Diphenyl Ethers from a Marine-Derived Aspergillus sydowii

 ,
,
Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction and Isolation
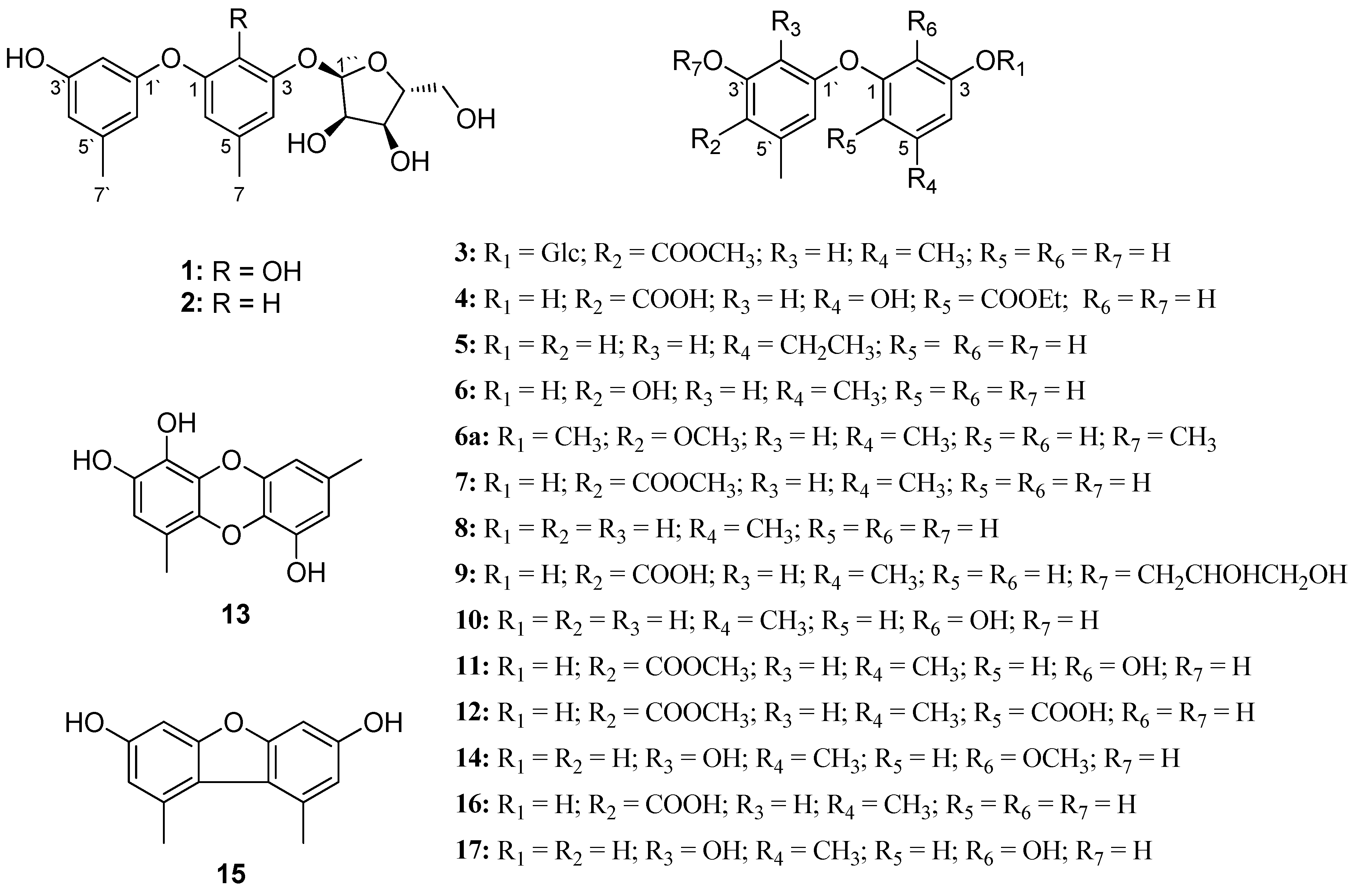
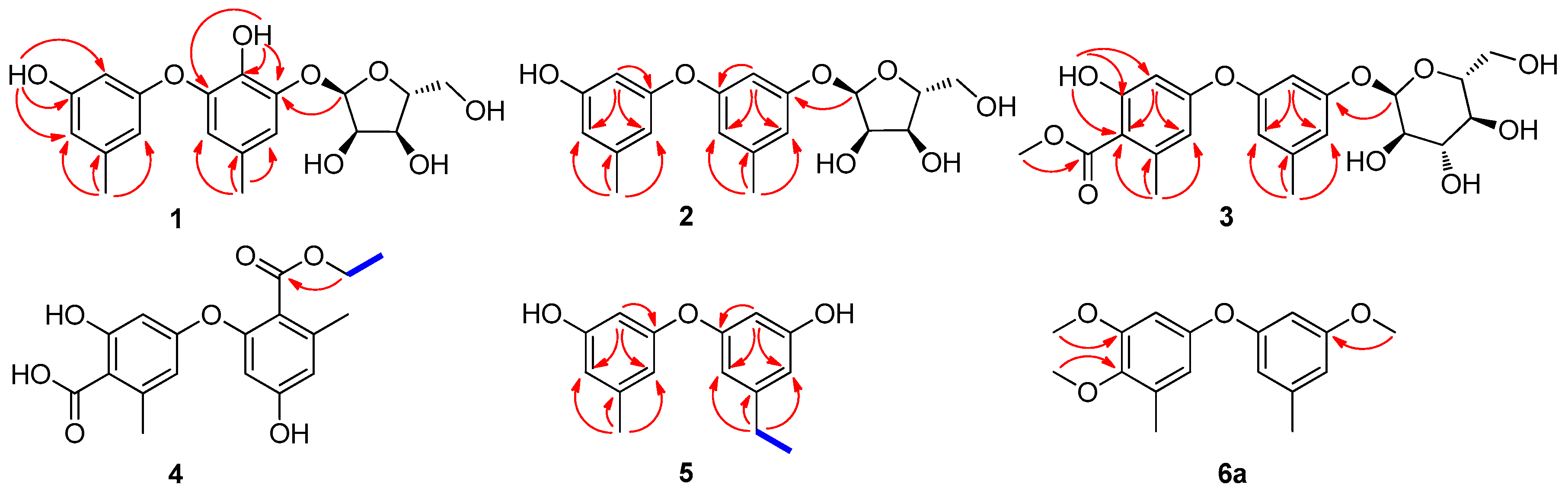
3.4.1. Cordyol C-3-O-α-d-ribofuranoside (1)
3.4.2. Diorcinol-3-O-α-d-ribofuranoside (2)
3.4.3. 4-Methoxycarbonyl Diorcinol-3-O-α-d-glucoside (3)
3.4.4. 2-(Ethoxycarbonyl)-4′-carboxydiorcinal (4)
3.4.5. 7-Ethyldiorcinol (5)
3.4.6. 3-Hydroxydiorcinol (6)
3.5. Determination of the Absolute Configuration of Sugar Moieties in 1–3
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, Y.; Potts, M.B.; Colosimo, D.; Herrera-Herrera, M.L.; Legako, A.G.; Yousufuddin, M.; White, M.A.; MacMillan, J.B. Discoipyrroles A–D: Isolation, structure determination, and synthesis of potent migration inhibitors from Bacillus hunanensis. J. Am. Chem. Soc. 2013, 135, 13387–13392. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, K.; MacMillan, J.B. Hunanamycin A, an antibiotic from a marine-derived Bacillus hunanensis. Org. Lett. 2013, 15, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xiong, Y.; Qi, X.; Tang, W.; Dai, J.; Gu, Q.; Li, J. Molecular Targets of Active Anticancer Compounds Derived from Marine Sources. Mar. Drugs 2018, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Moghadamtousi, S.; Nikzad, S.; Kadir, H.; Abubakar, S.; Zandi, K. Potential Antiviral Agents from Marine Fungi: An Overview. Mar. Drugs 2015, 13, 4520–4538. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.F.; Somoza, A.D.; Keller, N.P.; Wang, C.C.C. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat. Prod. Rep. 2012, 29, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Gao, H.; Zhang, X.; Wang, S.; Wu, C.; Gu, Q.; Guo, P.; Zhu, T.; Li, D. Psychrophilins E–H and Versicotide C, Cyclic Peptides from the Marine-Derived Fungus Aspergillus versicolor ZLN-60. J. Nat. Prod. 2014, 77, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Bladt, T.; Frisvad, J.; Knudsen, P.; Larsen, T. Anticancer and Antifungal Compounds from Aspergillus, Penicillium and Other Filamentous Fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.L.; Wang, X.J.; Xiang, Z.D.; Wang, J.D.; Zhang, H.; Liu, C.X.; Zhang, J.; Xiang, W.S. Diphenyl etheric metabolites from Streptomyces sp. neau50. J. Antibiot. 