for natural agonists of human FXR and PXR, most of the above isolated compounds were screened for their agonistic effects on these nuclear receptors. Chenodeoxycholic acid (CDCA) or rifampicin was used as the positive control at the concentration of 80.0 μM or 10.0 μM, respectively (Figure 12 and Figure 13). The results showed that 6 and 7 displayed significant agonistic effects on FXR at the concentration of 1.0 μM; while 5, 6, 7, and 9 exhibited significant agonistic effects on FXR at the concentration of 10.0 μM. Moreover, 1 displayed a moderate significant agonistic effect on FXR at the concentration of 10.0 μM (Figure 12). Compound 10 exhibited a significant agonistic effect on PXR at the concentration of 10.0 nM, and even a higher agonistic effect on PXR as compared to that of the positive control, rifampicin, at the same concentration of 10.0 μM (Figure 13).
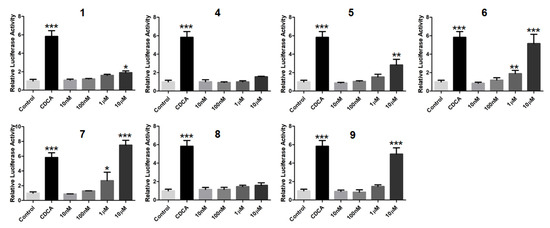
Figure 12.
Agonistic effects of 1, 4–9 on the farnesoid–X–receptor (FXR) in HepG2 cells. CDCA (80.0 µM) was used as the positive control. * p < 0.01, ** p < 0.001, *** p < 0.0001 compared to the control group.
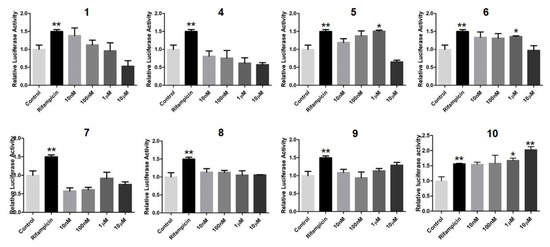
Figure 13.
Agonistic effects of 1, 4–10 on the pregnane–X–receptor (PXR) to modulate PXR target gene CYP3A4 transactivation in HepG2 cells. Rifampicin (10.0 µM) was used as the positive control. * p < 0.01, ** p < 0.001, *** p < 0.0001 compared to the control group.
3. Materials and Methods
3.1. General Methods
Optical rotations were recorded at room temperature on a MCP200 modular circular polarimeter (Anton Paar GmbH, Seelze, Germany). A GENESYS 10S UV–Vis spectrophotometer (Thermo Scientific, Shanghai, China) was used to obtain UV spectra. The NMR spectroscopic data were measured on a Bruker AV-400 NMR spectrometer (Bruker Scientific Technology Co. Ltd., Karlsruhe, Germany) using TMS as the internal standard. Single-crystal X-ray diffraction analyses were carried out on an Agilent Xcalibur Atlas Gemini Ultra-diffractometer with mirror monochromated Cu Kα radiation (λ = 1.54184 Å) at 100 K. An LC-ESI (Bruker Daltonics, Bremen, Germany) and an LC-ESI-QTOF mass spectrometer (SYNAPTTM G2 HDMS, Waters, Manchester, UK) were used to acquire HR-ESIMS data. For electronic circular dichroism (ECD) spectra, a Jasco 810 spectropolarimeter (JASCO Corporation, Tokyo, Japan) was applied with the solvent of acetonitrile. Semi-preparative HPLC was carried out on a Waters 2535 pump equipped with a 2489 UV detector (Waters Corporation, Milford, NY, USA) and an ODS column (YMC, 250 × 10 mm inner diameter, 5 µm). Silica gel (100–200 mesh, Qingdao Mar. Chem. Ind. Co. Ltd., Qingdao, China) and ODS silica gel (A-HG 12 nm, 50 mm, YMC Co. Ltd., Kyoto, Japan) were used for column chromatography.
