Three New Malyngamides from the Marine Cyanobacterium Moorea producens

 , and
, and
Abstract
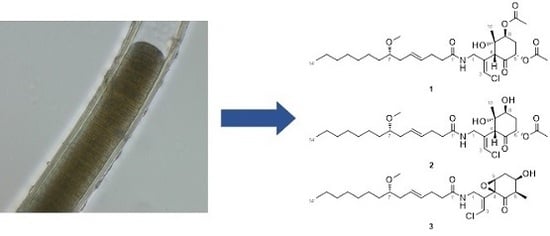
:
1. Introduction
2. Results
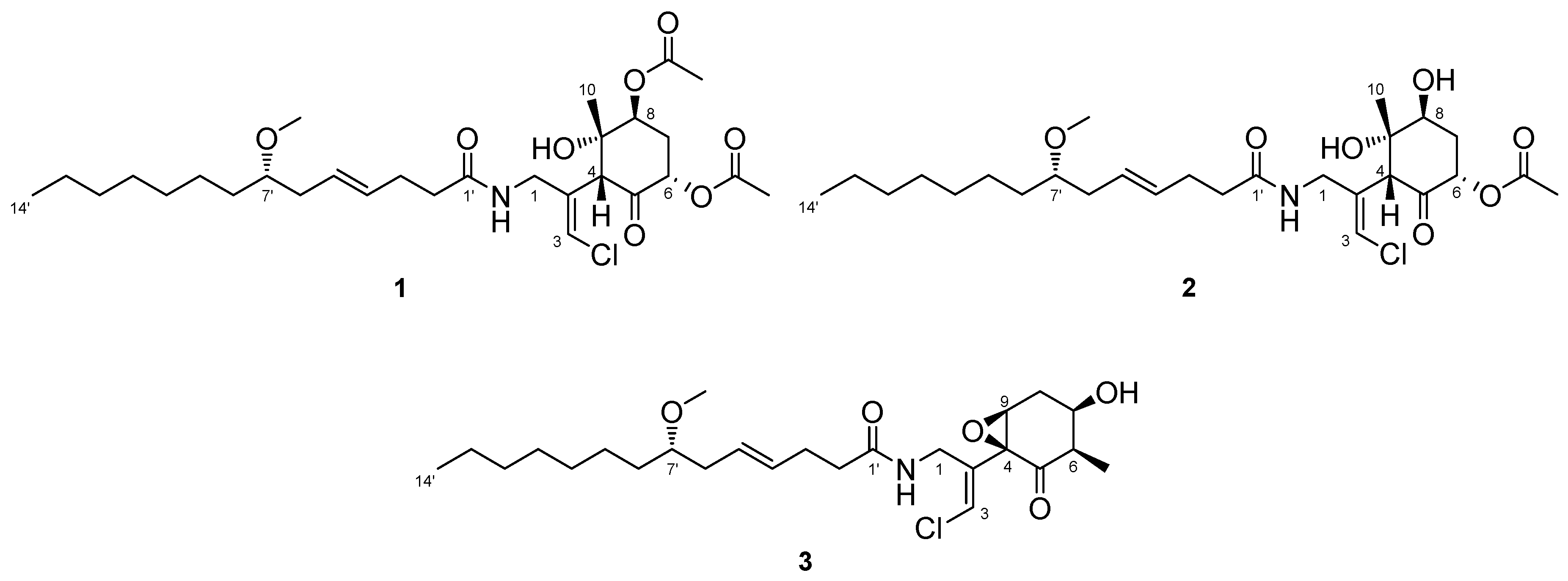
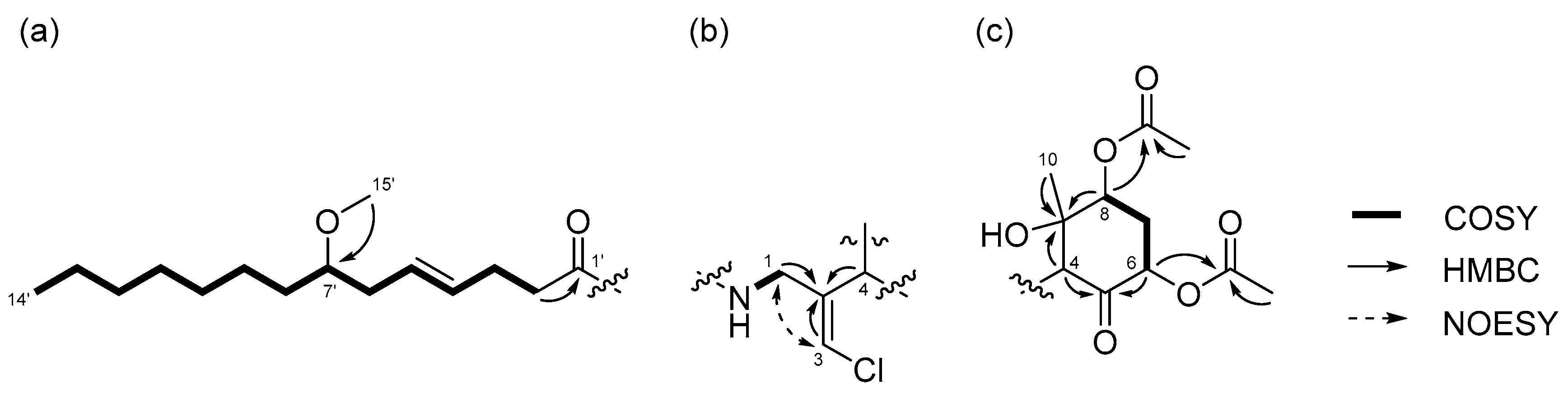