2011, 64, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xia, Z.; Tang, J.; Wu, J.; Tong, J.; Li, M.; Ju, J.; Chen, H.; Wang, L. Identification and Biological Evaluation of Secondary Metabolites from Marine Derived Fungi-Aspergillus sp. SCSIOW3, Cultivated in the Presence of Epigenetic Modifying Agents. Molecules 2017, 22, 1302. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Smetanina, O.F.; Kalinovsky, A.I.; Kirichuk, N.N.; Pivkin, M.V.; Ivanets, E.V.; Yurchenko, E.A.; Afiyatullov, S.S. New Metabolites from a Marine Sediment-Derived Fungus, Aspergillus carneus. Nat. Prod. Commun. 2015, 10, 1247–1250. [Google Scholar] [PubMed]
- Wu, Z.; Wang, Y.; Liu, D.; Proksch, P.; Yu, S.; Lin, W. Antioxidative phenolic compounds from a marine-derived fungus Aspergillus versicolor. Tetrahedron 2016, 72, 50–57. [Google Scholar] [CrossRef]
- Bunyapaiboonsri, T.; Yoiprommarat, S.; Intereya, K.; Kocharin, K. New diphenyl ethers from the insect pathogenic fungus Cordyceps sp. BCC 1861. Chem. Pharm. Bull. 2007, 55, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Qi, J.; Shao, C.L.; Zhao, D.L.; Hou, X.M.; Wang, C.Y. Bioactive Diphenyl Ethers and Isocoumarin Derivatives from a Gorgonian-Derived Fungus Phoma sp. (TA07-1). Mar. Drugs 2017, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, V.M.; Choudhary, M.I.; Andersen, R.J.; Karunaratne, V. Novel entry into 5-decarboxydibenzofurans via Smiles rearrangement of the lichen para-depside, erythrin. J. Chem. Res. 2010, 34, 154–157. [Google Scholar] [CrossRef]
- Yang, G.; Yun, K.; Nenkep, V.N.; Choi, H.D.; Kang, J.S.; Son, B.W. Induced production of halogenated diphenyl ethers from the marine-derived fungus Penicillium chrysogenum. Chem. Biodivers. 2010, 7, 2766–2770. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.L.; Shao, C.L.; Zhang, Q.; Wang, K.L.; Guan, F.F.; Shi, T.; Wang, C.Y. Azaphilone and Diphenyl Ether Derivatives from a Gorgonian-Derived Strain of the Fungus Penicillium pinophilum. J. Nat. Prod. 2015, 78, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Hu, J.Y.; Sun, H.D.; Zhao, X.; Zhong, W.T.; Duan, D.Z.; Wang, L.; Wang, X.L. Sinopestalotiollides A–D, cytotoxic diphenyl ether derivatives from plant endophytic fungus Pestalotiopsis palmarum. Bioorg. Med. Chem. Lett. 