3.2. Plant Material
The seeds of two mangrove plants, Xylocarpus granatum and Xylocarpus moluccensis, were collected in September 2007 at the mangrove swamps of Krishna estuary, Andhra Pradesh, India. The plant species were identified by Mr. Tirumani Satyanandamurty (Government Degree College at Amadala Valasa, India). The voucher samples (No. IXG-02 for X. granatum) and (No. IXM200701 for X. moluccensis) were kept in the Marine Drugs Research Center, College of Pharmacy, Jinan University.
The stems and twigs of the semi-mangrove plant, Excoecaria agallocha, were collected in December 2003 at the mangrove swamps of Hainan Island, China. The identification of the plant was done by Professor Jun Wu (School of Pharmaceutical Sciences, Southern Medical University). A voucher sample (EA-J1) was kept in Marine Drugs Research Center, College of Pharmacy, Jinan University.
3.3. Extraction and Isolation
The seeds of X. granatum were air-dried (22.0 kg), powdered, and then extracted with 95% EtOH (5 × 75 L) at room temperature. The resulting EtOH extract (4.0 kg) was partitioned between EtOAc/water (3:1, v/v) to afford the EtOAc portion (1100.0 g). The EtOAc portion (250.0 g) was subjected to silica gel column chromatography (120 × 10 cm inner diameter; chloroform/methanol, from 100:0 to 10:1) to give 210 fractions. Fractions 50–82 (20.0 g) were combined and separated by an ODS silica gel column (100 × 7 cm inner diameter; acetone/water, from 40:60 to 100:0) to afford 81 subfractions. The subfraction 52 (308.0 mg) was purified by semi-preparative HPLC (MeCN/H2O, 40:60, 3.0 mL/min) to give 1 (78.0 mg, tR = 54.0 min) and 5 (3.0 mg, tR = 62.0 min); whereas the subfraction 53 (128.0 mg) was separated by semi-preparative HPLC (MeCN/H2O, 40:60, 3.0 mL/min) to yield 4 (15.5 mg, tR = 35.8 min).
The seeds of X. moluccensis were air-dried (6.0 kg), powdered, and extracted with 95% EtOH (6 × 15 L) at room temperature. The resulting extract (824.6 g) was partitioned between EtOAc/water (3:1, v/v) to give the EtOAc portion (299.1 g), which was applied to silica gel column chromatography (150 × 8.5 cm inner diameter; chloroform/methanol, from 100:0 to 5:1) to afford 223 fractions. Fractions 9–11 (18.5 g) were combined and further separated by ODS silica gel column chromatography (60 × 3 cm inner diameter; acetone/water, from 50:50 to 100:0) to afford 90 subfractions. The subfraction 61 (800.0 mg) was purified by semi-preparative HPLC (MeOH/H2O, 82:18, 3.0 mL/min) to yield 2 (6.5 mg, tR = 45.0 min) and 3 (1.0 mg, tR = 52.0 min). Fractions 68–71 (22.9 g) were combined and separated by ODS silica gel column chromatography (60 × 3 cm inner diameter.; acetone/water, from 40:60 to 100:0) to give 111 subfractions. The subfraction 75 (210.0 mg) was purified by semi-preparative HPLC (MeCN/H2O, 58:42, 3.0 mL/min) to afford 10 (0.9 mg, tR = 46.0 min).
The stems and twigs of E. agallocha were air-dried (10.0 kg), powdered, and extracted with 95% EtOH (5 × 40.0 L) at room temperature. The resulting brown extract (370.0 g)was then suspended in water, and extracted with hexane (3:1, v/v) and EtOAc (3:1, v/v), successively, to give the EtOAc portion (192.0 g), which was separated by silica gel column chromatography (150 × 10.5 cm inner diameter; chloroform/methanol, from 100:0 to 5:1) to yield 285 fractions. Fractions 130 to 170 (17.5 g) were combined and subjected to ODS silica gel column chromatography (110 × 6 cm inner diameter; acetone/water, from 30:70 to 100:0) to give 76 subfractions. The subfraction 35 (1.3 g) was purified by semi-preparative HPLC (MeCN/H2O, 42:58, 3.0 mL/min) to yield 6 (4.0 mg, tR = 69.8 min) and 7 (4.0 mg, tR = 73.1 min).
