1. Introduction
Lung cancer is one of the most common causes of cancer-related death worldwide. Siegel et al. estimated the mortality rate of lung cancer-related cases of 2017 in USA could be more than one-quarter (26%) of all cancer deaths [
1]. Among these lung cancer cases, the majority of them are non-small cell lung cancer (NSCLC), which has a poor five-year survival rate [
2]. Lung cancer is usually detected after development of late stages, and drug resistance often appears later during the course of treatment, leading to unsatisfying outcomes and poor prognosis of patients [
3]. Therefore, there is a necessity for the development of novel therapeutic strategies against lung cancer, especially for NSCLC.
Marine natural products have emerged as the new era in discovering therapeutic drugs, including those can against cancer cell [
4,
5,
6]. Lobocrassin B is a natural cembrane-type compound diterpenoid firstly isolated from the soft coral
Lobophytum crassum in 2011 and has been shown to exhibit a wide variety of biological effects, such as anti-cancer and immunomodulatory activities [
7,
8]. In specific, Lobocrassin B could not only suppress the activation of immune cells via dampening toll-like receptor (TLR) pathway, but significantly inhibit the product of superoxide anion among other cembrane-type derivatives. Also, it was found to possess moderate cytotoxicity to certain cancer cells, including human leukemia and human hepatoma [
7]. However, the molecular mechanisms of this anti-cancer effect by Lobocrassin B remain unknown. In addition, to our knowledge, there has been no studies have been conducted on the anticancer effects of Lobocrassin B against human lung cancer cells.
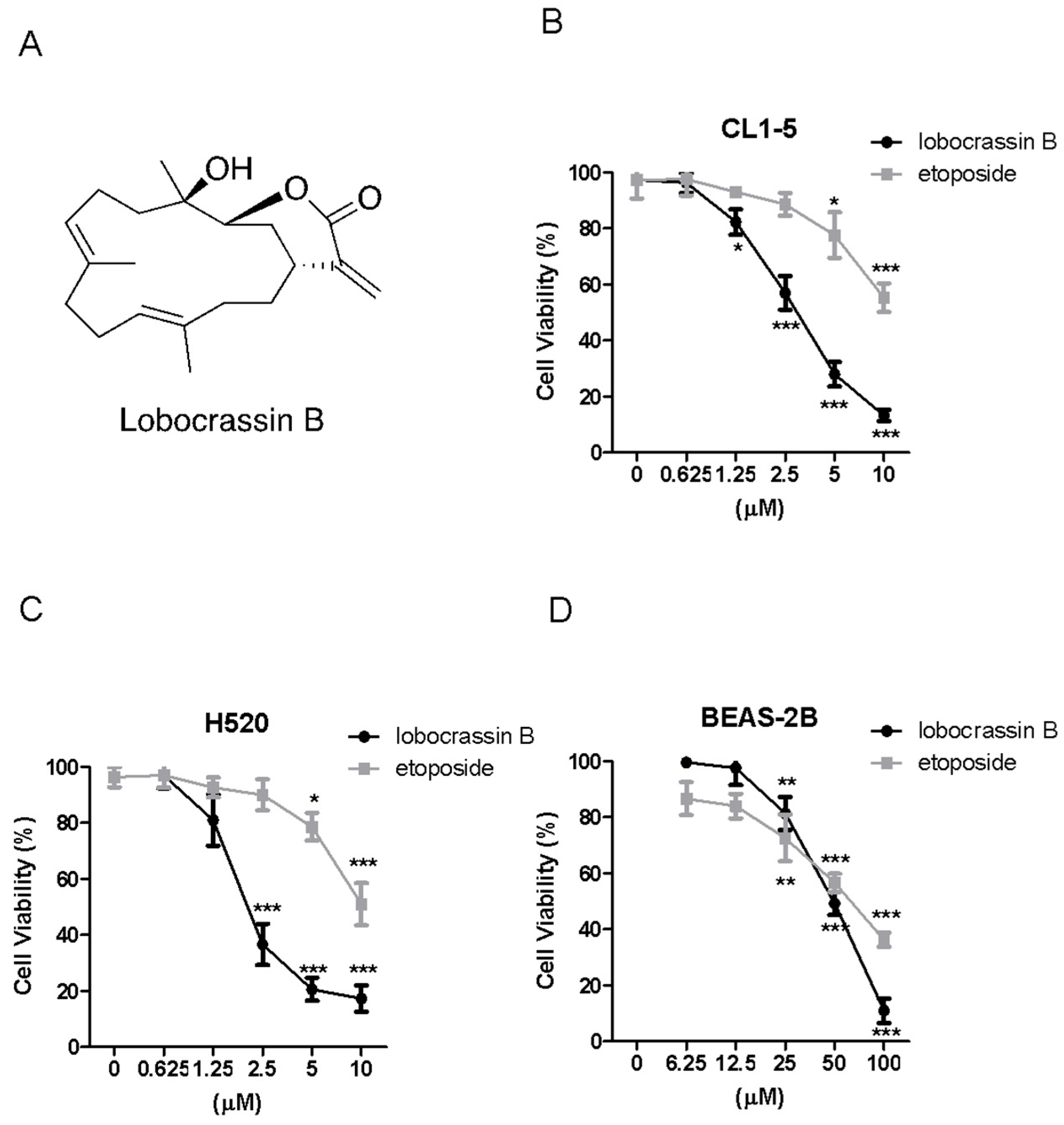
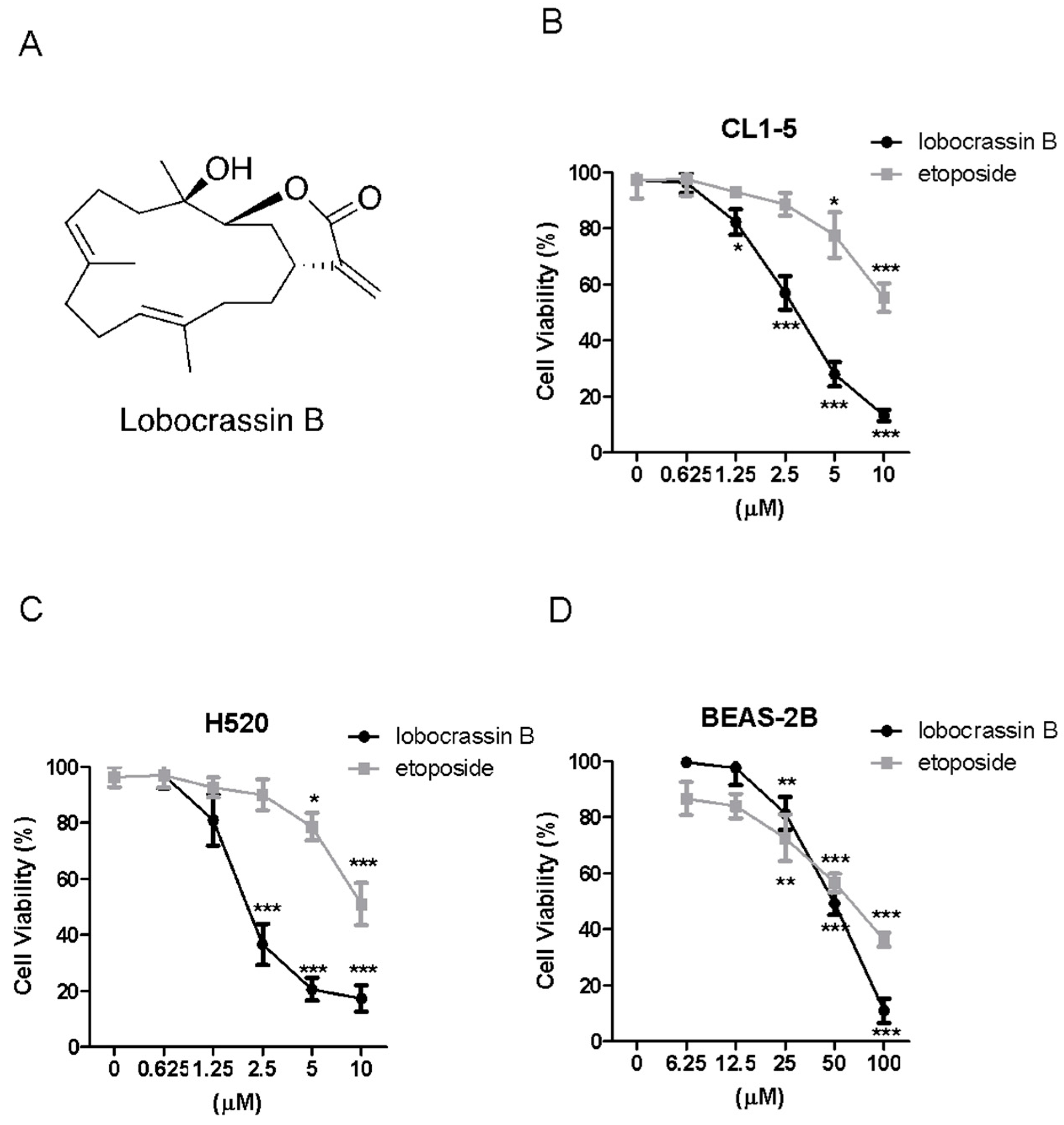
Thus, in this study, we determined the cytotoxic effects of Lobocrassin B in human CL1-5 and H520 NSCLC cancer cell lines and on xenograft CL1-5 tumor growth in nude mice. We also explore its potential mechanisms underlying the induced cancer cell death.