2018, 28, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.; Liu, P.; Wang, Y.; Li, J.; Hong, K.; Zhu, W. Cytotoxic polyphenols from the fungus Penicillium expansum 091 006 endogenous with the mangrove plant Excoecaria agallocha. Planta Med. 2012, 78, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; You, F.; Li, X.L.; Jia, M.; Zheng, C.J.; Han, T.; Qin, L.P. A new diphenyl ether from the endophytic fungus Verticillium sp. isolated from Rehmannia glutinosa. Chin. J. Nat. Med. 2013, 11, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Qin, X.; Lin, X.; Kaliyaperumal, K.; Zhou, X.; Liu, J.; Ju, Z.; Tu, Z.; Liu, Y. Sydoxanthone C and acremolin B produced by deep-sea-derived fungus Aspergillus sp. SCSIO Ind09F01. J. Antibiot. 2015, 68, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhu, H.; Fu, P.; Wang, Y.; Zhang, Z.; Lin, H.; Liu, P.; Zhuang, Y.; Hong, K.; Zhu, W. Cytotoxic polyphenols from the marine-derived fungus Penicillium expansum. J. Nat. Prod. 2010, 73, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, G.Q.; Tong, X.P.; Chen, G.D.; Huang, Y.F.; Cui, J.Y.; Kong, M.Z.; Guo, L.D.; Zheng, Y.Z.; Yao, X.S.; et al. Diphenyl ethers from Aspergillus sp. and their anti-Aβ42 aggregation activities. Fitoterapia 2014, 98, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Jang, J.H.; Oh, H.; Sohn, J.H.; Kim, J.W.; Moon, D.O.; Kwon, O.; Kawatani, M.; Osada, H.; Kim, B.Y.; et al. Violaceols Function as Actin Inhibitors Inducing Cell Shape Elongation in Fibroblast Cells. Biosci. Biotechnol. Biochem. 2012, 76, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, P.; Weaver, K.; Clark, M.T.; Bowyer, J.R.; Hallahan, B.J. Some Novel Diphenyl Ether Herbicides with Peroxidizing Activity. J. Agric. Food Chem. 1988, 36, 1061–1063. [Google Scholar] [CrossRef]
- Serianni, A.S.; Barker, R. [C-13]-Enriched tetroses and tetrofuranosides—An evaluation of the relationship between NMR parameters and furanosyl ring conformation. J. Org. Chem. 1984, 49, 3292–3300. [Google Scholar] [CrossRef]
- Gubica, T.; Szeleszczuk, L.; Pisklak, D.M.; Stepien, D.K.; Cyranski, M.K.; Kanska, M. Reliable evaluation of molecular structure of methyl 3-O-nitro-alpha-d-glucopyranoside and its intermediates by means of solid-state NMR spectroscopy and DFT optimization in the absence of appropriate crystallographic data. Tetrahedron 2014, 70, 1910–1917. [Google Scholar] [CrossRef]