Fractions 40 to 100 (18.0 g) were combined and further separated by ODS silica gel column chromatography (110 × 6 cm inner diameter; acetone/water, from 55:45 to 100:0) to yield 65 subfractions. The subfraction 33 (76.0 mg) was subjected to semi-preparative HPLC (MeCN/H2O, 70:30, 3.0 mL/min) to give 9 (3.3 mg, tR = 36.8 min). Fractions 101 to 129 (45.0 g) were combined and purified by ODS silica gel column chromatography (100 × 10 cm inner diameter; acetone/water, from 40:60 to 100:0) to yield 80 subfractions. The subfraction 44 (190.0 mg) was purified by semi-preparative HPLC (MeOH/H2O, 82:18, 3.0 mL/min) to afford 8 (4.0 mg, tR = 30.8 min).
Xylocarpol A (1): (5R,9R,10R,13S,14S,17S,20R,22S,23S,24S)-22,23,24,25-tetrahydroxytirucalla- 7-ene-3-one. Colorless crystals; = −68.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 193 (4.20) nm; 1H- and 13C-NMR spectroscopic data see Table 1 and Table 2, respectively; HR-ESIMS m/z 525.3356 [M + Cl]− (calculated for C30H50ClO5, 525.3352).
Xylocarpol B (2): (5R,9R,10R,13S,14S,17S,20R,22S)-22-hydroxytirucalla-7-ene-3,23-dione. Colorless crystals; = −8.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 191 (4.16) nm; 1H- and 13C-NMR spectroscopic data see Table 1 and Table 2, respectively; HR-ESIMS m/z 457.3670 [M + H]+ (calculated for C30H49O3, 457.3676).
Xylocarpol C (3): White amorphous power; = −82.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 193 (3.77) nm; 1H- and 13C-NMR spectroscopic data see Table 1 and Table 2, respectively; HR-ESIMS m/z 457.3654 [M + H]+ (calculated for C30H49O3, 457.3676).
Xylocarpol D (4): (3S,5R,9R,10R,13S,14S,17S,20R,22S,23R,24S)-3,23,24-trihydroxy-22,25- epoxytirucalla-7-ene. White amorphous power; = −70.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 203 (4.40) nm; 1H- and 13C-NMR spectroscopic data see Table 1 and Table 2, respectively; HR-ESIMS m/z 509.3402 [M + Cl]− (calculated for C30H50ClO4, 509.3403).
Xylocarpol E (5): White amorphous power; = −8.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 194 (3.70) nm; 1H- and 13C-NMR spectroscopic data see Table 1 and Table 2, respectively; HR-ESIMS m/z 507.3245 [M + Cl]− (calculated for C30H48ClO4, 507.3247).
Agallochol A (6): (3S,5R,9R,10R,13S,14S,17S,20S,24S)-3,24,25-trihydroxytirucalla-7-ene-6-one. White amorphous power; = −19.5 (c 0.1, acetone); UV (MeCN) λmax (logε) 204 (3.61), 246 (3.68) nm; 1H- and 13C-NMR spectroscopic data see Table 3 and Table 4, respectively; HR-ESIMS m/z 475.3790 [M + H]+ (calculated for C30H51O4, 475.3782).
Agallochol B (7): (3S,5R,9R,10R,13S,14S,17S,20S,24R)-3,24,25-trihydroxytirucalla-7-ene-6-one. White amorphous power; = −13.2 (c 0.1, acetone); UV (MeCN) λmax (logε) 204 (3.55), 246 (3.49) nm; ECD (0.21 mM, MeCN) λmax (∆ε) 209.0 (−16.1), 242.6 (+ 12.8) nm; 1H- and 13C-NMR spectroscopic data see Table 3 and Table 4, respectively; HR-ESIMS m/z 475.3792 [M + H]+ (calculated for C30H51O4, 475.3782).
Agallochol C (8): (3S,5R,9R,10R,13S,14S,17S,20S)-3,25-dihydroxytirucalla-7,23-diene-6-one. White amorphous power; = −22.4 (c 0.1, acetone); UV (MeCN) λmax (logε) 202 (3.07), 245 (3.82) nm; ECD (0.37 mM, MeCN) λmax (∆ε) 209.6 (−10.4), 242.4 (+ 7.5) nm; 1H- and 13C-NMR spectroscopic data see Table 3 and Table 4, respectively; HR-ESIMS m/z 457.3673 [M + H]+ (calculated for C30H49O3, 457.3682).