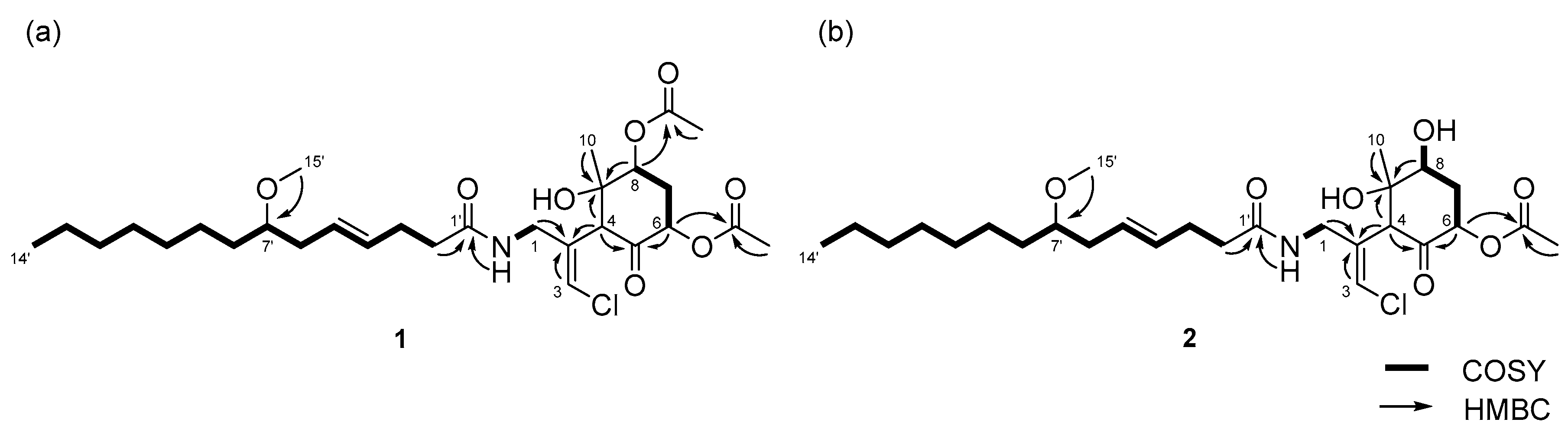
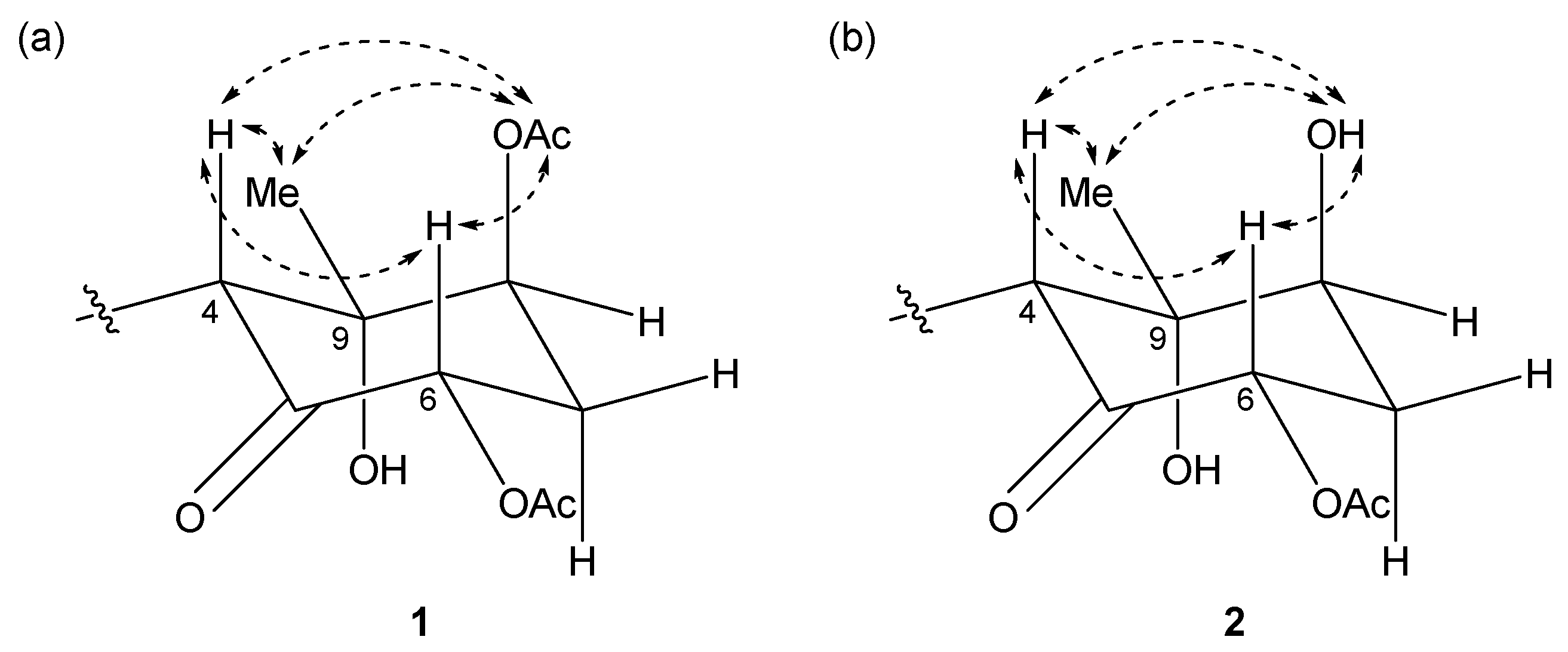
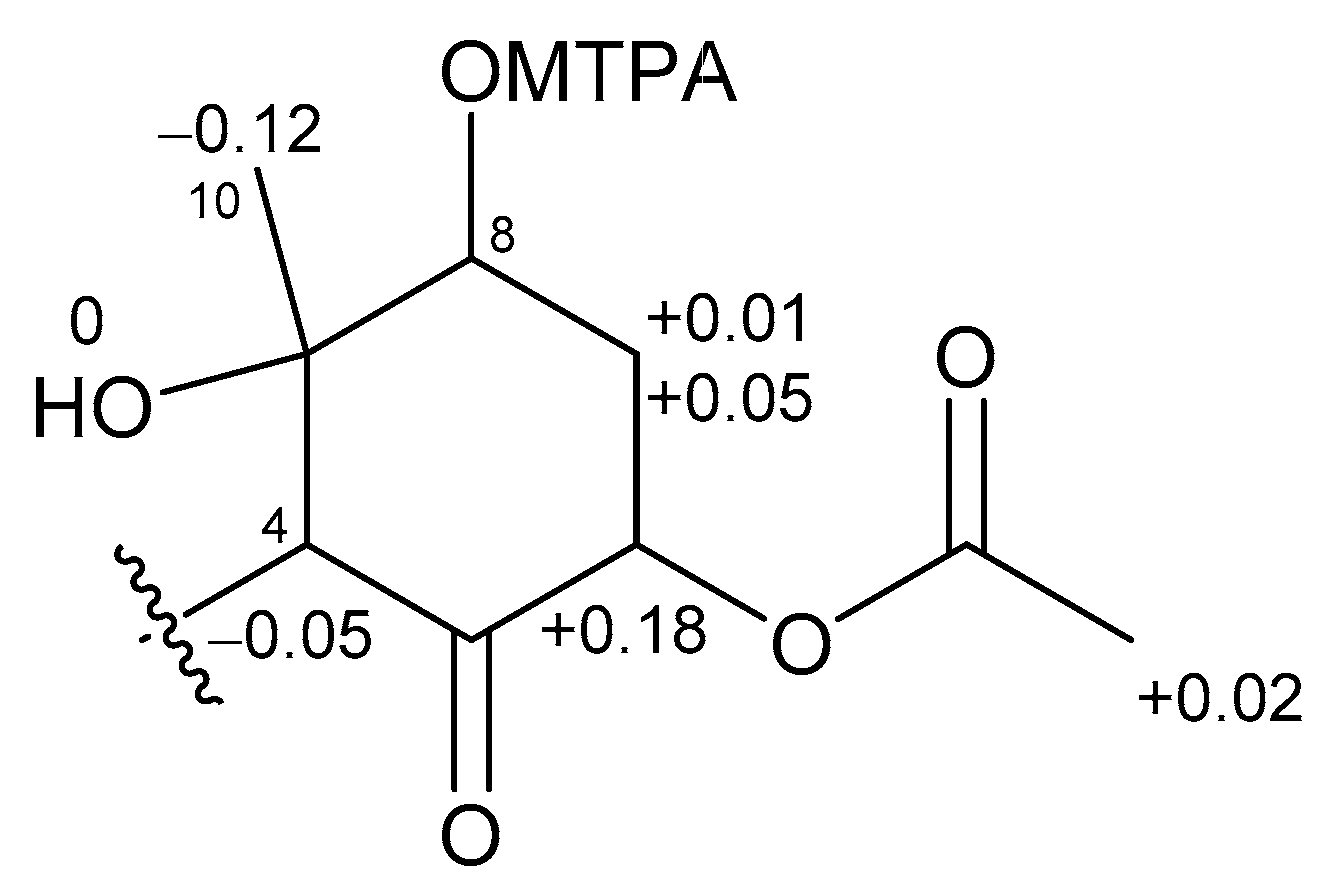
