Two Furanosesterterpenoids from the Sponge Luffariella variabilis



Abstract
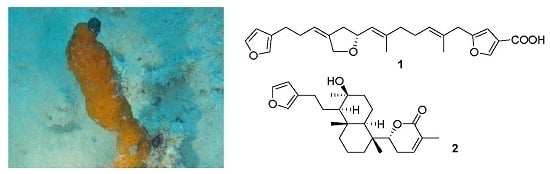
:
1. Introduction
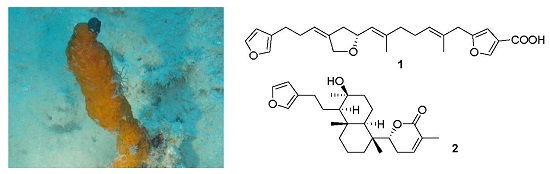
2. Results and Discussion
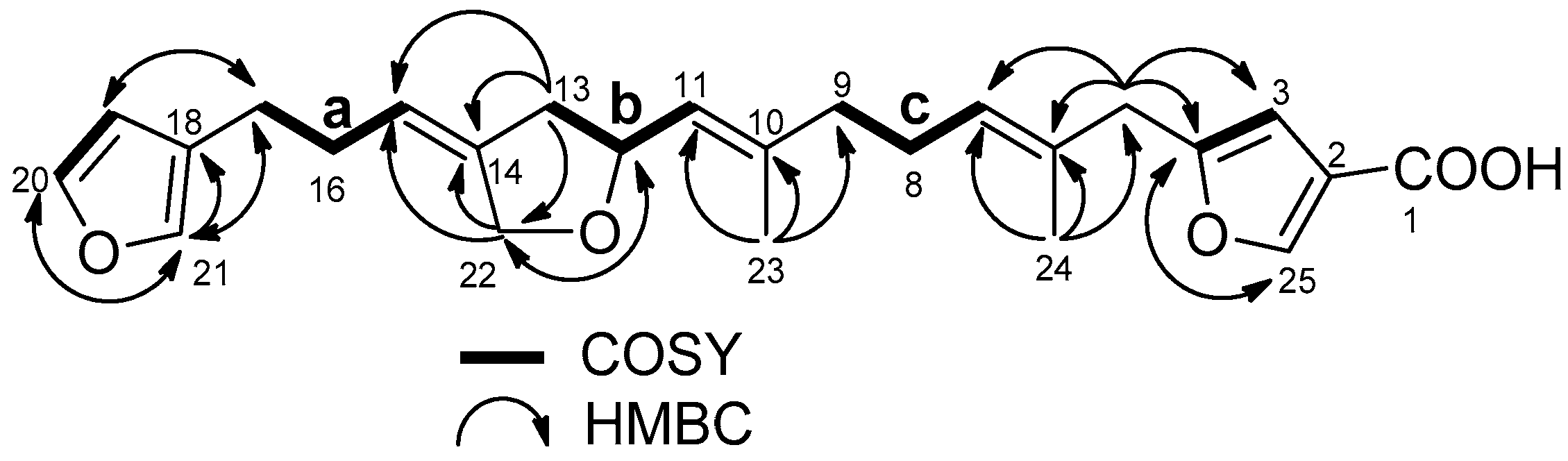
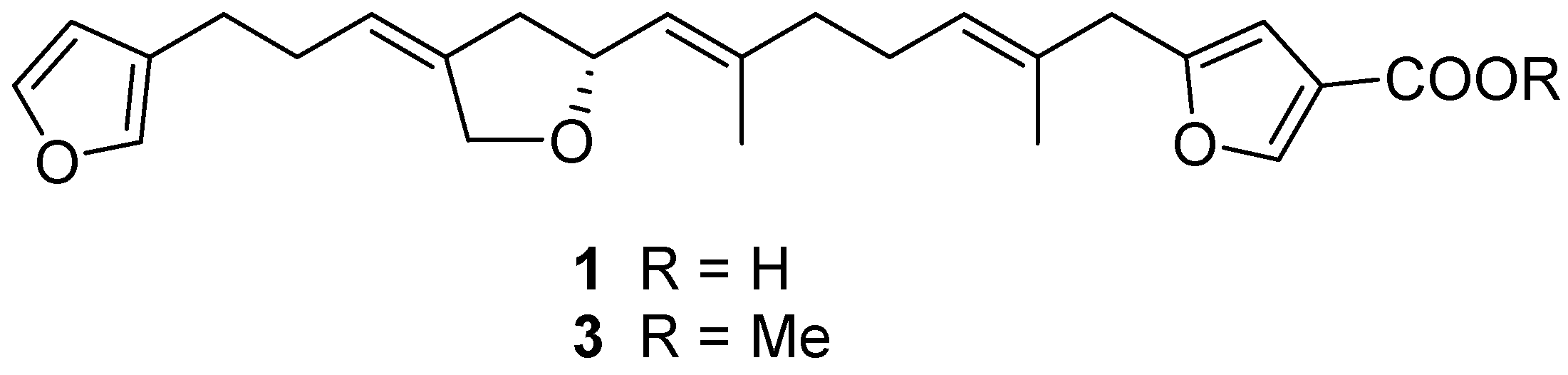
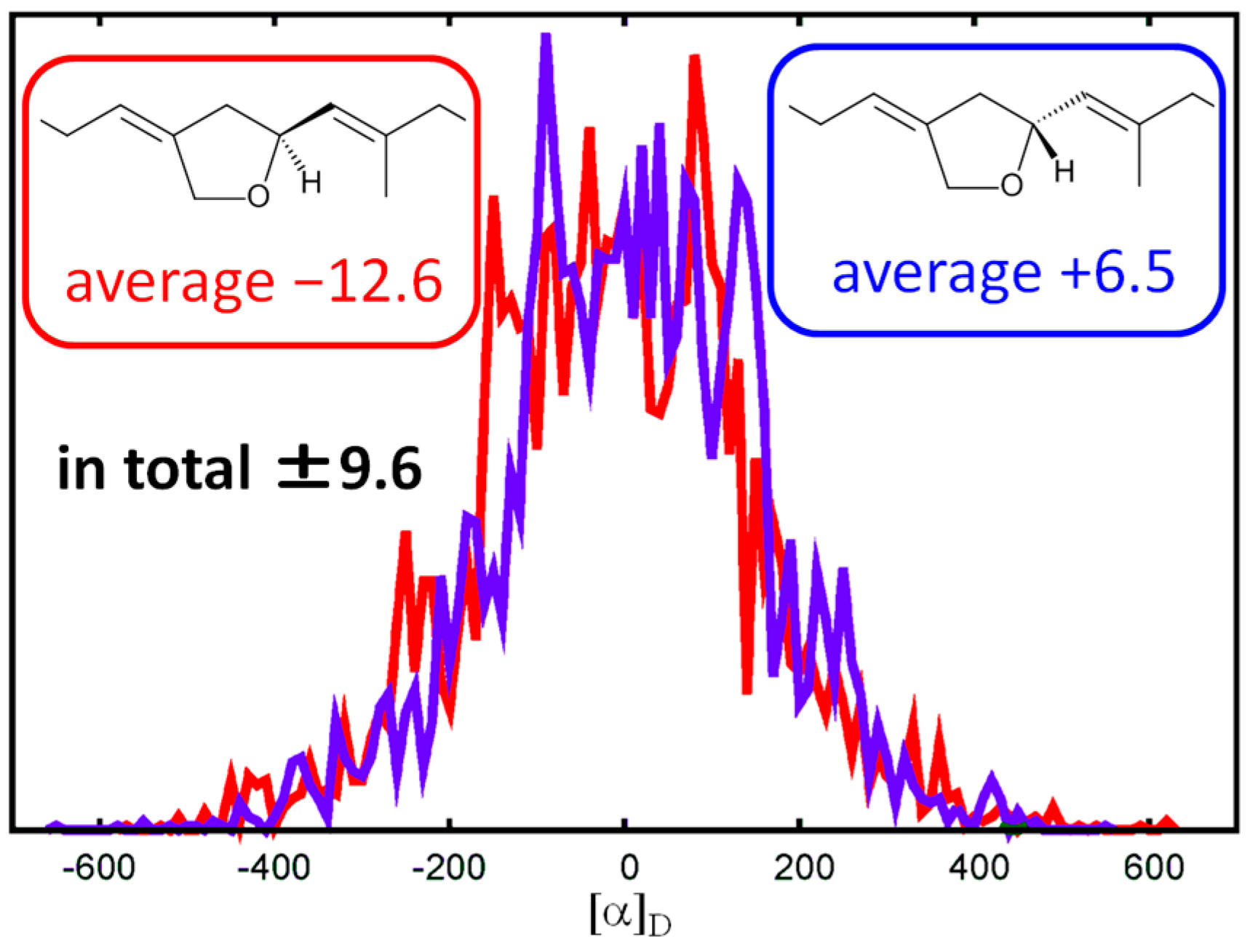
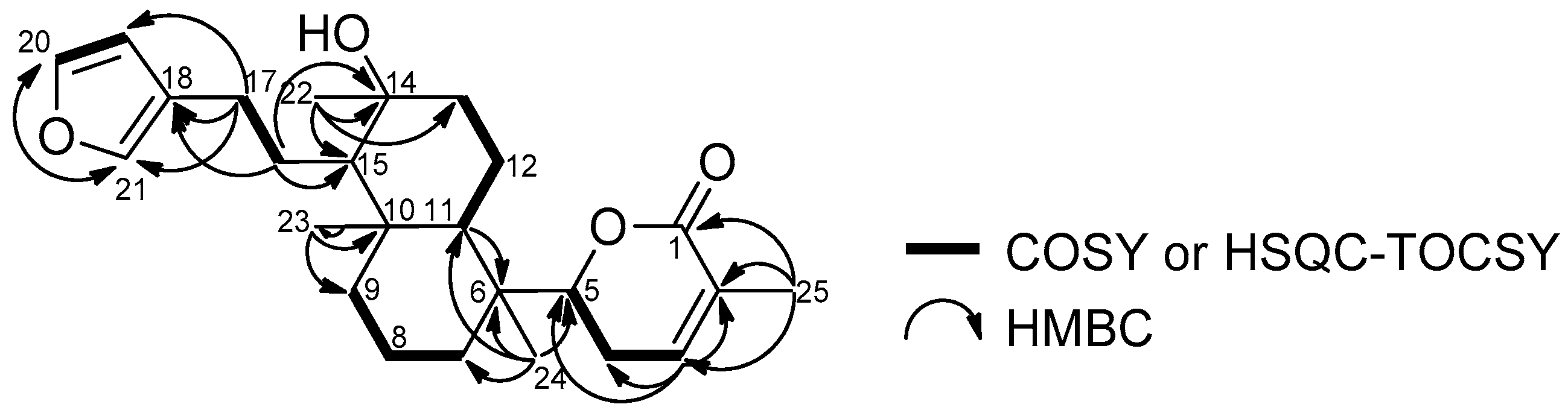
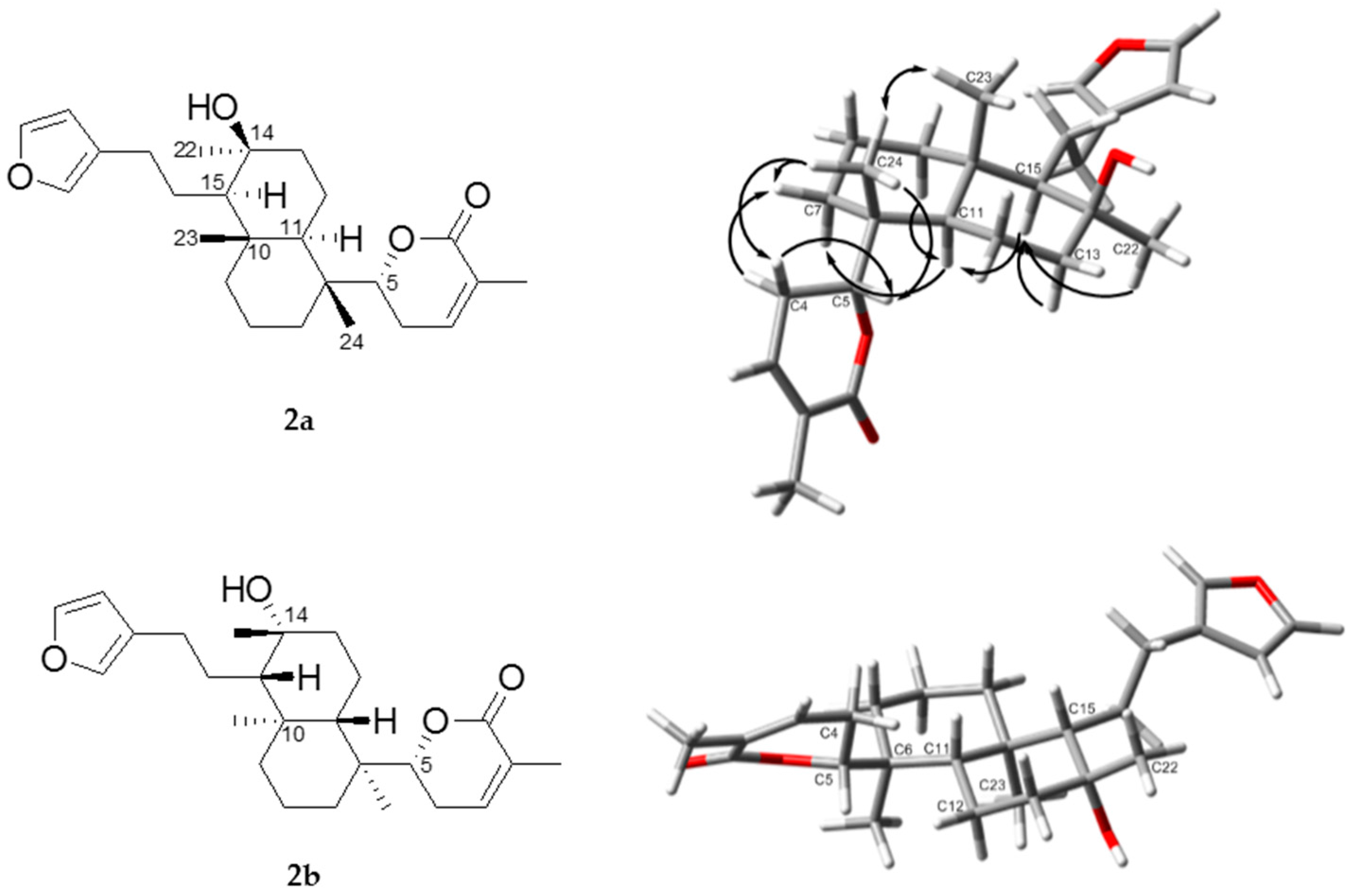
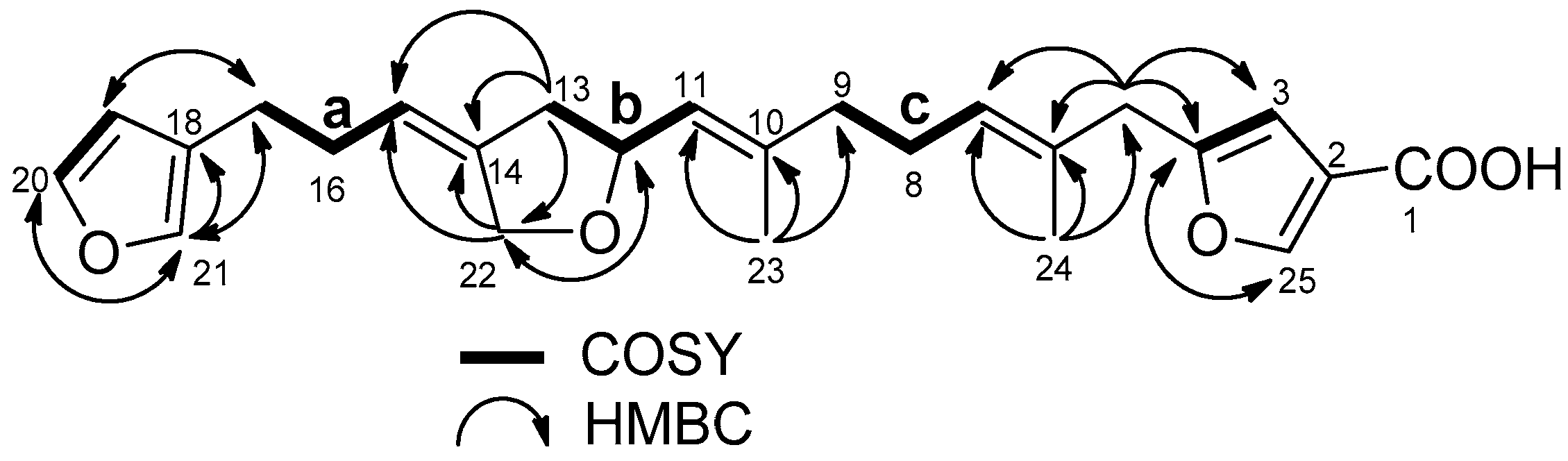
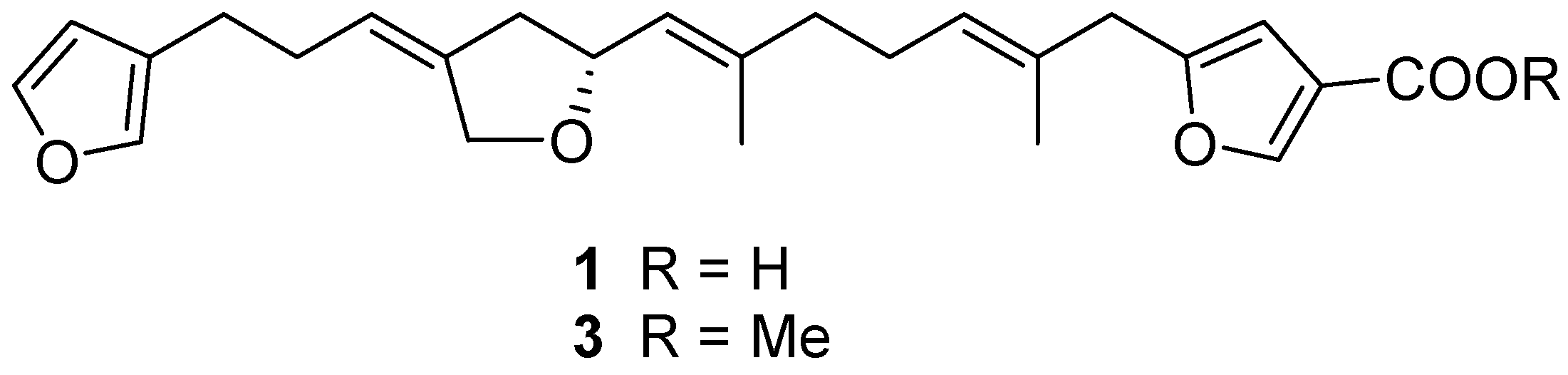
2.1. Structure Elucidation
2.2. Biological Activity
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Specimen
4.3. Extraction and Isolation
4.4. Methylation of Compound 1 to 3
4.5. Cytotoxicity Assay
4.6. Calculation of Specific Rotation Value
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- De Silva, E.D.; Scheuer, P.J. Manoalide, an antibiotic sesterterpenoid from the marine sponge Luffariella variabilis (polejaff). Tetrahedron Lett. 1980, 21, 1611–1614. [Google Scholar] [CrossRef]