3.4. Preparation of the (3,23,24)-Tri(S)- and (3,23,24)-Tri(R)-MTPA Esters of 4
Compound 4 (1.0 mg) was treated with (S)-MTPA acid (2.0 mg), 4-dimethylaminopyridine (DMAP) (1.0 mg), and N,N′-dicyclohexylcarbodiimide (DCC) (4.0 mg) in dried dichloromethane (0.5 mL) at room temperature for 72 h. The reaction mixture was concentrated and purified by C18 reversed-phase HPLC (YMC-Pack 250 × 10 mm i.d.) with acetonitrile (MeCN/H2O, 100:0) to afford the (3,23,24)-tri(S)-MTPA esters of 4, named 4s (1.7 mg). Similarly, the (3,23,24)-tri(R)-MTPA esters of 4, named 4r (1.8 mg), were prepared in the same way.
4s: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.252 (1H, m, H-1α), 1.705 (1H, m, H-1β), 1.637 (1H, m, H-2α), 1.797 (1H, m, H-2β), 4.730 (1H, dd, J = 11.2, 3.2 Hz, H-3), 1.454 (1H, m, H-5), 0.795 (3H, s, H3-18), 0.752 (3H, s, H3-19), 1.647 (1H, m, H-20), 0.910 (3H, overlapped, H3-21), 3.853 (1H, d, J = 5.2 Hz, H-22), 5.365 (1H, t, J = 5.2 Hz, H-23), 5.000 (1H, d, J = 6.0 Hz, H-24), 1.055 (3H, s, H3-26), 1.288 (3H, s, H3-27), 0.891 (3H, overlapped, H3-28), 0.900 (3H, s, H3-29), 0.967 (3H, s, H3-30).
4r: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.264 (1H, m, H-1α), 1.736 (1H, m, H-1β), 1.751 (1H, m, H-2α), 1.868 (1H, m, H-2β), 4.757 (1H, dd, J = 11.6, 4.4 Hz, H-3), 1.442 (1H, m, H-5), 0.820 (3H, s, H3-18), 0.778 (3H, s, H3-19), 1.803 (1H, m, H-20), 0.929 (3H, d, J = 6.0 Hz, H3-21), 4.056 (1H, d, J = 4.8 Hz, H-22), 5.403 (1H, t, J = 5.2 Hz, H-23), 4.926 (1H, d, J = 6.4 Hz, H-24), 0.989 (3H, s, H3-26), 1.216 (3H, s, H3-27), 0.884 (3H, overlapped, H3-28), 0.806 (3H, s, H3-29), 0.968 (3H, s, H3-30).
3.5. Preparation of the (3,24)-Di(S)- and (3,24)-Di(R)-MTPA Esters of 6 and 7
Compound 6 (1.0 mg) was treated with (S)-MTPA acid (2.0 mg), DMAP (1.0 mg), and DCC (4.0 mg) in dried dichloromethane (0.5 mL) at room temperature for 72 h. The reaction mixture was concentrated and purified by C18 reversed-phase HPLC (YMC-Pack 250 × 10 mm i.d.) with aqueous acetonitrile (MeCN/H2O, 84:16) to afford the (3,24)-di(S)-MTPA esters of 6, named 6s (0.5 mg). The (3,24)-di(R)-MTPA esters of 6, named 6r (0.7 mg), was prepared in the same way.
6s: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.497 (1H, m, H-1α), 1.717 (1H, m, H-1β), 1.619 (1H, m, H-2α), 1.780 (1H, m, H-2β), 4.672 (1H, dd, J = 11.2, 4.0 Hz, H-3), 2.220 (1H, s, H-5), 5.699 (1H, d, J = 2.8 Hz, H-7), 0.806 (3H, s, H3-18), 0.867 (3H, s, H3-19), 1.358 (1H, m, H-20), 0.784 (3H, d, J = 6.0 Hz, H3-21), 1.353 (1H, m, H-22a), 0.876 (1H, m, H-22b), 1.613 (1H, m, H-23a), 1.547 (1H, m, H-23b), 4.964 (1H, dd, J = 10.0, 2.4 Hz, H-24), 1.163 (3H, s, H3-26), 1.228 (3H, s, H3-27), 1.220 (3H, s, H3-28), 1.139 (3H, s, H3-29), 1.032 (3H, s, H3-30).