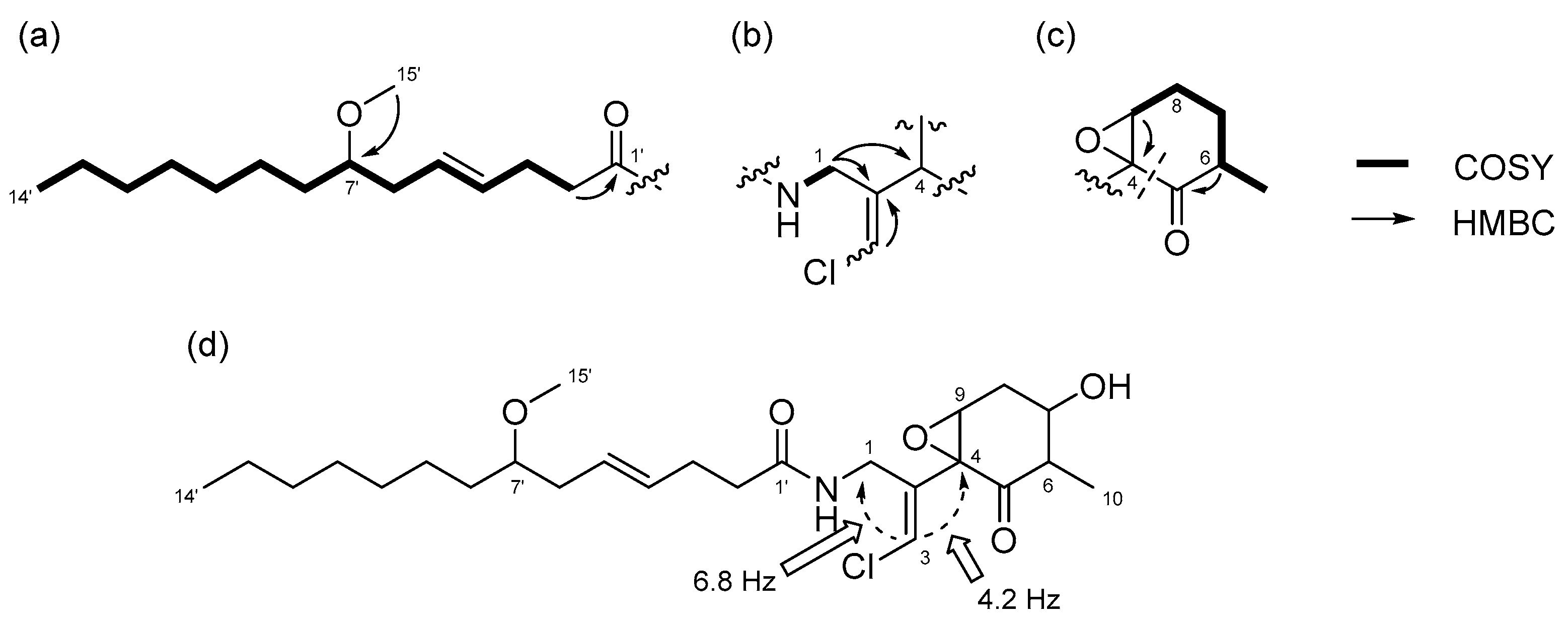
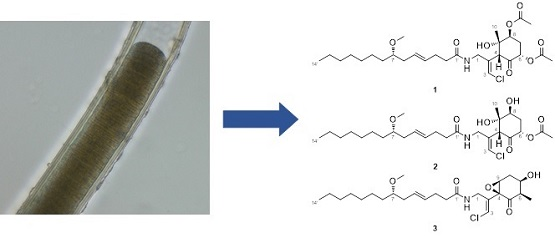
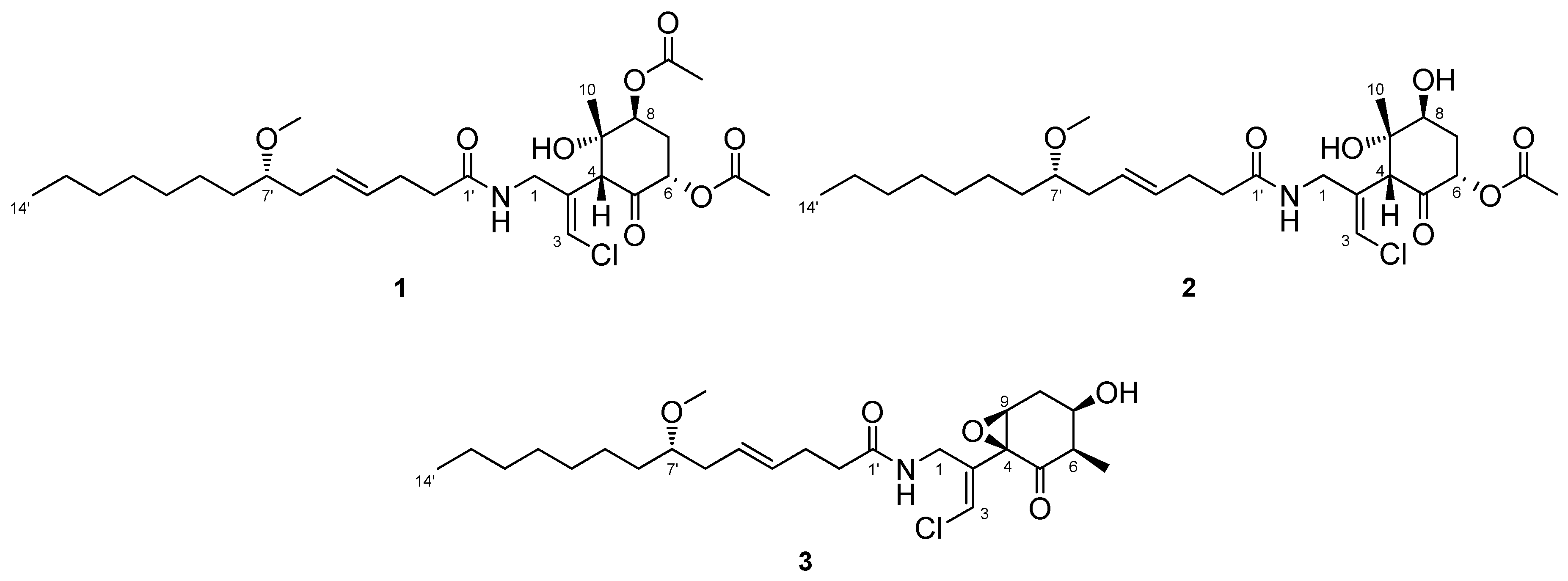