- De Silva, E.D.; Scheuer, P.J. Three new sesterterpenoids antibiotic from the marine sponge Luffariella variabilis (polejaff). Tetrahedron Lett. 1981, 22, 3147–3150. [Google Scholar] [CrossRef]
- Kernan, M.R.; Faulkner, D.J.; Jacobs, R.S. The luffariellins, novel anti-inflammatory sesterterpenes of chemotaxonomic importance from the marine sponge Luffariella variabilis. J. Org. Chem. 1987, 52, 3081–3083. [Google Scholar] [CrossRef]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakanoshi, M.; Tsubuki, M.; Tani, H.; et al. Inhibition of hepatitis C virus NS3 helicase by manoalide. J. Nat. Prod. 2012, 75, 650–654. [Google Scholar] [CrossRef] [PubMed]
- König, G.M.; Wright, A.D.; Sticher, O. Four new antibacterial sesterterpenes from a marine sponge of the genus Luffariella. J. Nat. Prod. 1992, 55, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.M.; Capon, R.J.; Faulkner, D.J. Luffalactone and (4E,6E)-dehydromanoalide from the sponge Luffariella variabilis. J. Org. Chem. 1992, 57, 2965–2967. [Google Scholar] [CrossRef]
- Tsuda, M.; Shigemori, H.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Luffariolides A-E, new cytotoxic sesterterpenes from the Okinawan marine sponge Luffariella sp. J. Org. Chem. 1992, 57, 3503–3507. [Google Scholar] [CrossRef]
- Kobayashi, J.; Zeng, C.-M.; Ishibashi, M. Luffariolides F and G, new manoalide derivatives from the Okinawan marine sponge Luffariella sp. J. Nat. Prod. 1993, 56, 435–439. [Google Scholar] [CrossRef]
- Tsuda, M.; Endo, T.; Mikami, Y.; Fromont, J.; Kobayashi, J. Luffariolides H and J, new sesterterpenes from a marine sponge Luffariella species. J. Nat. Prod. 2002, 65, 1507–1508. [Google Scholar] [CrossRef] [PubMed]
- Ettinger-Epstein, P.; Motti, C.A.; de Nys, R.; Wright, A.D.; Battershill, C.N.; Tapiolas, D.M. Acetylated sesterterpenes from the Great Barrier reef sponge Luffariella variabilis. J. Nat. Prod. 2007, 70, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Gauvin-Bialecki, A.; Aknin, M.; Smadja, J. 24-O-ethylmanoalide, a manoalide-related sesterterpene from the marine sponge Luffariella cf. variabilis. Molecules 2008, 13, 3148–3191. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, P.; Haruyama, T.; Kobayashi, N.; de Voogd, N.J.; Tanaka, J. Spongian diterpenes from the sponge Hyatella aff. intestinalis. Chem. Pharm. Bull. 2017. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Scheuer, P.J. Isolation and structure determination of two furanosesquiterpenes from the soft coral Sinularia iochmodes. J. Korean Chem. Soc. 1994, 38, 749–752. [Google Scholar]
- Liu, J.-S.; Huang, M.-F.; Ayer, W.A.; Bigam, G. Schisanlactone B, a new triterpenoid from a Schisandra sp. Tetrahedron Lett. 1983, 24, 2355–2358. [Google Scholar] [CrossRef]
- Lakornwong, W.; Kanokmedhakul, K.; Kanokmedhakul, S.; Kongsaeree, P.; Prabpai, S.; Sibounavong, P.; Soytong, K. Triterpene lactones from cultures of Ganoderma sp. KM01. J. Nat. Prod. 2014, 77, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.F. The CD of αβ-unsaturated lactones. Tetrahedron 1972, 28, 5543–5554. [Google Scholar] [CrossRef]
- Faricha, A.; Ahmadi, P.; de Voogd, N.J.; Tanaka, J. Two isospongian diterpenes from the sponge Luffariella sp. Nat. Prod. Commun. 2017, 12, 1011–1012. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Calculation of optical rotation using density functional theory. J. Phys. Chem. A 2001, 105, 5356–5371. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| 13C | 1H (J in Hz) | 13C | 1H (J in Hz) | |||||
| 1 | 167.0 | s | - | - | 166.5 | s | - | - |
| 2 | 119.0 | s | - | - | 128.2 | s | - | - |
| 3 | 106.0 | d | - | 6.38 brs | 140.0 | d | - | 6.63 brd (6.7) |
| 4 | 156.4 | s | - | - | 23.8 | t | a | 2.34 m |
| - | - | - | - | - | - | - | b | 2.21 ddd (3.1, 6.7, 17.7) |
| 5 | 38.1 | t | - | 3.28 s (2H) | 81.9 | d | - | 4.30 dd (3.1, 12.9) |
| 6 | 130.9 | s | - | - | 38.9 | s | - | - |
| 7 | 127.2 | d | - | 5.24 m | 31.8 | t | α | 1.56 m |
| - | - | - | - | - | - | - | β | 1.30 m |
| 8 | 26.3 | t | - | 2.16 m (2H) | 17.4 | t | - | 1.51 m (2H) |
| 9 | 39.2 | t | - | 2.06 m (2H) | 37.8 | t | α | 0.97 dt (3.1, 12.6) |