6r: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.520 (1H, m, H-1α), 1.762 (1H, m, H-1β), 1.749 (1H, m, H-2α), 1.850 (1H, m, H-2β), 4.703 (1H, dd, J = 11.6, 4.4 Hz, H-3), 2.222 (1H, s, H-5), 5.697 (1H, d, J = 2.4 Hz, H-7), 0.825 (3H, s, H3-18), 0.895 (3H, s, H3-19), 1.416 (1H, m, H-20), 0.854 (3H, d, J = 6.0 Hz, H3-21), 1.445 (1H, m, H-22a), 1.009 (1H, m, H-22b), 1.710 (1H, m, H-23a), 1.645 (1H, m, H-23b), 4.981 (1H, dd, J = 10.0, 2.4 Hz, H-24), 1.148 (3H, overlapped, H3-26), 1.180 (3H, s, H3-27), 1.148 (3H, overlapped, H3-28), 1.148 (3H, overlapped, H3-29), 1.035 (3H, s, H3-30).
Compound 7 (1.0 mg) was treated with (S)-MTPA acid (2.0 mg), DMAP (1.0 mg), and DCC (4.0 mg) in dried dichloromethane (0.5 mL) at room temperature for 72 h. The reaction mixture was concentrated and purified by C18 reversed-phase HPLC (YMC-Pack 250 × 10 mm i.d.) with aqueous acetonitrile (MeCN/H2O, 90:10) to afford (3,24)-di(S)-MTPA esters of 7, named 7s (1.5 mg). The (3,24)-di(R)-MTPA esters of 7, named 7r (1.4 mg), was prepared in the same way.
7s: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.505 (1H, m, H-1α), 1.722 (1H, m, H-1β), 1.625 (1H, m, H-2α), 1.787 (1H, m, H-2β), 4.673 (1H, dd, J = 11.6, 4.0 Hz, H-3), 2.220 (1H, s, H-5), 5.694 (1H, d, J = 2.4 Hz, H-7), 0.815 (3H, s, H3-18), 0.864 (3H, s, H3-19), 1.333 (1H, m, H-20), 0.890 (3H, d, J = 6.4 Hz, H3-21), 1.853 (1H, m, H-22a), 1.052 (1H, m, H-22b), 1.861 (1H, m, H-23a), 1.444 (1H, m, H-23b), 4.951 (1H, dd, J = 9.6, 2.4 Hz, H-24), 1.151 (3H, s, H3-26), 1.183 (3H, s, H3-27), 1.228 (3H, s, H3-28), 1.137 (3H, s, H3-29), 1.029 (3H, s, H3-30).
7r: amorphous powder; 1H NMR (CDCl3, 400 MHz): δH 1.523 (1H, m, H-1α), 1.747 (1H, m, H-1β), 1.747 (1H, m, H-2α), 1.859 (1H, m, H-2β), 4.699 (1H, dd, J = 11.2, 4.0 Hz, H-3), 2.214 (1H, s, H-5), 5.692 (1H, d, J = 2.8 Hz, H-7), 0.791 (3H, s, H3-18), 0.893 (3H, s, H3-19), 1.272 (1H, m, H-20), 0.845 (3H, d, J = 6.4 Hz, H3-21), 1.743 (1H, m, H-22a), 0.945 (1H, m, H-22b), 1.716 (1H, m, H-23a), 1.397 (1H, m, H-23b), 4.948 (1H, dd, J = 10.0, 2.4 Hz, H-24), 1.162 (3H, s, H3-26), 1.224 (3H, s, H3-27), 1.148 (3H, overlapped, H3-28), 1.145 (3H, overlapped, H3-29), 1.028 (3H, s, H3-30).