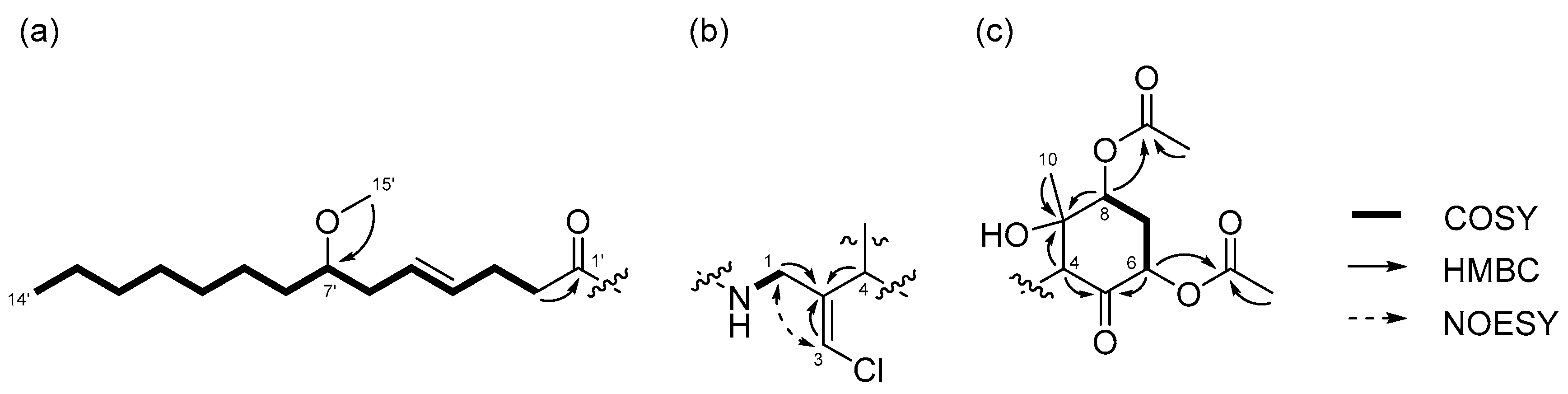
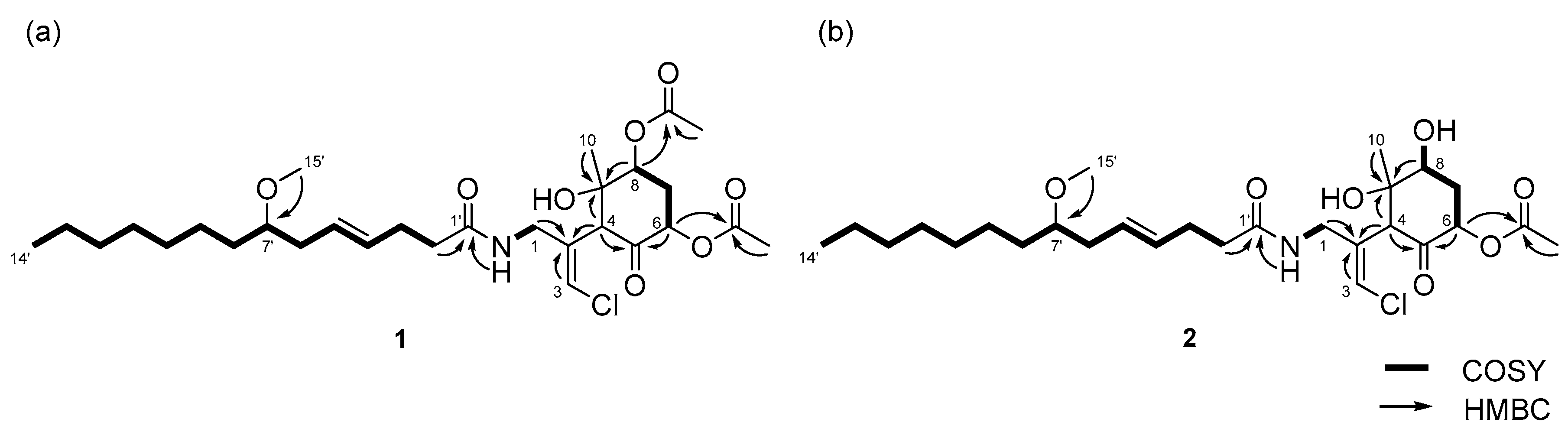
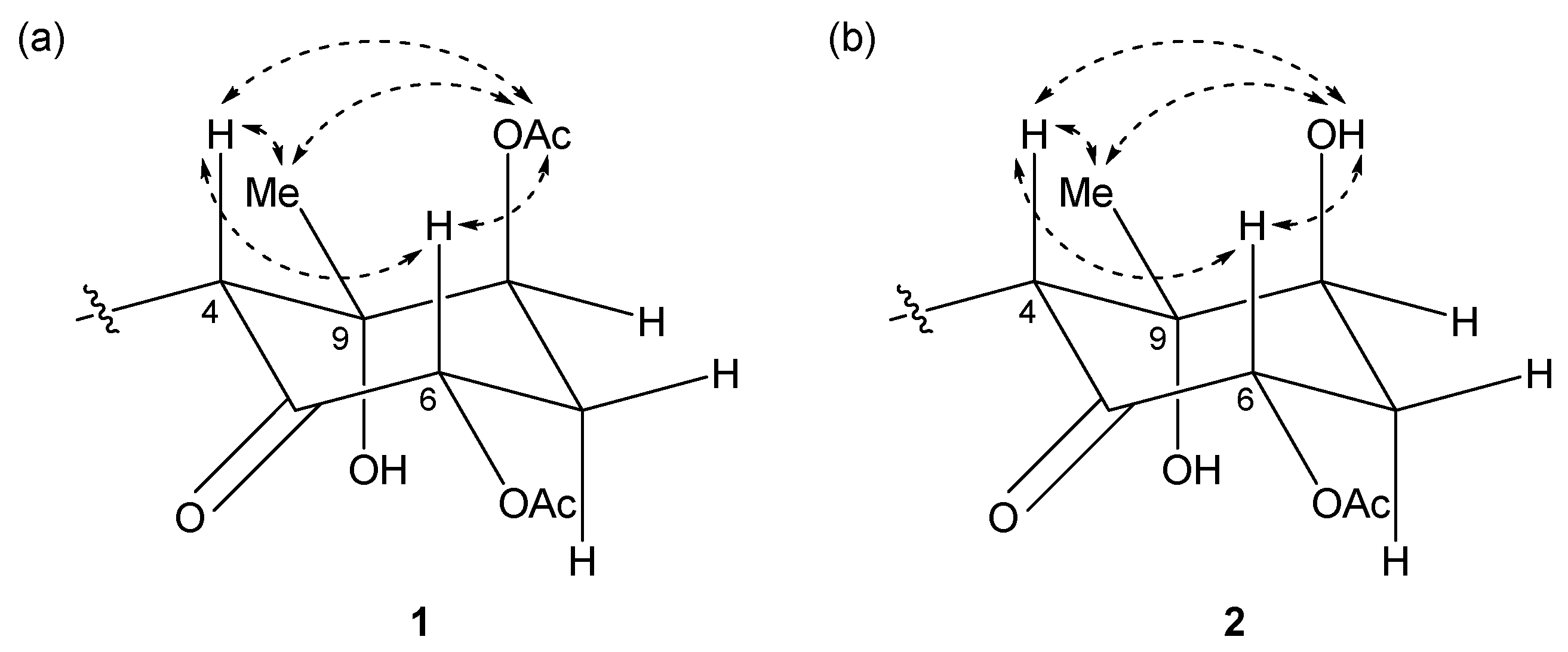