3. Discussion
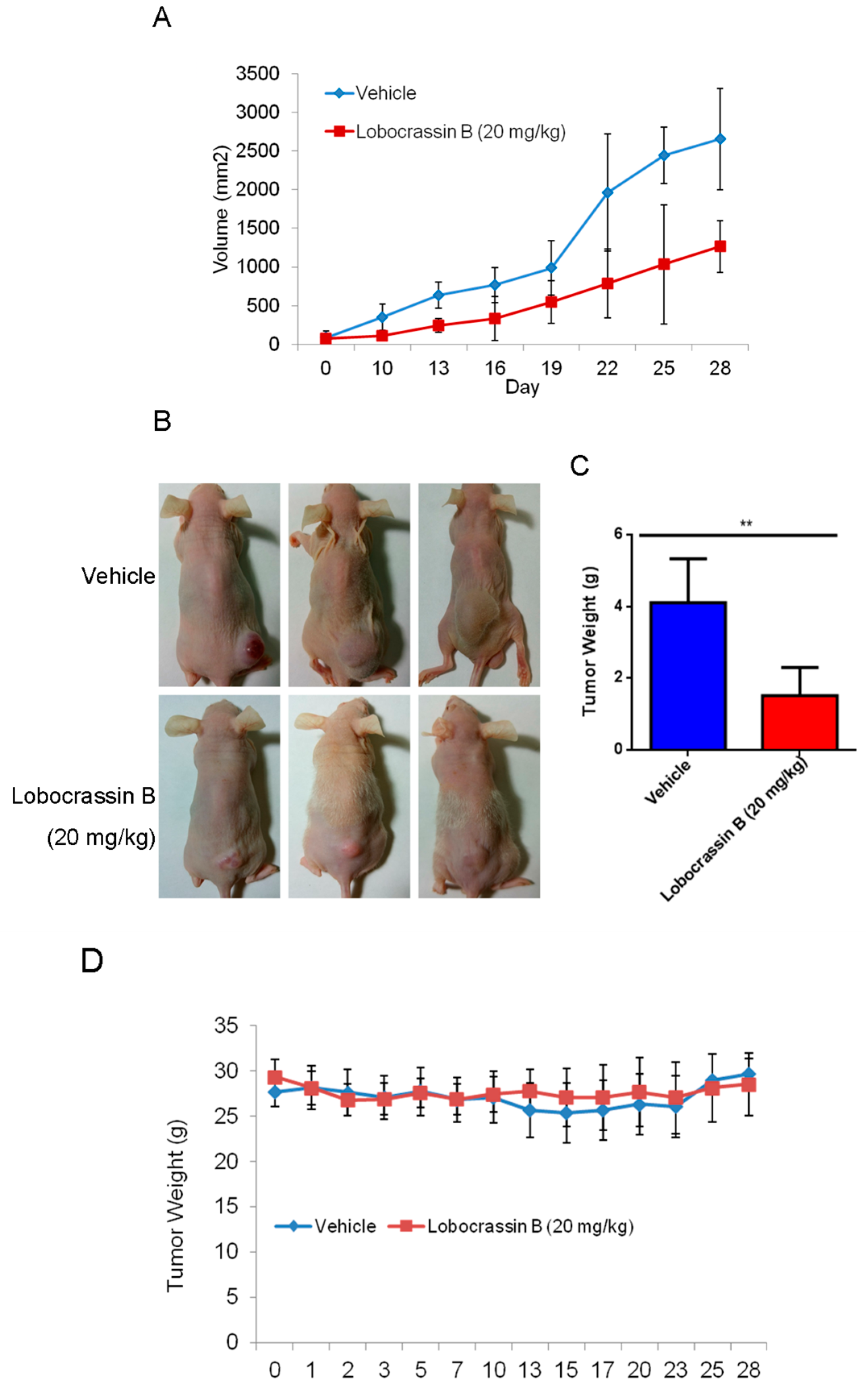
In the present study, the cytotoxic effects of Lobocrassin B on human lung cancer were confirmed in CL1-5 and H520 cells and tumor xenograft on athymic nude mice. The Lobocrassin B–induced cell death on the human lung cancer cell lines, as evidenced by a decrease in cell viability, and an increase levels of ROS, as well as the activation of caspase-8 and induction of mitochondrial apoptotic pathway, revealed the possible mechanisms involved.
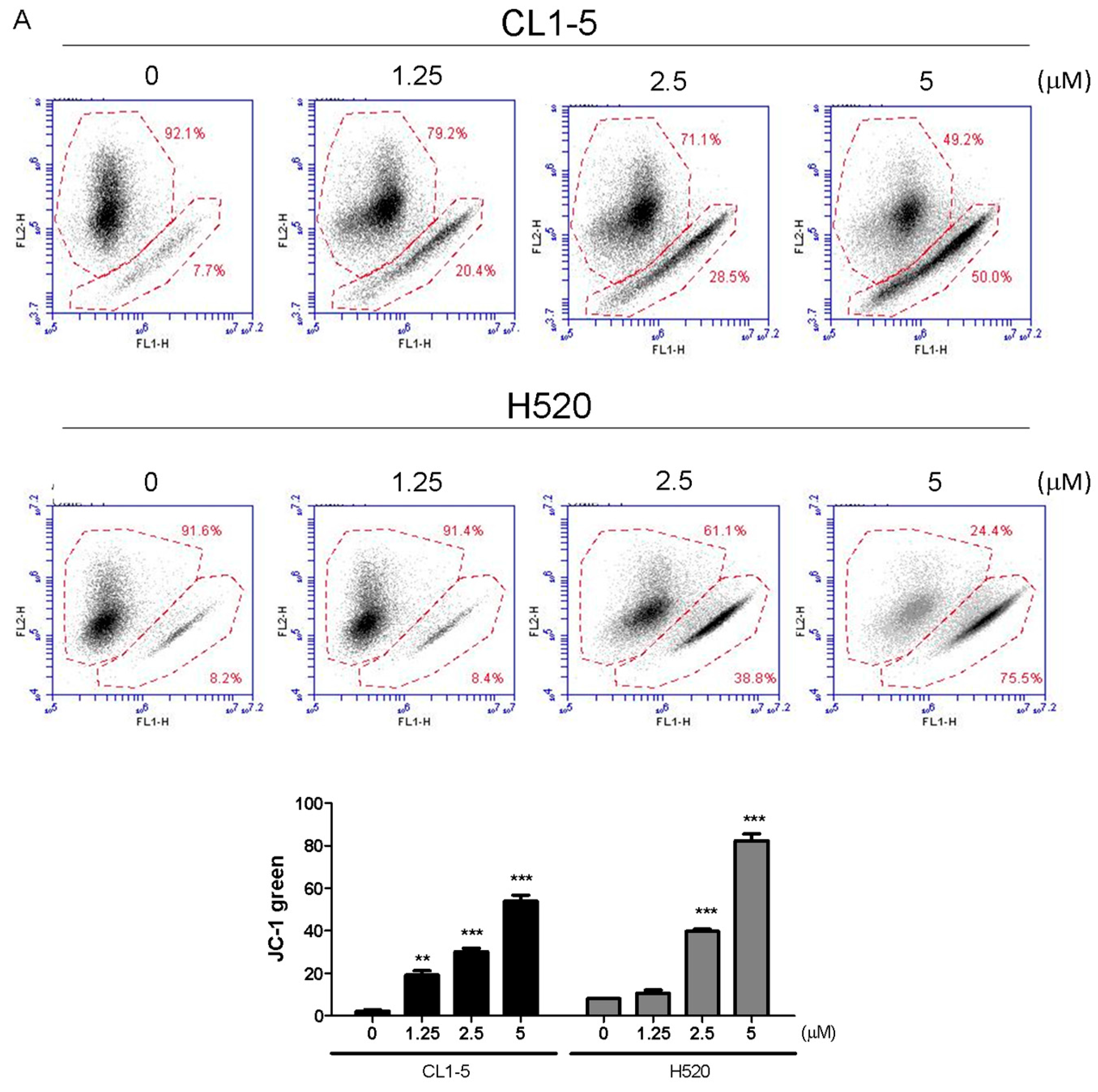
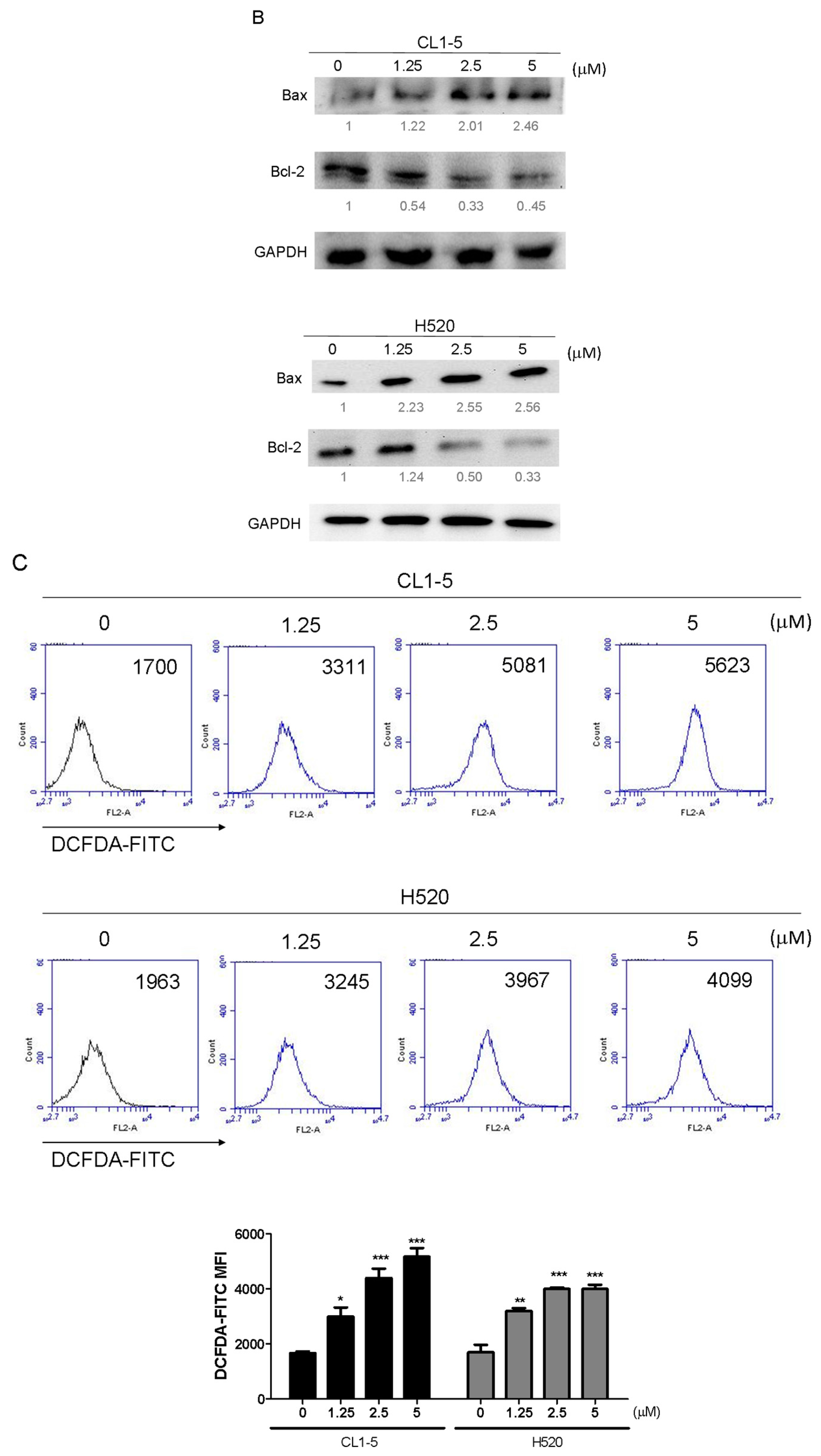
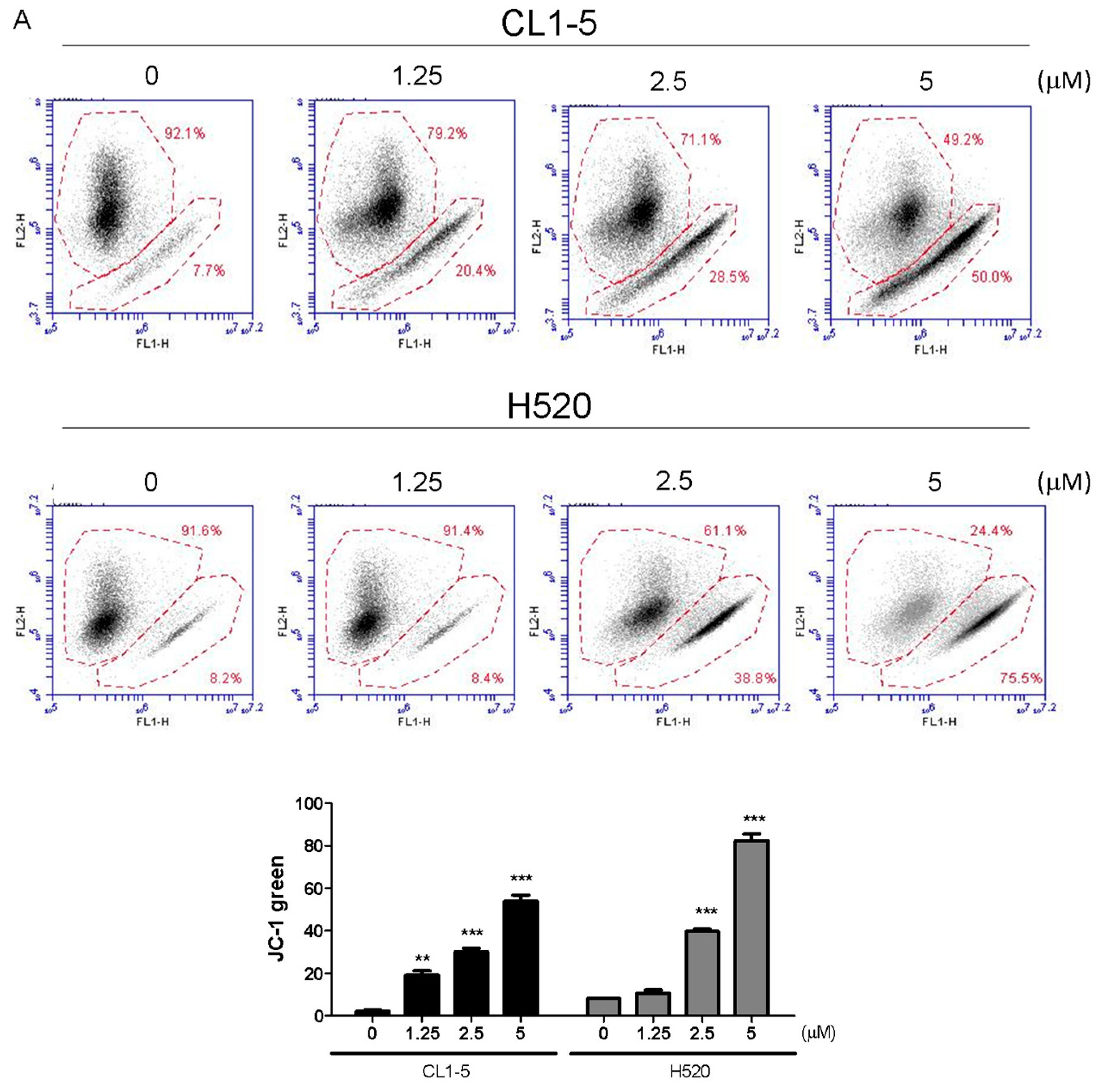
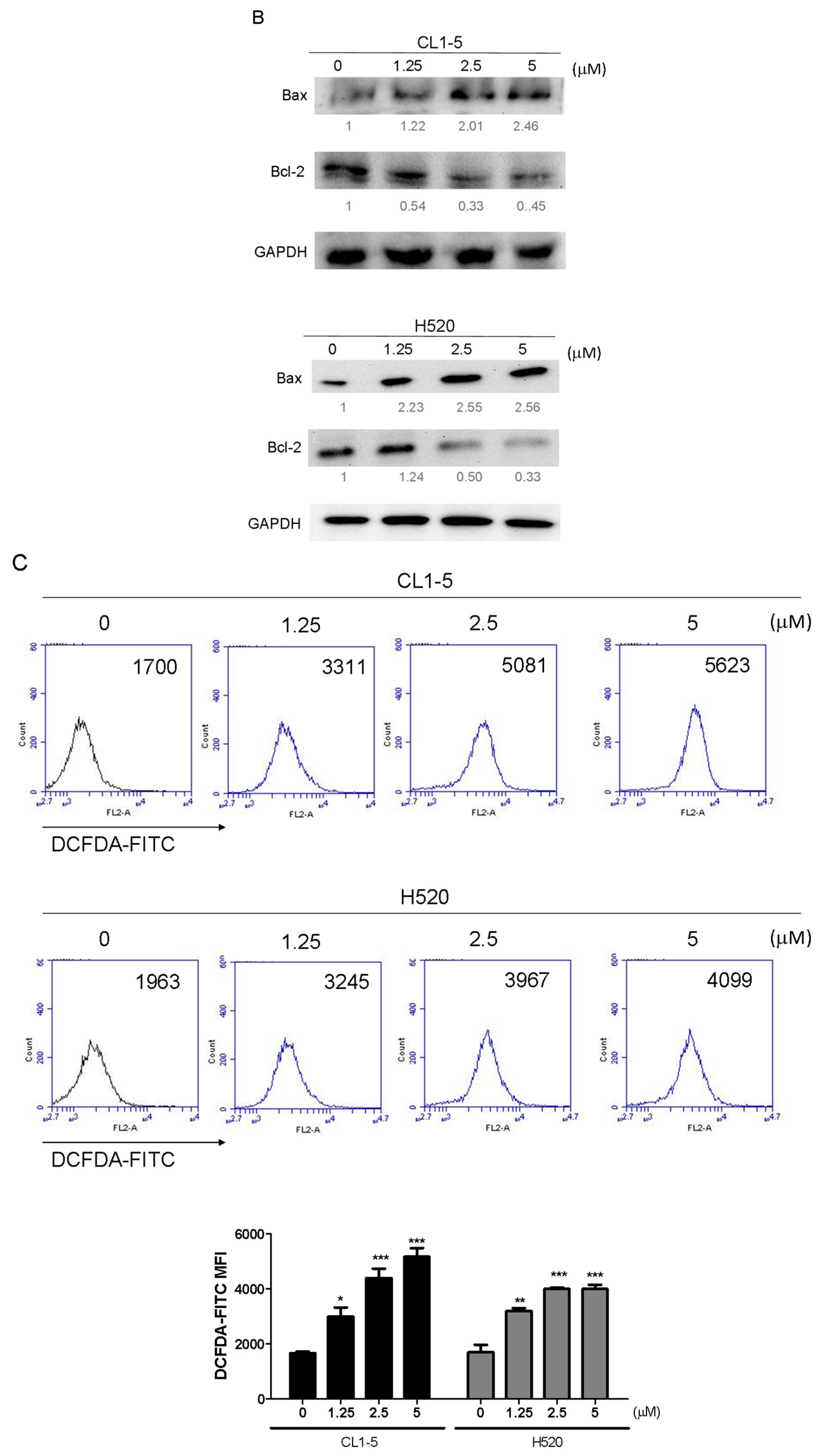