- Tvaroska, I.; Taravel, F.R. Carbon-proton coupling constants in the conformational analysis of sugar molecules Adv. Carbohydr. Chem. Biochem. 1995, 51, 15–61. [Google Scholar] [CrossRef]
- Li, X.B.; Zhou, Y.H.; Zhu, R.X.; Chang, W.Q.; Yuan, H.Q.; Gao, W.; Zhang, L.L.; Zhao, Z.T.; Lou, H.X. Identification and Biological Evaluation of Secondary Metabolites from the Endolichenic Fungus Aspergillus versicolor. Chem. Biodivers. 2015, 12, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, T.; Takenaka, Y.; Nagakura, N.; Hamada, N. Dibenzofurans from the cultured lichen mycobionts of Lecanora cinereocarnea. Phytochemistry 2001, 58, 1129–1134. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Su, M.; Hwang, G.J.; Hong, J.; Jung, J.H. New metabolites from the sponge-derived fungus Aspergillus sydowii J05B-7F-4. Nat. Prod. Res. 2017, 31, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, Y.; Tanahashi, T.; Nagakura, N.; Hamada, N. Phenyl ethers from cultured lichen mycobionts of Graphis scripta var. serpentina and G. rikuzensis. Chem. Pharm. Bull. 2003, 51, 794–797. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, J.; Yao, X.J.; Yang, J.; Liu, L.; Xie, T.G.; Jiang, P.C.; Jiang, Z.H.; Zhu, G.Y. Phenolic Constituents Isolated from the Twigs of Cinnamomum cassia and Their Potential Neuroprotective Effects. J. Nat. Prod. 2018, 81, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Han, J.W.; Ngo, M.T.; Dang, Q.L.; Kim, J.-C.; Kim, H.; Choi, G.J. Identification of novel compounds, oleanane- and ursane-type triterpene glycosides, from Trevesia palmata: Their biocontrol activity against phytopathogenic fungi. Sci. Rep. 2018, 8, 14522. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Hao, L.J.; Zhou, Z.B.; Zhu, X.L.; Shi, Z.H.; Miyamoto, T.; Pan, K. Lycodine-type alkaloids and their glycosides from Lycopodiastrum casuarinoides. Phytochemistry 2018, 154, 63–72. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NO. | δH (J in Hz) | ||||||
|---|---|---|---|---|---|---|---|
| 1 a | 2 b | 3 a | 4 b | 5 b | 6 b | 6a b | |
| 2 | 6.20 t (2.2) | 6.57 t (2.4) | 6.24 d (2.3) | 6.21 brs | 6.13 t (2.4) | 6.30 d (2.2) | |