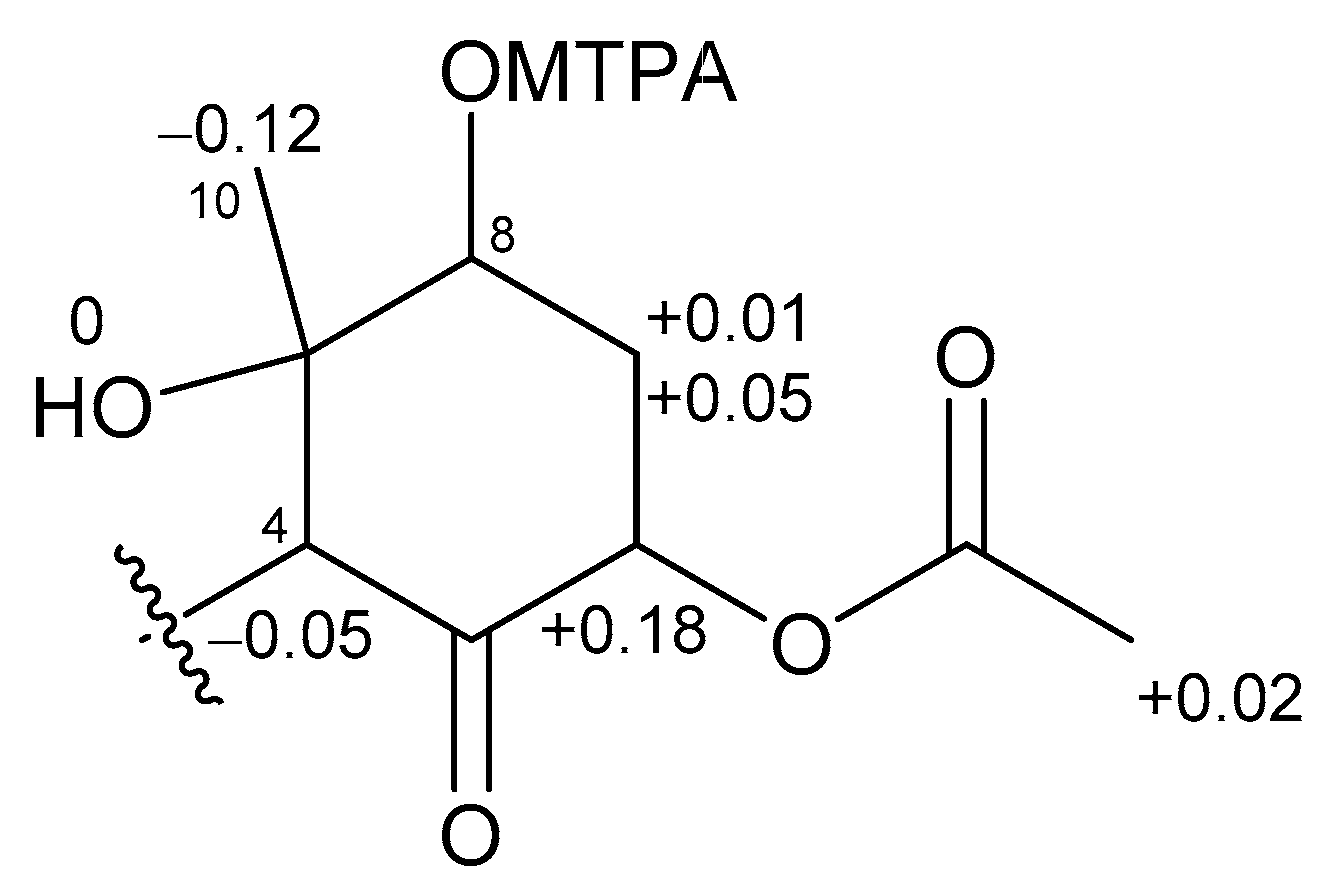
2.1. 6,8-Di-O-Acetylmalyngamide 2 (1) and 6-O-Acetylmalyngamide 2 (2)