3.6. X-Ray Crystal Data for Xylocarpols A–B (1–2)
Xylocarpol A (1): orthorhombic, C31H54O6 (C30H50O5·CH3OH), space group P21, a = 35.3234(5) Å, b = 11.41820(10) Å, c = 7.38890(10) Å, α = 90°, β = 90°, γ = 90°, V = 2980.16(6) Å3, Z = 4, Dcalcd = 1.165 g cm−3, μ = 0.624 mm−1. Crystal size: 0.14 × 0.13 × 0.12 mm3, 33,526 measured reflections, 5916 (Rint = 0.0475) independent reflections, 351 parameters, 0 restraints, F (000) = 1152.0, R1 = 0.0429, wR2 = 0.1120 (all data), R1 = 0.0426, wR2 = 0.1118 (I > 2σ(I)), and goodness-of-fit (F2) = 1.030. The absolute structural parameter was 0.01 (6), the Flack x parameter was 0.01(6), and the Hooft y was 0.01(5).
Xylocarpol B (2): orthorhombic, C30H48O3, space group P21, a = 6.61860 (11) Å, b = 18.2616 (3) Å, c = 21.8783 (3) Å, α = 90°, β = 90°, γ = 90°, V = 2644.36 (7) Å3, Z = 4, Dcalcd = 1.147 g cm−3, μ = 0.551 mm−1, crystal size: 0.14 × 0.12 × 0.11 mm3, 13,817 measured reflections, 5179 (Rint = 0.0367) independent reflections, 307 parameters, 0 restraints, F(000) = 1008.0, R1 = 0.0467, wR2 = 0.1178 (all data), R1 = 0.0443, wR2 = 0.1147 (I > 2σ(I)), and goodness-of-fit (F2) = 1.025. The absolute structural parameter was 0.08 (13), the Flack x parameter was 0.09(14), and the Hooft y was 0.02(13).
CCDC-1872275 (1) and 1872277 (2) contained the supplementary crystallographic data for this paper (excluding structure factors). These data were provided free of charge by The Cambridge Crystallographic Data Centre.
3.7. FXR Activation Bioassay
By cloning genes encoding FXR into pCI-neomammalian expression vector, the human FXR expression plasmid was constructed. By cloning a genomic DNA fragment upstream of the transcription start site into the luciferase vector pGL4.14 (luc2/Hygro) based on the previous method [29], bile salt export pump (BSEP) promoter reporter was constructed. Human hepatoma HepG2 cells were transiently transfected with the expression plasmid of FXR (100.0 ng), the reporter vector of BSEP promoter luciferase (100.0 ng), and the null-Renilla luciferase plasmid (10.0 ng) as an internal control. After the incubation for 24 h, cells were treated with vehicle DMSO (0.1%), compounds 1, and 4–9, respectively, at different concentrations (10.0, 100.0 nM, 1.0, or 10.0 µM) for 24 h. Then, cells were harvested for the determination of luciferase activity. Chenodeoxycholic acid (CDCA) was used as the positive control at the final concentration of 80.0 μM.
3.8. PXR Activation Bioassay
As described previously [30], the expression vector of human PXR and the human PXR XREM-driven luciferase reporter plasmid (CYP3A4XREM-luciferase) were constructed. By using the lipofectamine 3000 (Invitrogen, Carlsbad, CA., USA), human hepatoma HepG2 cells were transfected with expression and reporter plasmids, along with pGL4.74 (Promega, Beijing, China) as an internal standard. Then, cells were treated with vehicle DMSO (0.1%), compounds 1, and 4–10, respectively, at different concentrations (10.0, 100.0 nM, 1.0, or 10.0 µM) for 24 h. By using the Dual-luciferase® Reporter Assay System (Promega, Beijing, China), the luciferase activities of the above compounds were recorded. By dividing the Firefly luciferase signal by the Renilla luciferase signal, the co-transfected plasmid results were normalized. Rifampicin, the well-known human PXR agonist, was used as the positive control at the final concentration of 10.0 μM.