| - | - | - | - | - | - | - | β | 1.67 m |
| 10 | 139.7 | s | - | - | 38.9 | s | - | - |
| 11 | 124.9 | d | - | 5.24 m | 46.6 | d | - | 1.73 dd (2.6, 11.3) |
| 12 | 75.9 | d | - | 4.56 dt (8.4, 5.9) | 17.5 | t | α | 1.55 m |
| - | - | - | - | - | - | - | β | 1.47 m |
| 13 | 39.4 | t | a | 2.58 dd (5.9, 15.0) | 41.6 | t | α | 1.65 m |
| - | - | - | b | 2.23 m | - | - | β | 1.70 m |
| 14 | 139.8 | s | - | - | 73.0 | s | - | - |
| 15 | 119.2 | d | - | 5.33 m | 58.8 | d | - | 1.05 brs |
| 16 | 29.9 | t | - | 2.16 m (2H) | 26.0 | t | a | 1.68 m |
| - | - | - | - | - | - | - | b | 1.54 m |
| 17 | 24.6 | t | - | 2.48 t (7.5) (2H) | 28.4 | t | a | 2.44 m |
| - | - | - | - | - | - | - | b | 2.39 m |
| 18 | 124.5 | s | - | - | 125.4 | s | - | - |
| 19 | 110.9 | d | - | 6.26 brs | 110.9 | d | - | 6.29 brs |
| 20 | 142.7 | d | - | 7.34 brs | 142.6 | d | - | 7.35 brt (1.5) |
| 21 | 138.9 | d | - | 7.21 brs | 138.6 | d | - | 7.25 brs |
| 22 | 68.3 | t | a | 4.39 d (13.0) | 30.5 | q | - | 1.18 s |
| - | - | - | b | 4.20 d (13.0) | - | - | - | - |