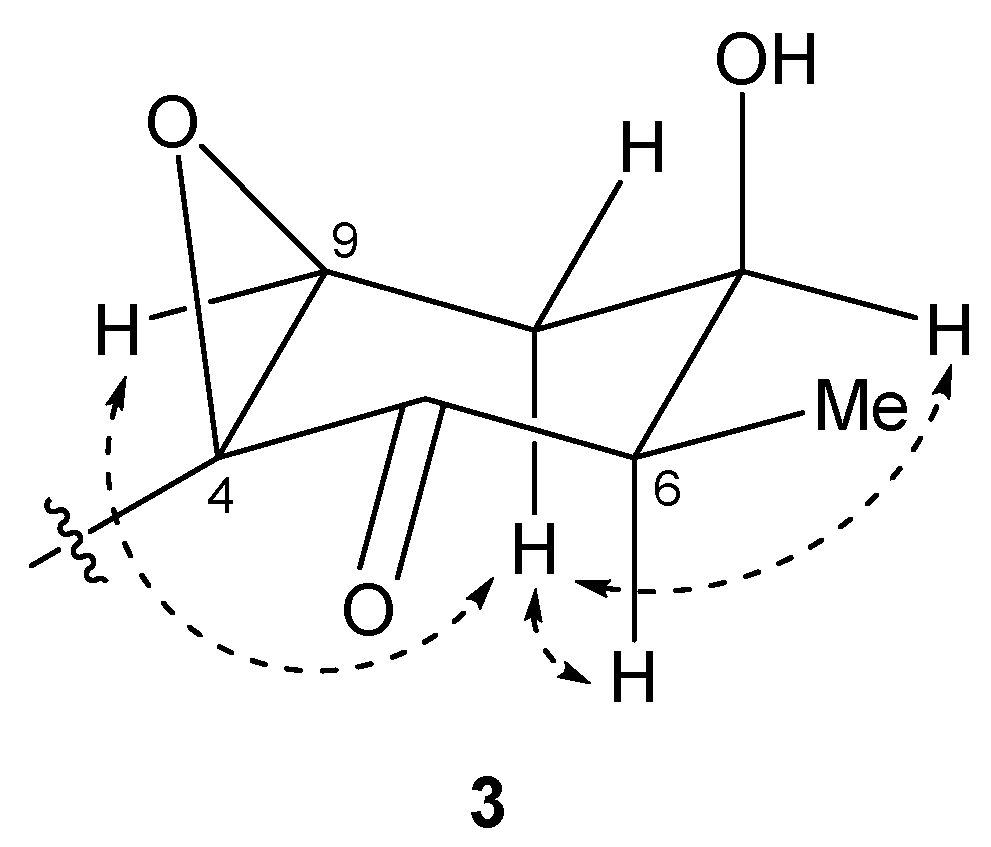
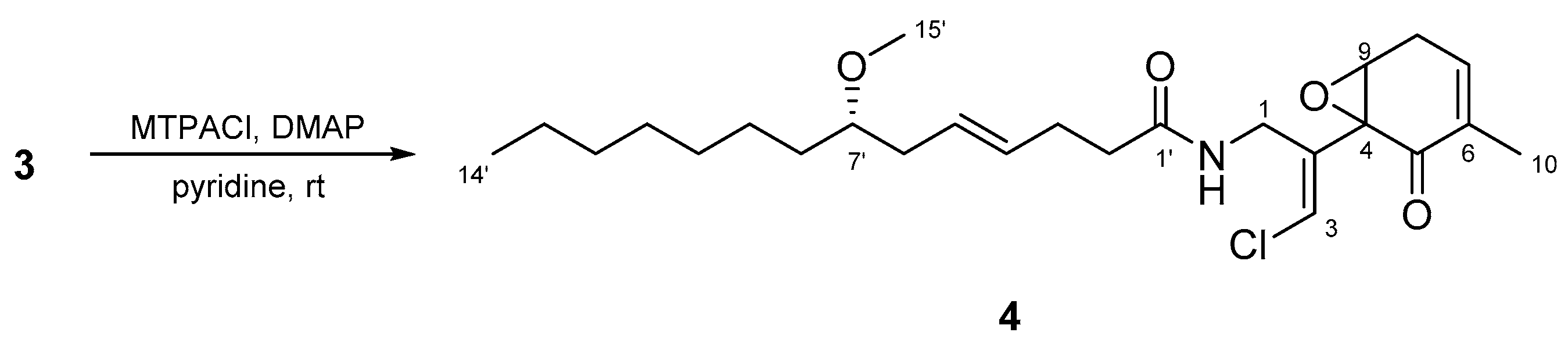
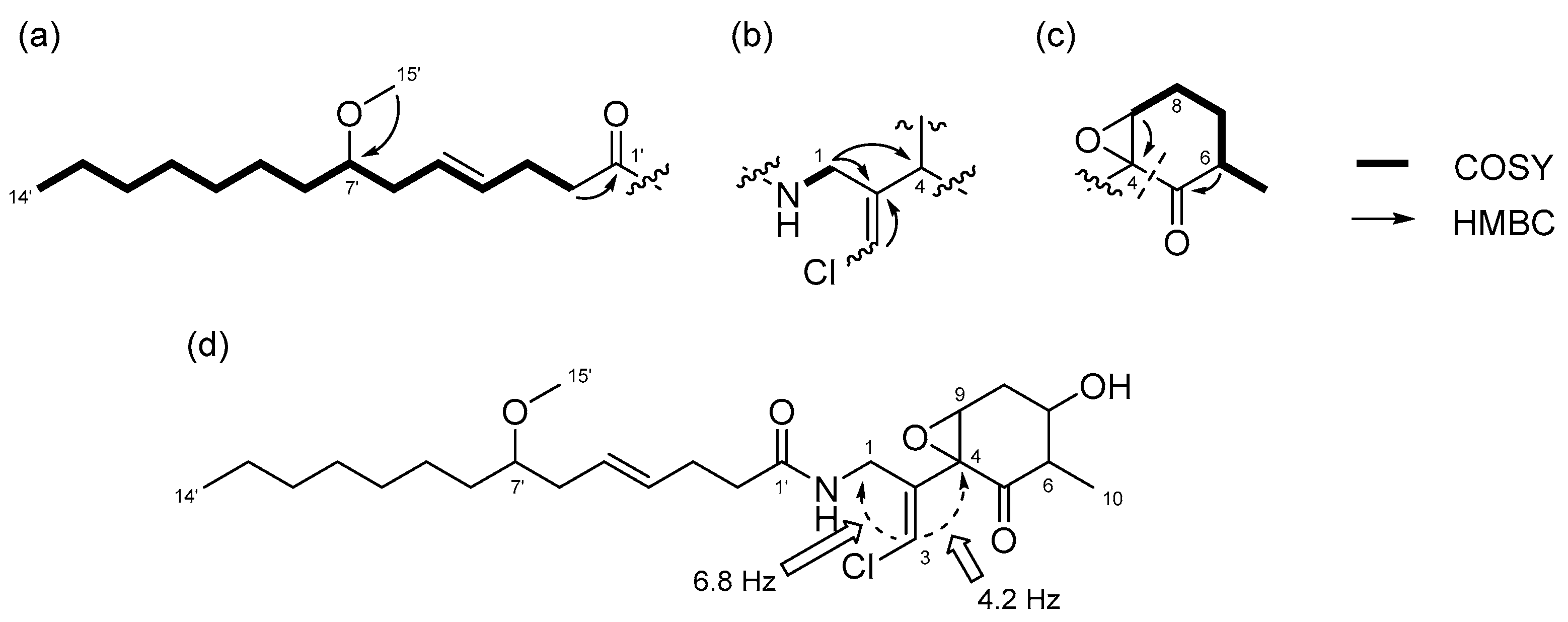