4. Conclusions
In summary, 10 new triterpenoid compounds, including nine tirucallanes (1–9) and one apotirucallane (10), were obtained from mangrove plants X. granatum, X. moluccensis, and E. agallocha. The absolute configurations of 1, 2, 4, and 6–8 were unequivocally determined by single-crystal X-ray diffraction analysis (Cu Kα), modified Mosher’s method, and the comparison of experimental ECD spectra. Compounds 5, 6, 7, and 9 displayed potent activation effects on FXR at the concentration of 10.0 μM; whereas 10 exhibited very significant agonistic effects on PXR at nanomolar concentration. This work demonstrated that mangrove tirucallane- and apotirucallane-type triterpenoid compounds are valuable natural products for the discovery of leading compounds with potent activation effects on human FXR and PXR.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/12/488/s1, Copies of HR-ESIMS of compounds 1–10, 4s/r, 6s/r, and 7s/r; 1D and 2D NMR spectra of 1–10; and 1H NMR spectra of 4s/r, 6s/r, and 7s/r.
Author Contributions
L.S. and J.W. conceived and designed the experiments; Z.-P.J., R.-X.L. and Q.Z. performed the chemistry part of experiments and analyzed the data; Z.-L.L. performed the bioactivity part of experiments and analyzed the data; Z.-P.J. wrote the draft; X.-C.M., L.S. and J.W. revised the paper. All authors have read and approved the manuscript.
Funding
This work was financially supported by grants from the National Natural Science Foundation of China (NSFC) (U1501221, 31770377, 81661148049, and 81601174) and the Fundamental Research Funds for the Central Universities, P.R. China (21617474).
Acknowledgments
We thank Tirumani Satyanandamurty (Government Degree College at Amadala Valasa, India) for the providing of mangrove materials in this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jonker, J.W.; Liddle, C.; Downes, M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2011, 130, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.K.; Liu, J.; Yua, Z.L.; Wang, Y.F.; Wang, C.; Tian, X.G.; Ning, J.; Feng, L.; Sun, C.P.; Zhang, B.J.; et al. Alisma orientale extract exerts the reversing cholestasis effect by activation of farnesoid X receptor. Phytomedicine 2018, 42, 34–42. [Google Scholar] [CrossRef]
- Olefsky, J.M. Nuclear receptor minireview series. J. Biol. Chem. 2001, 276, 36863–36864. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Champagne, D.E.; Koul, O.; Isman, M.B.; Scudder, G.E.; Towers, G.H.N. Biological activity of limonoids from the Rutales. Phytochemistry 1992, 31, 377–394. [Google Scholar] [CrossRef]
- Tan, Q.G.; Luo, X.D. Meliaceous limonoids: Chemistry and biological activities. Chem. Rev. 2011, 111, 7437–7522. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xiao, Q.; Xu, J.; Li, M.Y.; Pan, J.Y.; Yang, M.H. Natural products from true mangrove flora: Source, chemistry and bioactivities. Nat. Prod. Rep. 2008, 25, 955–981. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Li, X.W.; Guo, Y.W. Recent progress on the mangrove plants: Chemistry and bioactivity. Curr. Org. Chem. 2016, 20, 1923–1942. [Google Scholar] [CrossRef]
- Li, M.Y.; Xiao, Q.; Pan, J.Y.; Wu, J. Natural products from semi-mangrove flora: Source, chemistry and bioactivities. Nat. Prod. Rep. 2008, 26, 281–298. [Google Scholar] [CrossRef]
- Wiriyachitra, P.; Hajiwangoh, H.; Boonton, P.; Adolf, W.; Opferkuch, H.J.; Hecker, E. Investigations of Medicinal plants of Euphorbiaceae and Thymelaeaceae occurring and used in Thailand; II. Cryptic irritants of the diterpene ester type from three Excoecaria species. Planta Med. 1985, 51, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Lin, P. Mangrove Vegetation; China Ocean Press: Beijing, China, 1988; pp. 51–52. [Google Scholar]