Mitochondrial functions play central roles in activating apoptosis in mammalian cells. The functional impairment of mitochondria is associated to the loss of MMP [
15], which was noted in this study following treatment with the Lobocrassin B for 24 h. The increased apoptotic Bax expression and decreased anti-apoptotic Bcl-2 expression were also observed in the Lobocrassin B -treated cells. The Bcl-2 family is made up of outer mitochondrial membrane proteins that can control mitochondrial-mediated apoptosis [
16]. Furthermore, in this study, the increased of intracellular ROS level was detected in the cells treated with the Lobocrassin B. The increased of ROS causes oxidative stress, which in turn can induce mitochondrial apoptosis and cellular dysfunction [
10]. It has been reported that ROS challenge can cause mitochondrial permeability transition pore (mPTP) opening, which leads to mitochondrial inner membrane permeabilization, membrane potential dissipation and Cyto c release [
17]. Taken together, the effects of Lobocrassin B on mitochondrial function are involved in its anti-lung cancer effects.
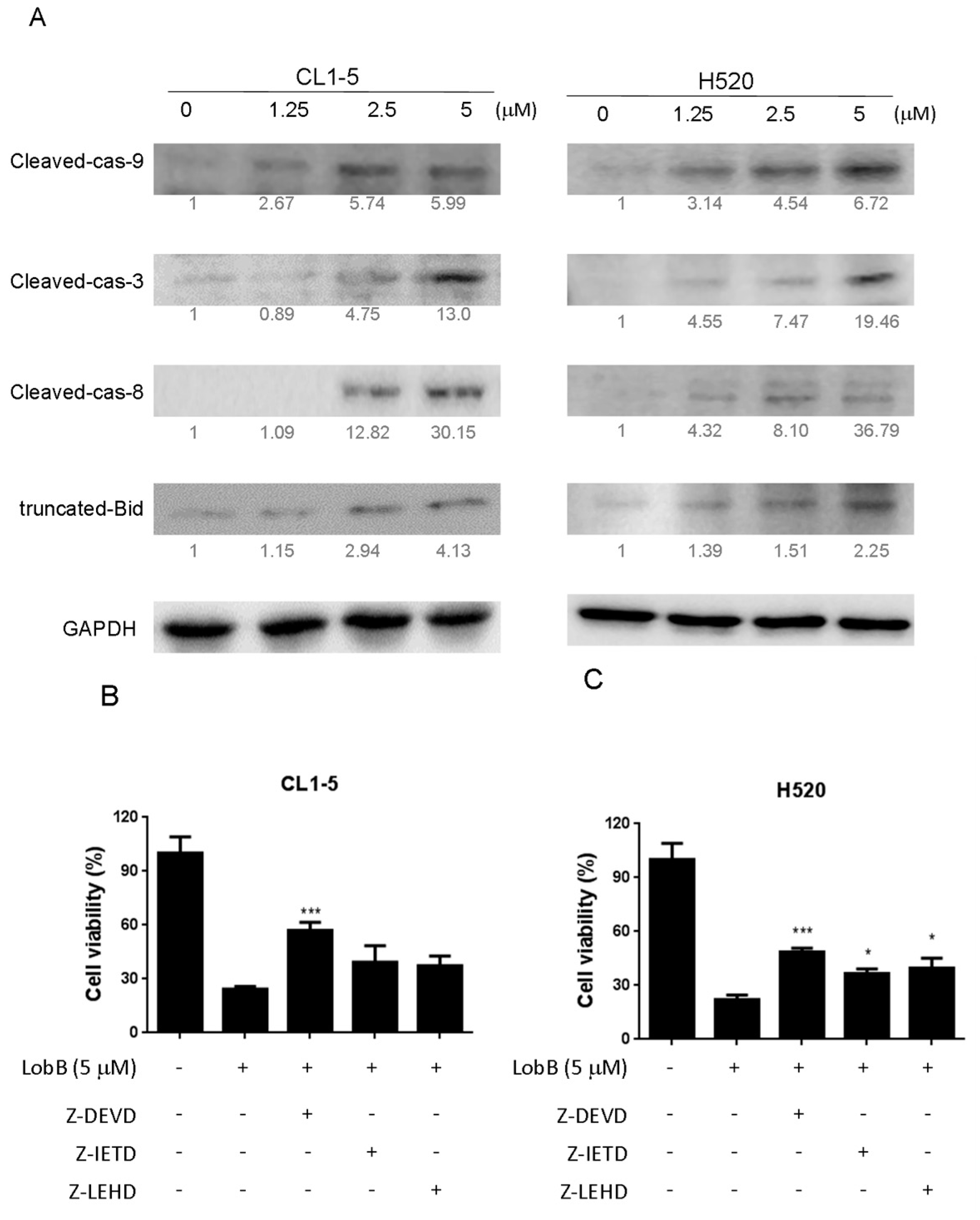
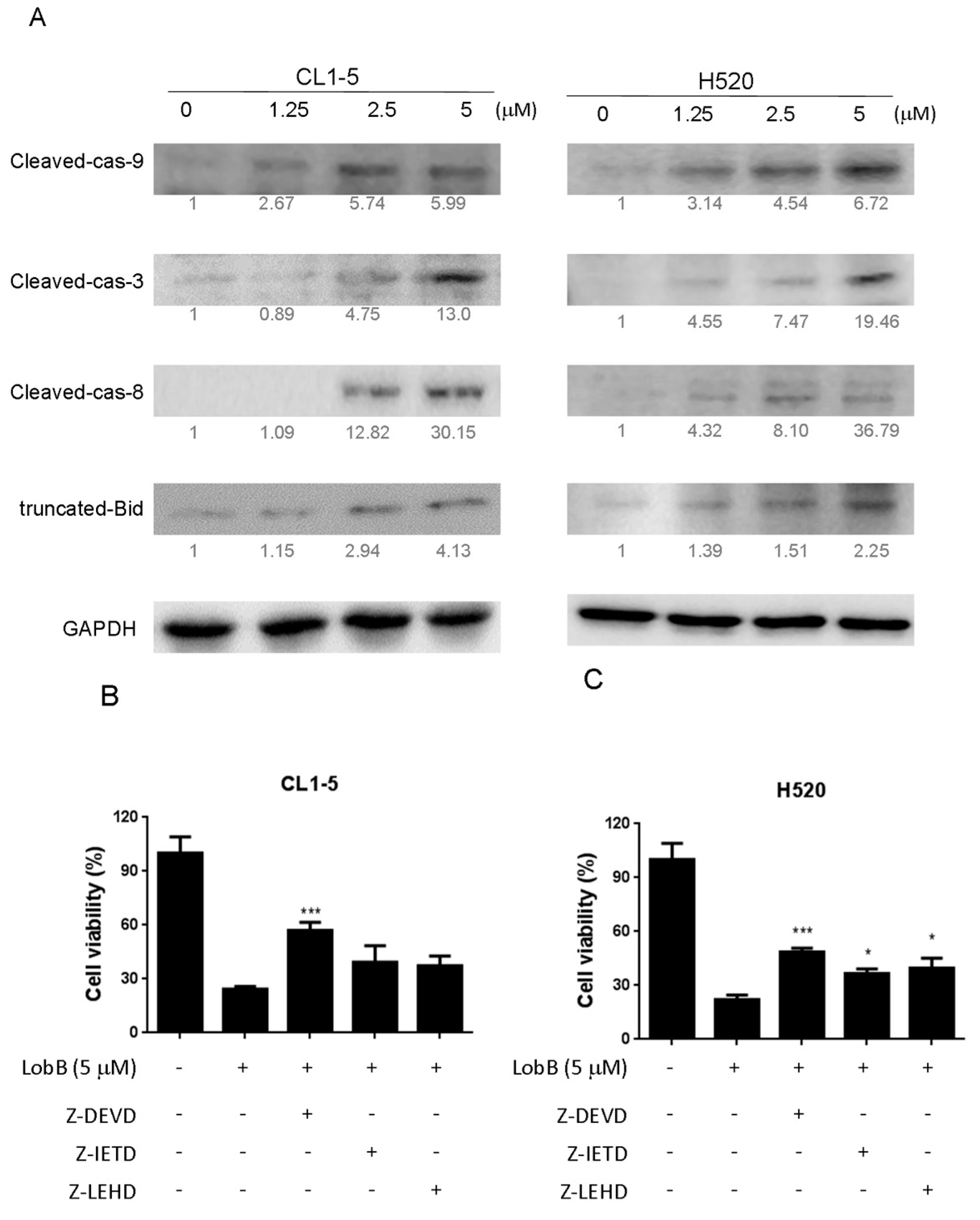
On the other hand, apoptosis can be induced through two main apoptotic pathways: the extrinsic (death receptor pathway) and the intrinsic (mitochondrial pathway). Caspase-3 is considered to be the most important of the executioner caspase that can be activated by a mitochondrial pathway involving initiator caspase-9 or a death receptor pathway involving initiator caspase-8 [