| 23 | 16.7 | q | - | 1.70 brs | 15.9 | q | - | 1.03 s |
| 24 | 15.9 | q | - | 1.60 brs | 17.4 | q | - | 0.87 s |
| 25 | 147.7 | d | - | 7.96 brs | 16.9 | q | - | 1.92 s |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmadi, P.; Higashi, M.; Voogd, N.J.d.; Tanaka, J. Two Furanosesterterpenoids from the Sponge Luffariella variabilis. Mar. Drugs 2017, 15, 249. https://doi.org/10.3390/md15080249
Ahmadi P, Higashi M, Voogd NJd, Tanaka J. Two Furanosesterterpenoids from the Sponge Luffariella variabilis. Marine Drugs. 2017; 15(8):249. https://doi.org/10.3390/md15080249
Chicago/Turabian StyleAhmadi, Peni, Masahiro Higashi, Nicole J. de Voogd, and Junichi Tanaka. 2017. "Two Furanosesterterpenoids from the Sponge Luffariella variabilis" Marine Drugs 15, no. 8: 249. https://doi.org/10.3390/md15080249
APA StyleAhmadi, P., Higashi, M., Voogd, N. J. d., & Tanaka, J. (2017). Two Furanosesterterpenoids from the Sponge Luffariella variabilis. Marine Drugs, 15(8), 249. https://doi.org/10.3390/md15080249