| 4 | 6.81 brs | 6.27 brs | 6.75 brs | 6.35 d (2.3) | 6.30 m | 6.28 brs | 6.48 brs |
| 6 | 6.46 brs | 6.37 brs | 6.53 brs | 6.39 m | 6.20 brs | 6.43 brs | |
| 7 | 2.18 s | 2.27 s | 2.26 s | 2.47 s | 2.53 q (7.5) | 2.19 s | 2.26 s |
| 8 | 1.18 t (7.5) | 3.73 s | |||||
| 9 | 4.40 q (7.5) | ||||||
| 10 | 1.40 t (7.5) | ||||||
| 2′ | 6.02 brs | 6.57 t (2.8) | 6.28 d (2.3) | 6.28 d (2.7) | 6.21 brs | 6.33 d (2.8) | 6.52 d (2.8) |
| 4′ | 6.21 brs | 6.45 brs | 6.28 brs | ||||
| 6′ | 6.12 brs | 6.72 brs | 6.37 d (2.3) | 6.56 d (2.7) | 6.36 brs | 6.26 d (2.8) | 6.37 d (2.8) |
| 7′ | 2.15 s | 2.22 s | 2.21 s | 2.35 s | 2.22 s | 2.17 s | 2.20 s |
| 8′ | 3.78 s | ||||||
| 9′ | 3.78 s | 3.75 s | |||||
| 1″ | 5.50 d (4.6) | 5.57 d (4.5) | 5.35 d (3.6) | ||||
| 2″ | 4.07 ddd (10.7, 6.8, 4.1) | 4.15 dd (6.5, 4.5) | 3.33 m | ||||
| 3″ | 3.93 ddd (11.7, 5.9, 2.7) | 4.06 dd (6.5, 3.2) | 3.58 t (9.2) | ||||
| 4″ | 3.98 q (4.0) | 4.12 dd (6.9, 3.5) | 3.17 t (9.2) | ||||
| 5″ | 3.46 brt (5.1) | 3.63 dd (12.1, 3.9) 3.69 dd (11.7, 3.4) | 3.42 m | ||||
| 6″ | 3.47 dd (11.7, 5.2) | ||||||
| 3.55 dd (11.8, 1.8) | |||||||
| 2′-OH | 8.41 s | ||||||
| 3″-OH | 9.26 s | 10.26 s | |||||
| 2′-OH | 5.14 d (6.0) | ||||||
| 3″-OH | 5.16 brs | ||||||
| 5″-OH | 4.81 t (5.6) | ||||||
| NO. | δC, Type | ||||||
|---|---|---|---|---|---|---|---|
| 1 a | 2 b | 3 a | 4 b | 5 b | 6 b | 6a b | |
| 1 | 142.0 (C) | 159.6 (C) | 156.1 (C) | 163.2 (C) | 159.5 (C) | 161.2 (C) | 160.2 (C) |
| 2 | 137.3 (C) | 106.3 (CH) | 105.5 (CH) | 103.9 (C) | 104.5 (CH) | 102.8 (CH) | 102.6 (CH) |
| 3 | 145.7 (C) | 159.5 (C) | 158.3 (C) | 164.9 (C) | 159.7 (C) | 159.4 (C) | 162.3 (C) |
| 4 | 112.9 (CH) | 111.8 (CH) | 113.5 (CH) | 113.3 (CH) | 111.7 (CH) | 111.0 (CH) | 110.3 (CH) |
| 5 | 127.3 (C) | 141.6 (C) | 140.5 (C) | 144.1 (C) | 148.2 (C) | 141.4 (C) | 141.7 (C) |
| 6 | 115.9 (CH) | 112.1 (CH) | 113.6 (CH) | 109.7 (CH) | 110.8 (CH) | 110.4 (CH) | 112.2 (CH) |
| 7 | 20.6 (CH3) | 21.6 (CH3) | 21.1 (CH3) | 23.9 (CH3) | 29.8 (CH2) | 21.6 (CH3) | 21.7 (CH3) |
| 8 | 172.4 (C) | 15.9 (CH3) | 55.7 (CH3) | ||||
| 9 | 62.5 (CH2) | ||||||
| 10 | 14.5 (CH3) | ||||||
| 1′ | 159.1 (C) | 159.7 (C) | 158.9 (C) | 160.8 (C) | 159.6 (C) | 150.1 (C) | 154.3 (C) |
| 2′ | 100.3 (CH) | 104.3 (CH) | 102.6 (CH) | 106.4 (CH) | 104.2 (CH) | 106.1 (CH) | 103.4 (CH) |