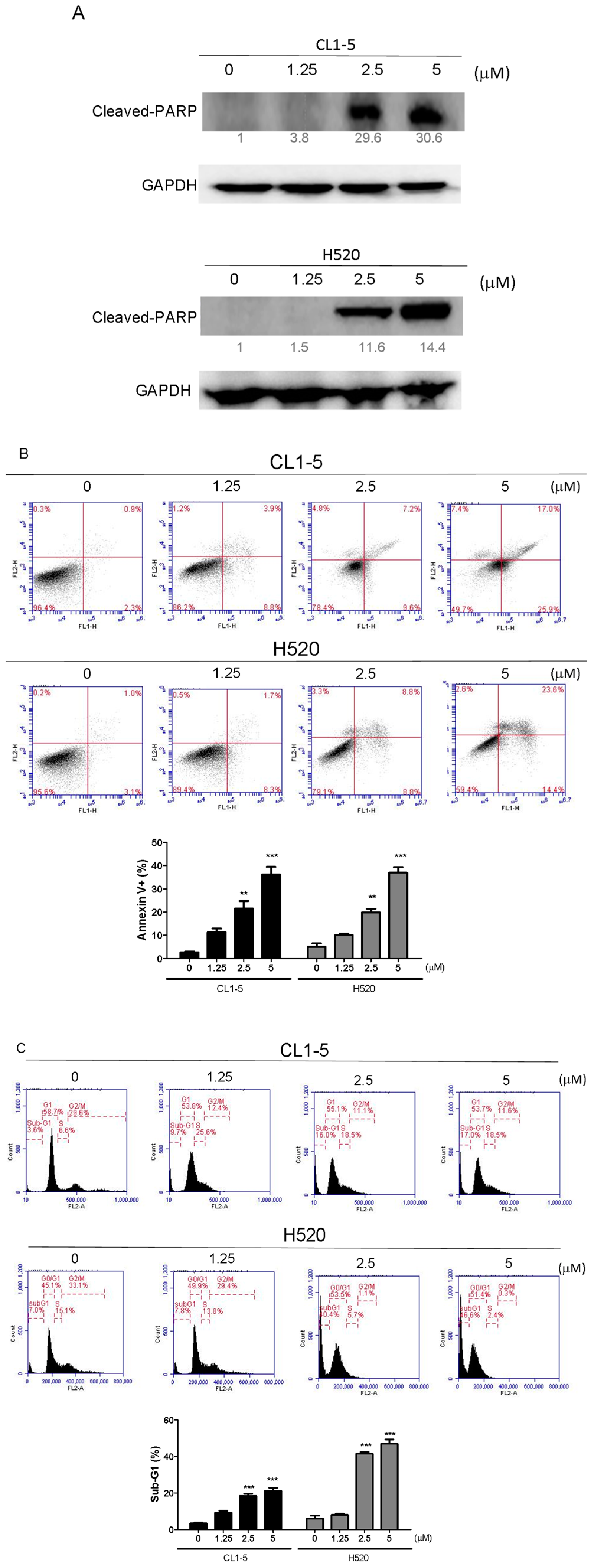
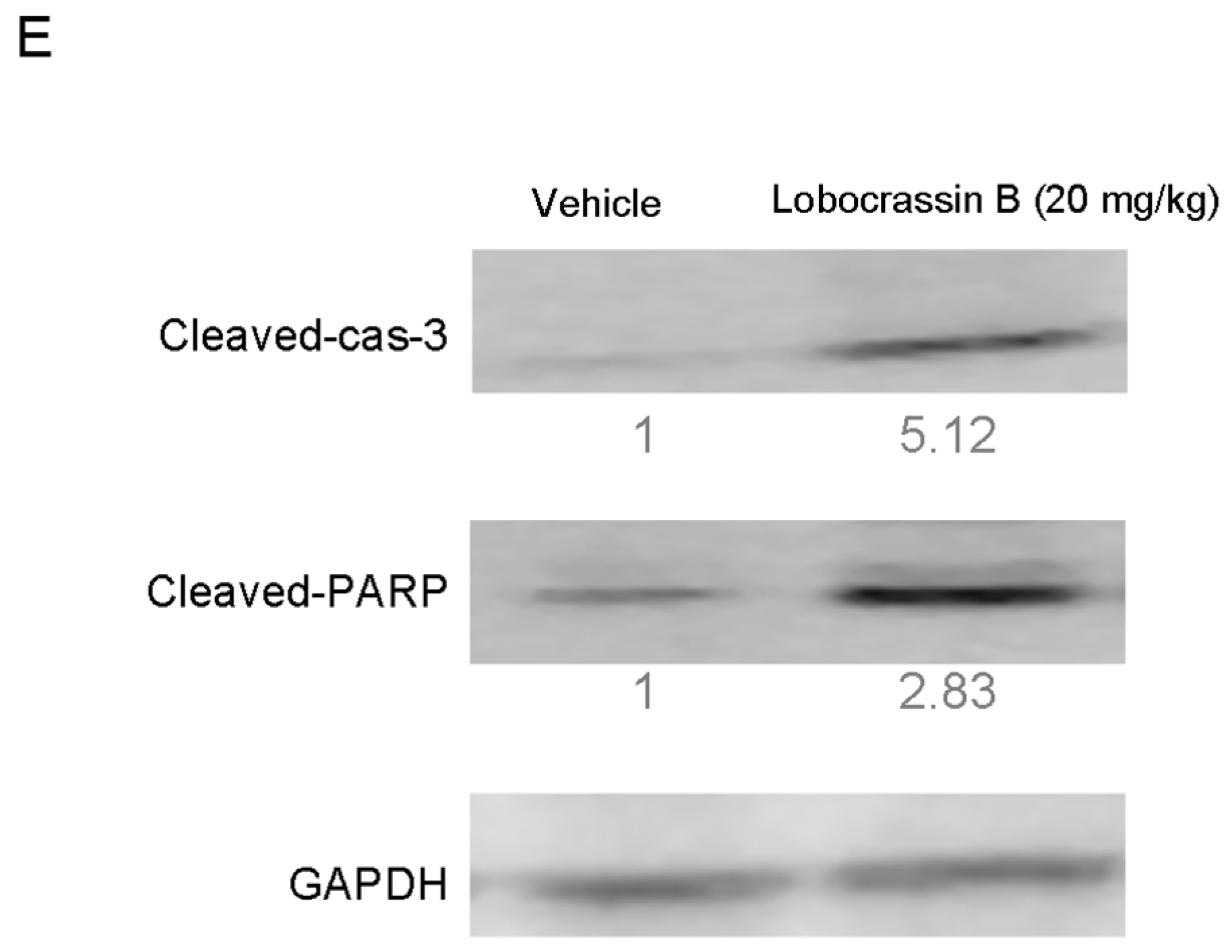
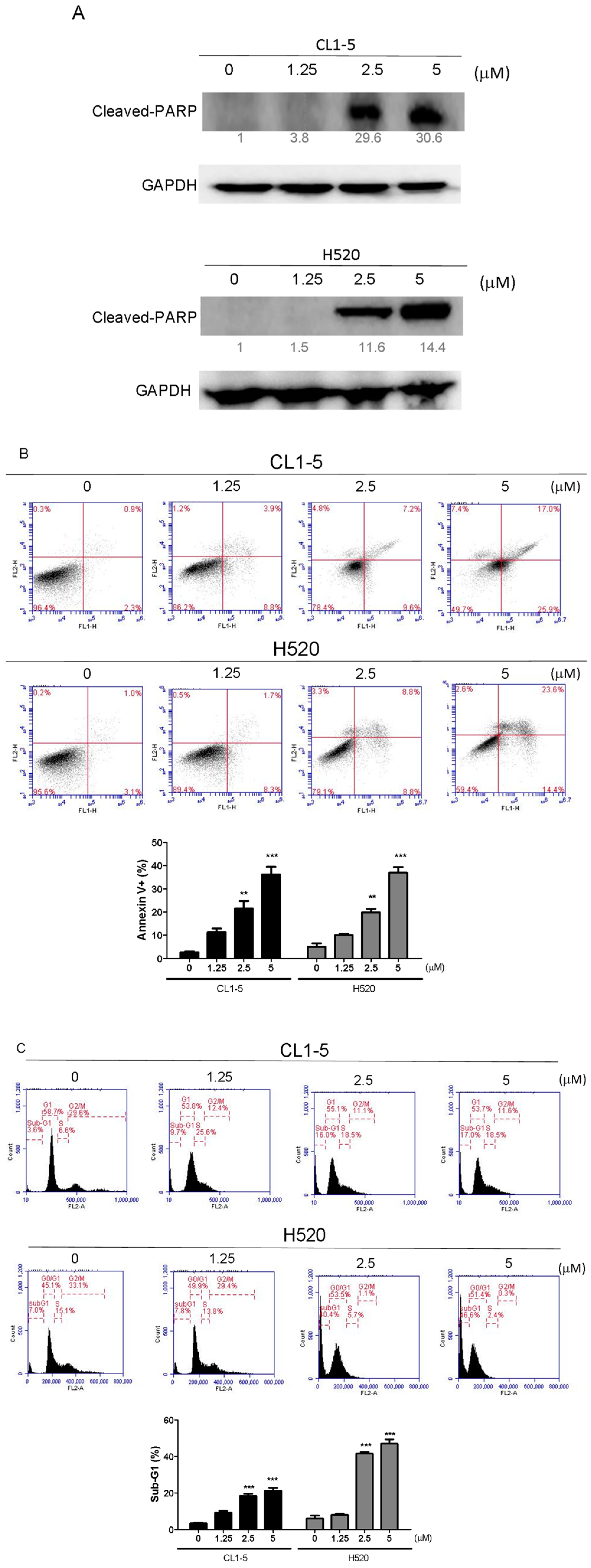
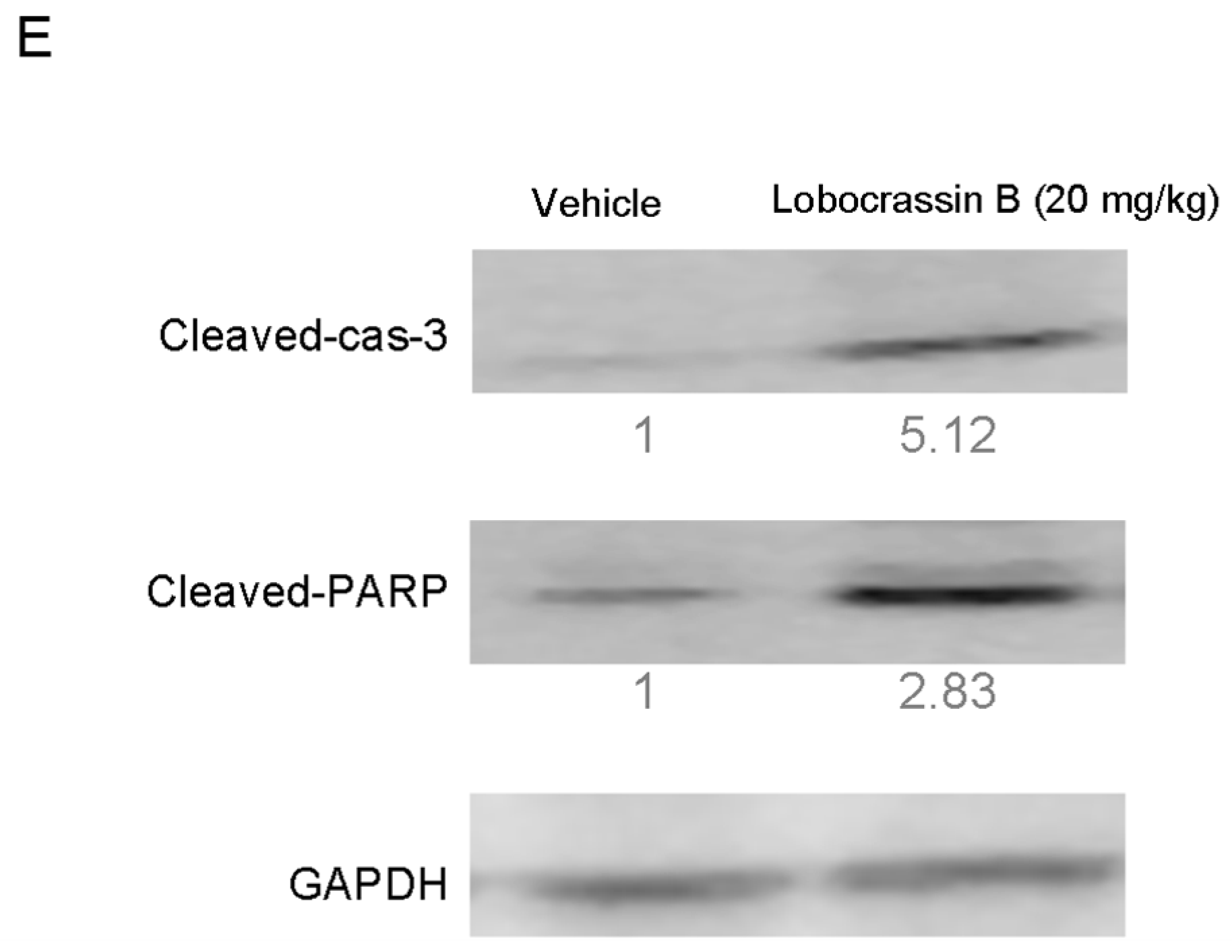
18]. Activation of caspase-3 activation leads to the cleavage of number important substrates, including PARP. Since caspase-8 cleaves Bcl-2 family protein, Bid into tBid cause mitochondria membrane potential disruption and then also lead to trigger activation of caspase-9, a cross-talk exists between the extrinsic and intrinsic apoptotic pathways [
19]. In the present study, we found the cleavage of PARP and the activation of caspase-3 were induced after lobocrassin B treatment, and caspase-3 inhibitor effectively prevented lobocrassin B—induced cell viability reduction. These dates suggest that apoptosis induced by lobocrassin B may be caspase-dependent. At the same time, we also found the activation of initiator caspase-8 and -9 in CL1-5 and H520 cells after lobocrassin B treatment. Furthermore, either Z-IETD-FMK (caspase-8 inhibitor) or Z-LEHD-FMK (caspase-9 inhibitor) only partially reverses lobocrassin B -induced reduction in cell viability in CL1-5 and H520 cells. Therefore, we suggest that combine activation of extrinsic (death receptor pathway) and the intrinsic (mitochondrial pathway) are involved in lobocrassin B -induced apoptosis in human lung cancer cells.