- Kumarasinghe, S.; Seneviratne, R. Skin and eye injury due to latex of Excoecaria agallocha. Aust. J. Dermatol. 1998, 39, 275–276. [Google Scholar] [CrossRef]
- Kawashima, T.; Takahashi, T.; Inoue, Y.; Kodama, M.; Ito, S. Constituents of Excoecaria agallocha. Phytochemistry 1971, 10, 3308–3309. [Google Scholar] [CrossRef]
- Ohigashi, H.; Katsumata, H.; Kawazu, K.; Koshimizu, K.; Mitsui, T. A piscicidal constituent of Excoecaria agallocha. Agric. Biol. Chem. 1974, 38, 1093–1095. [Google Scholar] [CrossRef]
- Prakash, S.; Khan, M.; Khan, H.; Zaman, A. A piperidine alkaloid from Excoecharia agallocha. Phytochemistry 1983, 22, 1836–1837. [Google Scholar] [CrossRef]
- Yin, B.W.; Shen, L.R.; Zhang, M.L.; Zhao, L.; Wang, Y.L.; Huo, C.H.; Shi, Q.W. Chemical constituents of plants from the genus Excoecaria. Chem. Biodivers. 2008, 5, 2356–2371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Liu, J.; Yu, S.J.; Proksch, P.; Gu, J.; Lin, W.H. TNF-α inhibitory diterpenoids from the Chinese mangrove plant Excoecaria agallocha L. Phytochemistry 2010, 71, 2124–2131. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, W.; Deng, Z.W.; Lin, W.H. Assignment of the absolute stereochemistry of an unusual diterpenoid from the mangrove plant Excoecaria agallocha L. J. Chin. Pharm. Sci. 2010, 19, 387–392. [Google Scholar] [CrossRef]
- Ponnapalli, M.G.; Ankireddy, M.; Annam, S.C.; Ravirala, S.; Sukki, S.; Tuniki, V.R. Unusual ent-isopimarane-type diterpenoids from the wood of Excoecaria agallocha. Tetrahedron. Lett. 2013, 54, 2942–2945. [Google Scholar] [CrossRef]
- Annam, S.C.; Ankireddy, M.; Sura, M.B.; Ponnapalli, M.G.; Sarma, A.V. Epimeric excolides from the stems of Excoecaria agallocha and structural revision of rhizophorin A. Org. Lett. 2015, 17, 2840–2843. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.S.; Gu, Y.C.; Kong, L.Y. Cytotoxic and anti-inflammatory triterpenoids from Toona ciliate. J. Nat. Prod. 2012, 75, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Zhang, Y.; Luo, J.; Kong, L.Y. Complete 1H and 13C NMR data assignment of protolimonoids from the stem barks of Aphanamixis grandifolia. Magn. Reson. Chem. 2011, 49, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, B.S.; Afshan, F.; Gulzar, T.; Sultana, R.; Naqvi, S.N.H.; Tariq, R.M. Tetracyclic triterpenoids from the leaves of Azadirachta indica and their insecticidal activities. Chem. Pharm. Bull. 2003, 51, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Chen, H.; Ma, S.G.; Fang, Z.F.; Bai, J.; Yu, S.S.; Chen, X.G.; Hou, Q.; Yuan, S.P.; Chen, X. Tirucallane triterpenoids from the stems of Brucea mollis. Chem. Biodivers. 2013, 10, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Peng, W.B.; Li, Z.L.; Wang, X.; Wei, J.G.; Liu, K.C.; Yang, Y.P.; Li, X.L. Chemical constituents from the aerial parts of Euphorbia sikkimensis and their bioactivities. Nat. Prod. Bioprospect. 2013, 3, 112–116. [Google Scholar] [CrossRef]
- Hu, W.M.; Wu, J. Protoxylogranatin B, a key biosynthetic intermediate from Xylocarpus granatum: Suggesting an oxidative cleavage biogenetic pathway to limonoid. Open. Nat. Prod. J. 2010, 3, 1–5. [Google Scholar] [CrossRef]
- Soisson, S.M.; Parthasarathy, G.; Adams, A.D.; Sahoo, S.; Sitlani, A.; Sparrow, C.; Cui, J.S.; Becker, J.W. Identification of a potent synthetic FXR agonist with an unexpected mode of binding and activation. Proc. Natl. Acad. Sci. USA 2008, 105, 5337–5342. [Google Scholar] [CrossRef]
- Zhou, C.; Poulton, E.J.; Grun, F.; Bammler, T.K.; Blumberg, B.; Thummel, K.E.; Eaton, D.L. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007, 71, 220–229. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).