| 3′ | 158.2 (C) | 159.4 (C) | 157.6 (C) | 155.0 (C) | 159.7 (C) | 146.8 (C) | 154.8 (C) |
| 4′ | 109.6 (CH) | 114.2 (CH) | 114.9 (C) | 120.1(C) | 111.9 (CH) | 140.8 (C) | 144.5 (C) |
| 5′ | 139.5 (C) | 141.7 (C) | 139.1 (C) | 140.6 (C) | 141.6 (C) | 126.7 (C) | 133.5 (C) |
| 6′ | 107.5 (CH) | 113.5 (CH) | 110.7 (CH) | 114.9 (CH) | 110.6 (CH) | 113.6 (CH) | 113.7 (CH) |
| 7′ | 21.2 (CH3) | 21.5 (CH3) | 20.1 (CH3) | 20.2 (CH3) | 21.5 (CH3) | 16.2 (CH3) | 16.0 (CH3) |
| 8′ | 168.5 (C) | 170.7 (C) | 56.3 (CH3) | ||||
| 9′ | 51.9 (CH3) | 60.6 (CH3) | |||||
| 1″ | 101.3 (CH) | 102.3 (CH) | 97.9 (CH) | ||||
| 2″ | 72.3 (CH) | 73.4 (CH) | 71.5 (CH) | ||||
| 3″ | 69.4 (CH) | 71.2 (CH) | 73.0 (CH) | ||||
| 4″ | 86.7 (CH) | 87.5 (CH) | 69.8 (CH) | ||||
| 5″ | 61.6 (CH2) | 63.2 (CH2) | 73.8 (CH) | ||||
| 6″ | 60.6 (CH2) | ||||||
| Compounds | A549 | U937 | HL-60 | K562 |
|---|---|---|---|---|
| 1 | 8.97 ± 0.48 | 4.64 ± 0.35 | / | / |
| 5 | 16.13 ± 1.24 | / | / | / |
| 6 | / | / | 11.98 ± 0.73 | 18.89 ± 1.14 |
| 8 | 15.51 ± 1.59 | / | / | / |
| 9 | 3.36 ± 0.68 | / | 21.22 ± 1.25 | / |
| 10 | / | / | 16.52 ± 0.99 | 20.88 ± 1.60 |
| 11 | / | / | 13.33 ± 0.87 | 23.03 ± 1.34 |
| DOX | 0.19 ± 0.04 | <0.125 | <0.125 | 0.49 ± 0.08 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-N.; Mou, Y.-H.; Dong, Y.; Wu, Y.; Liu, B.-Y.; Bai, J.; Yan, D.-J.; Zhang, L.; Feng, D.-Q.; Pei, Y.-H.; et al. Diphenyl Ethers from a Marine-Derived Aspergillus sydowii. Mar. Drugs 2018, 16, 451. https://doi.org/10.3390/md16110451
Wang Y-N, Mou Y-H, Dong Y, Wu Y, Liu B-Y, Bai J, Yan D-J, Zhang L, Feng D-Q, Pei Y-H, et al. Diphenyl Ethers from a Marine-Derived Aspergillus sydowii. Marine Drugs. 2018; 16(11):451. https://doi.org/10.3390/md16110451
Chicago/Turabian StyleWang, Ya-Nan, Yan-Hua Mou, Yu Dong, Yan Wu, Bing-Yu Liu, Jian Bai, Dao-Jiang Yan, Le Zhang, Dan-Qing Feng, Yue-Hu Pei, and et al. 2018. "Diphenyl Ethers from a Marine-Derived Aspergillus sydowii" Marine Drugs 16, no. 11: 451. https://doi.org/10.3390/md16110451
APA StyleWang, Y.-N., Mou, Y.-H., Dong, Y., Wu, Y., Liu, B.-Y., Bai, J., Yan, D.-J., Zhang, L., Feng, D.-Q., Pei, Y.-H., & Hu, Y.-C. (2018). Diphenyl Ethers from a Marine-Derived Aspergillus sydowii. Marine Drugs, 16(11), 451. https://doi.org/10.3390/md16110451