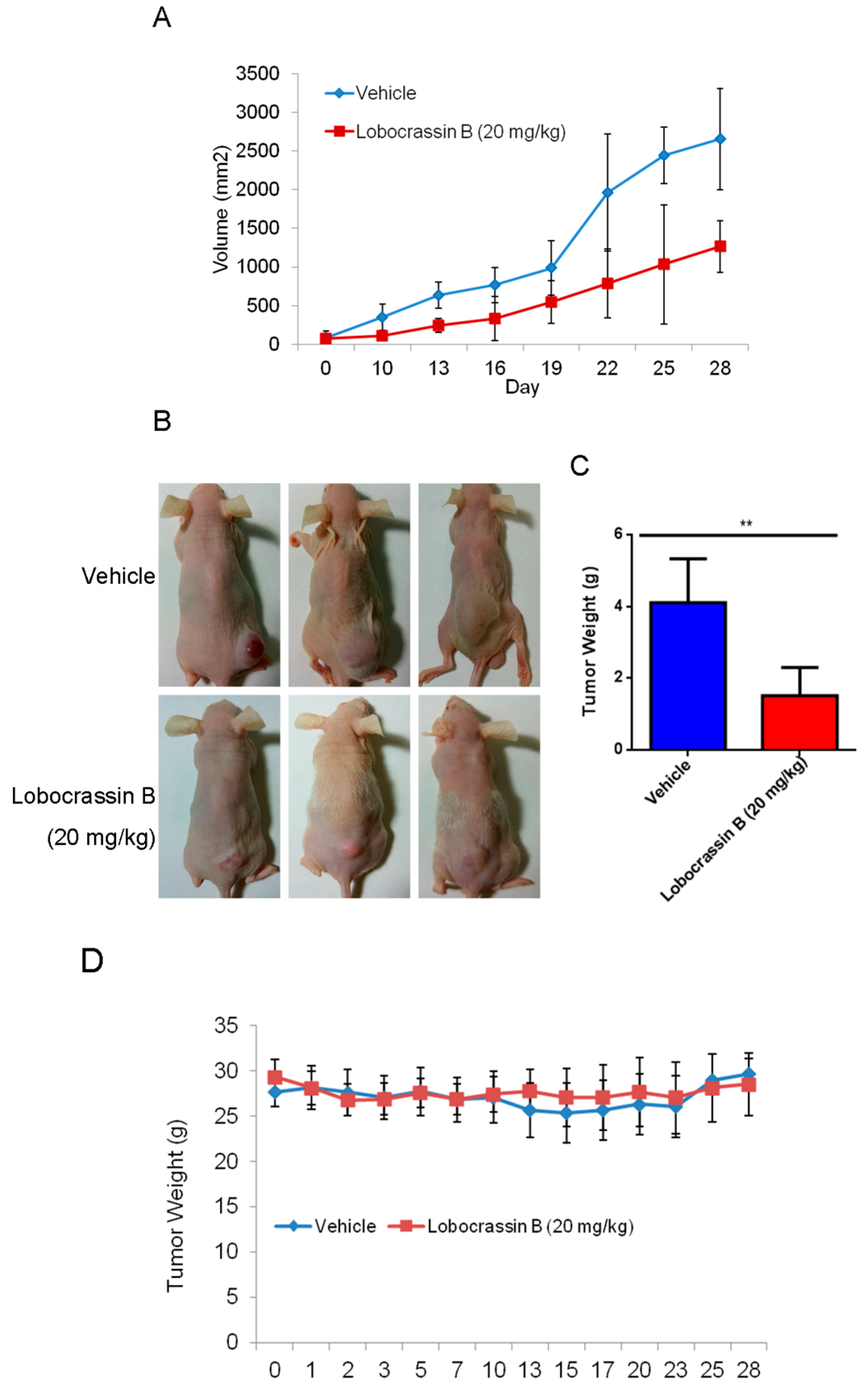
In conclusion, our study showed for the first time that Lobocrassin B exerts an anti–lung cancer effect in cell lines and in tumor xenograft in nude mice. Lobocrassin B treatment increases the ROS generation, MMP dissipation, and caused caspase cascade. These findings reveal that Lobocrassin B could efficiently induce lung cancer death through mitochondrial-dependent apoptotic pathway and it may be a potential chemotherapeutic agent.
4. Material and Methods
4.1. Cell Culture
The CL1-5 lung adenocarcinoma cell line was provided by Dr. Jeremy J. W. Chen (University of National Chung Hsing University, Taichung City, Taiwan). The lung squamous cell carcinoma cell line H520 and the immortalized bronchial epithelial cell line BEAS-2B were purchased from Food Industry Research and Development Institute (Hsinchu City, Taiwan). All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with a 10% heat inactive fetal bovine serum (FBS), 100 U/mL penicillin and 100 g/mL streptomycin. Cells were incubated at 37 °C in a humidified atmosphere containing 5%/95% of CO2/air. Cell culture reagents were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
4.2. Chemicals
Lobocrassin B was isolated from the wild-type soft coral
Lobophytum crassum as previously described [
8]. Etoposide was purchased from Sigma-Aldrich (Ann Arbor, MI, USA) as a positive control for cell viability assays. Both of Lobocrassin B and etoposide were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) at 100 mM as a stock solution prior to performing further assays.
4.3. MTT Cell Viability Assay
The cells were seeded into 24-well plates at 2 × 104 cells/well and treated with increasing concentrations of Lobocrassin B or etoposide or DMSO (0.1%) as vehicle control for 24. Then, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO, USA) solution (0.5 mg/mL final concentration) was added to each well. Following a 4-h reaction, the supernatant was aspirated, and then 600 μL DMSO were added. The absorbance was examined at 540 nm using a microplate reader (TECAN, Durham, NC, USA). Data were showed as a percentage of absorbance of Lobocrassin B treated cells relative to DMSO-treated cells. The IC50 values are calculated using SPSS 16.0 software (IBM Corporation, Armonk, NY, USA).
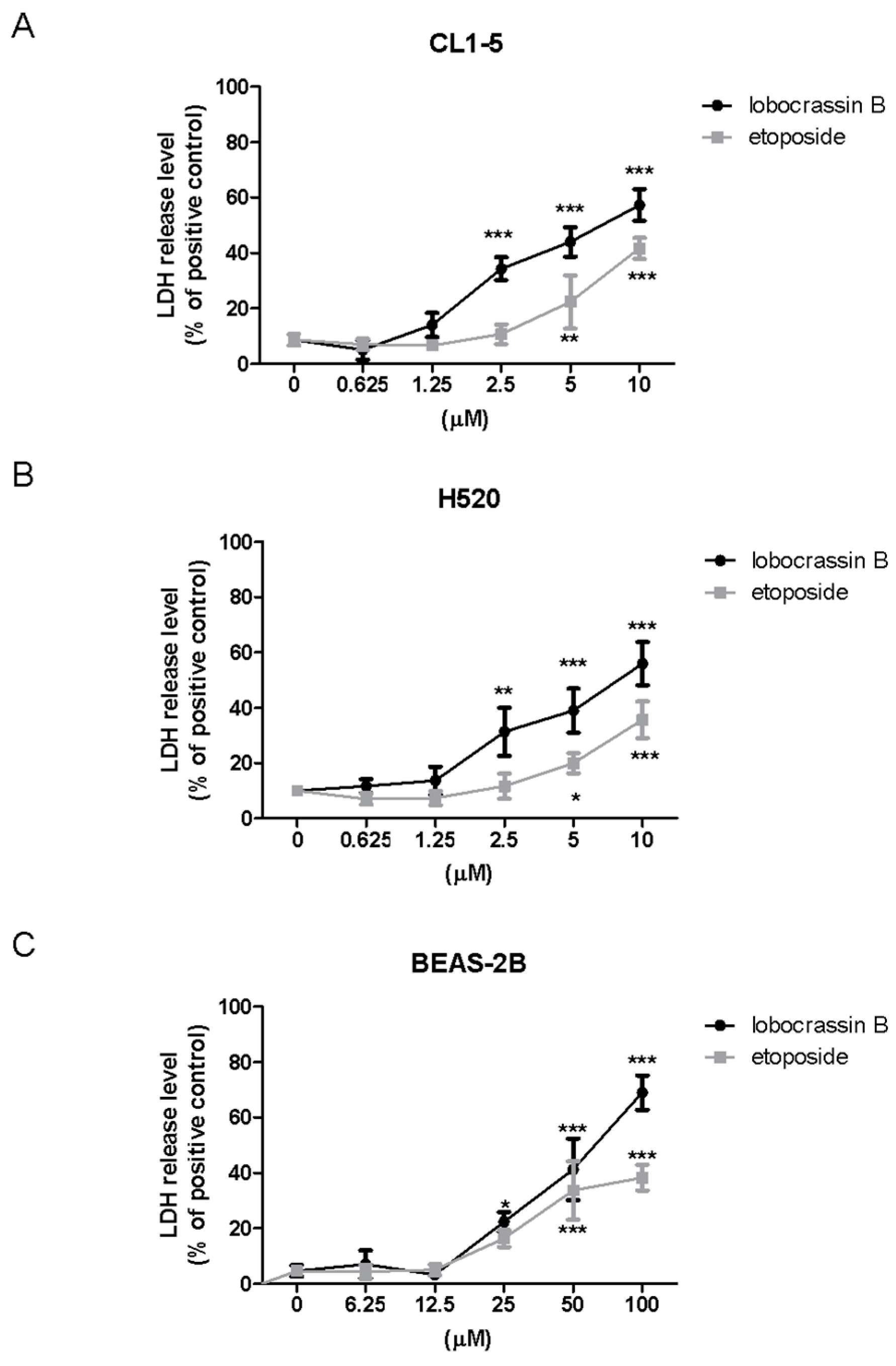
4.4. Lactate Dehydrogenase (LDH) Assay
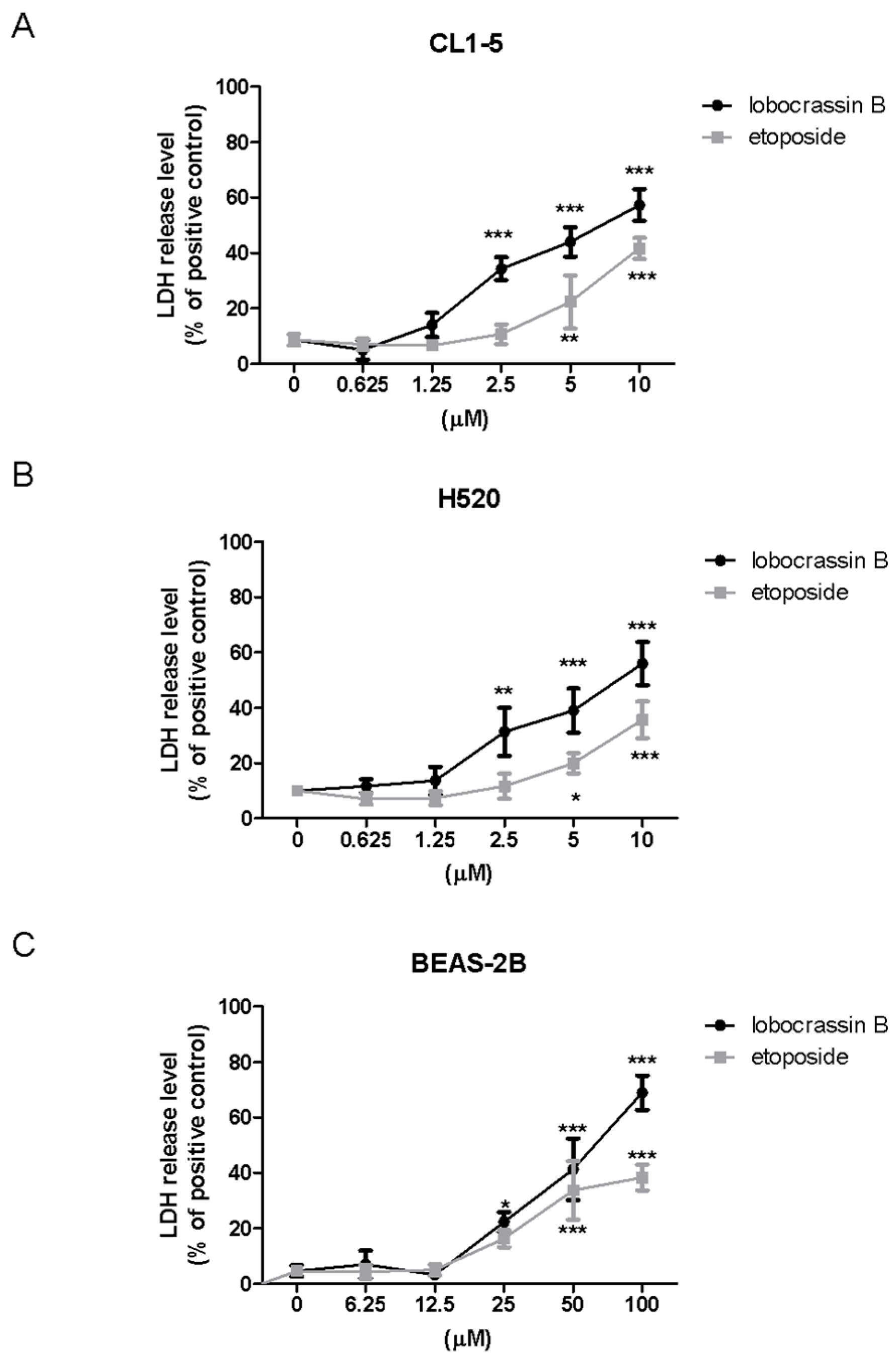
LDH cytotoxicity assay kit (Cayman Chemical Co., Ann Arbor, MI, USA) was employed to assess the cellular toxicity induced by compounds. Lung cells were treated with different concentrations of Lobocrassin B or etoposide for 24 h followed by incubating with Triton X-100, and centrifuging at 1200 rpm for 5 min to obtain the supernatant. 100 µL/well of the supernatant was transferred to a new 96-well plate and equal volume of LDH reagent was added to each well. After incubating for 30 min at room temperature, the concentration of LDH was measured with a microplate reader (TECAN, Durham, NC, USA) at wavelength of 490 nm. The LDH release level (% of positive control) was recorded as the percentage of (ODtest − ODblank)/(ODpositive − ODblank), where ODtest is the optical density of cells treated with either 0.1% DMSO or Lobocrassin B; ODpositive is the optical density of etoposide-treated cells and ODblank is the background optical density of the wells without cells.
4.5. Measurement of Apoptosis by Annexin V-FITC/PI Staining
The cells (2 × 105/well) were seeded into 6-well plates and treated with 1.25 and 2.5, 5 μM Lobocrassin B for 24 h and then harvested and washed with phosphate buffered saline (PBS). Staining went along with with 5 μL Annexin V-FITC (20 μg/mL) and 5 μL propidium iodide (PI; 50 μg/mL) (BD Biosciences, Franklin Lakes, NJ, USA) in the dark at room temperature for 15 min. The apoptotic cells were measured using an AccuriTM C5 cytometer (BD Biosciences, San Jose, CA, USA).
4.6. Caspase Inhibitor Assay
The cells were seeded into 24-well plates at 2 × 104 cells/well and were grown overnight and then pre-treated with inhibitors of caspase-3 (Z-DEVD-FMK, 50 µM, BioVision, San Francisco, CA, USA), caspase-8 (Z-IETD-FMK; 50 µM, San Francisco, CA, USA), and caspase-9 (Z-LEHD-FMK; 50 µM, San Francisco, CA, USA) for 2 h. Cells were then treated with 5 μM Lobocrassin B for 24 h. The cell viability were examined by MTT assay.
4.7. DNA Content by Flow Cytometric Analysis
Cells (2 × 105/well) were subcultured into 6-well plates and treated with 1.25 and 2.5, 5 μM Lobocrassin B for 24 h. After challenge, cells were harvested by trypsinization and washed with PBS, fixed in 70% (v/v) ethanol at −20 °C overnight. Afterward, cells were stained went along with 500 μL PBS with 0.1% (v/v) Triton X-100, 100 μg/mL RNase A and 50 μg/mL propidium iodide (PI) (Sigma-Aldrich, St. Louis, MO, USA) in constant darkness at room temperature for 20 min. Sub-G1 population containing apoptotic cells was analyzed using an AccuriTM C5 cytometer.
4.8. Detection of Reactive Oxygen Species (ROS) and Mitochondrial Membrane Potential (MMP)
Cells (2 × 105/well) were subcultured into 6-well plates and treated with 1.25 and 2.5, 5 μM Lobocrassin B for 12 h. Thereafter, cells were suspended and incubated in 500 μL of dichlorodihydrofluorescein diace-tate (DCFH-DA, Molecular probes, Eugene, OR, USA) (10 μM) for ROS examination and 2 μM 5,5′,6,6′-tetra chloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Sigma-Aldrich, St. Louis, MO, USA) (10 µg/mL) for mitochondrial membrane potential in constant darkness at room temperature for 30 min. The samples were analyzed using a using an AccuriTM C5 cytometer.
4.9. Western Blot Analysis
Cells (2 × 105/well) were seeded into 6-well plates and treated with indicated concentrations of Lobocrassin B for 24 h. The cells and collected tumor tissues were lysed by RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) containing 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and 2% phenylmethanesulfonyl fluoride (PMSF; Sigma-Aldrich, St. Louis, MO, USA). Equal loading was verified by using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), to examine protein concentration. The samples of cell lysates were separated by 12% SDS-PAGE gel, and then electroblotted onto Immobilon-P Transfer Membrane (Merck Millipore, Billerica, MA, USA).The membranes were hybridized with anti-Bcl-2 (ab32124), anti-Bax (ab7977), anti-cleaved caspase-3 (ab2302), anti- caspase-8 (ab220171), anti-cleaved caspase-9 (ab2324), anti-cleaved PARP (ab32064) and glyceralde-hyde 3-phosphate dehydrogenase (GAPDH) (ab8245) (Abcam, Cambridge, MA, USA) antibodies at 4 °C overnight after being blocked with PBS containing 0.1% (v/v) Tween 20 and 5% (w/v) nonfat dry milk for 3 h at room temperature. The membranes were then incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at 4 °C overnight. Band detection was visualized utilizing the enhanced chemiluminescence detection kit reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA). All bands in the blots were normalized to the level of GAPDH for each lane. The intensity of the bands was quantified using ImageJ 1.47 program for Windows from the National Institute of Health (NIH) (Bethesda, MD, USA).
4.10. Animals’ Experimentation
Sex-week-old female BALB/c athymic nude mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan). The all animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University (Taichung, Taiwan). The mice were housed at a constant room temperature and maintained on a 12 h light/dark cycle with fed a standard rodent diet and water.
4.11. Tumor Xenograft Model
CL1-5 cells (1 × 107/mouse) in 0.2 mL at 1:1 mixture of cultural medium and BD Matrigel Basement Membrane Matrix (BD Biosciences, Bedford, MA, USA) was inoculated subcutaneously into the flank of nude mice. When the tumor reached approximately 2 mm3 (at day 10 after cell inoculation), the mice were randomly divided into two groups (n = 6 each), and intraperitoneal (i.p.) injection included 20mg/kg Lobocrassin B or vehicle (10% DMSO + 90% glyceryl trioctanoate) (Sigma-Aldrich, St. Louis, MO, USA) every other day continuously for three weeks. During the Lobocrassin B administration, the body weight and tumor sizes (mm3) were recorded. The formula of length × (width)2 × 0.5 was applied to calculate the tumor size. At the end of treatment, all the mice were sacrificed after 28 days. The tumor tissues were measured weighted after being removed. The tumors were dissected and evaluated by immunoblotting as mentioned earlier.
4.12. Statistical Analysis
The results are expressed as the means ± standard deviation (SD) in triplicate. For comparison between two groups, we used unpaired two-tailed t test (Student’s t test). One-way or two-way ANOVA was used to compare multiple groups according to the experiments (GraphPad Prism Version 5.0 (San Diego, CA, USA). The level of statistical significance was considered as a probability (p) value b 0.05.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}