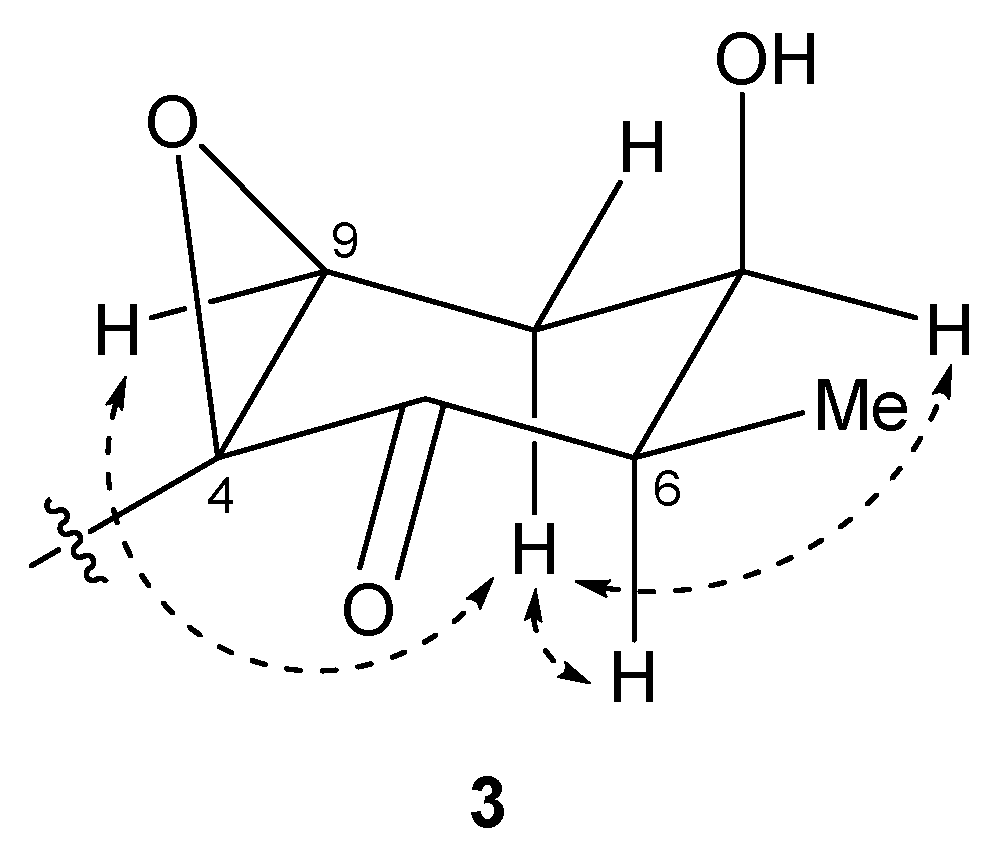
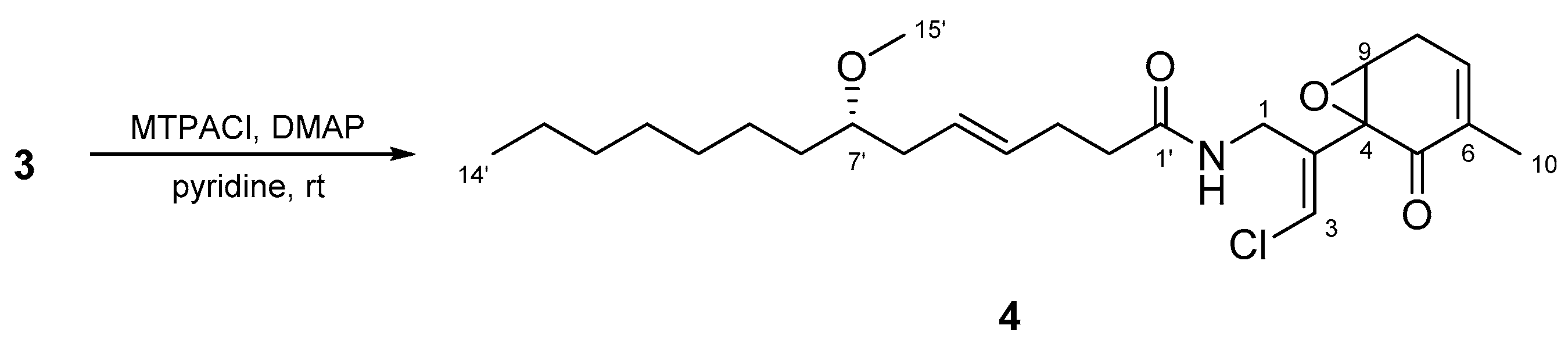
2.2. N-Demethyl-isomalyngamide I (3)
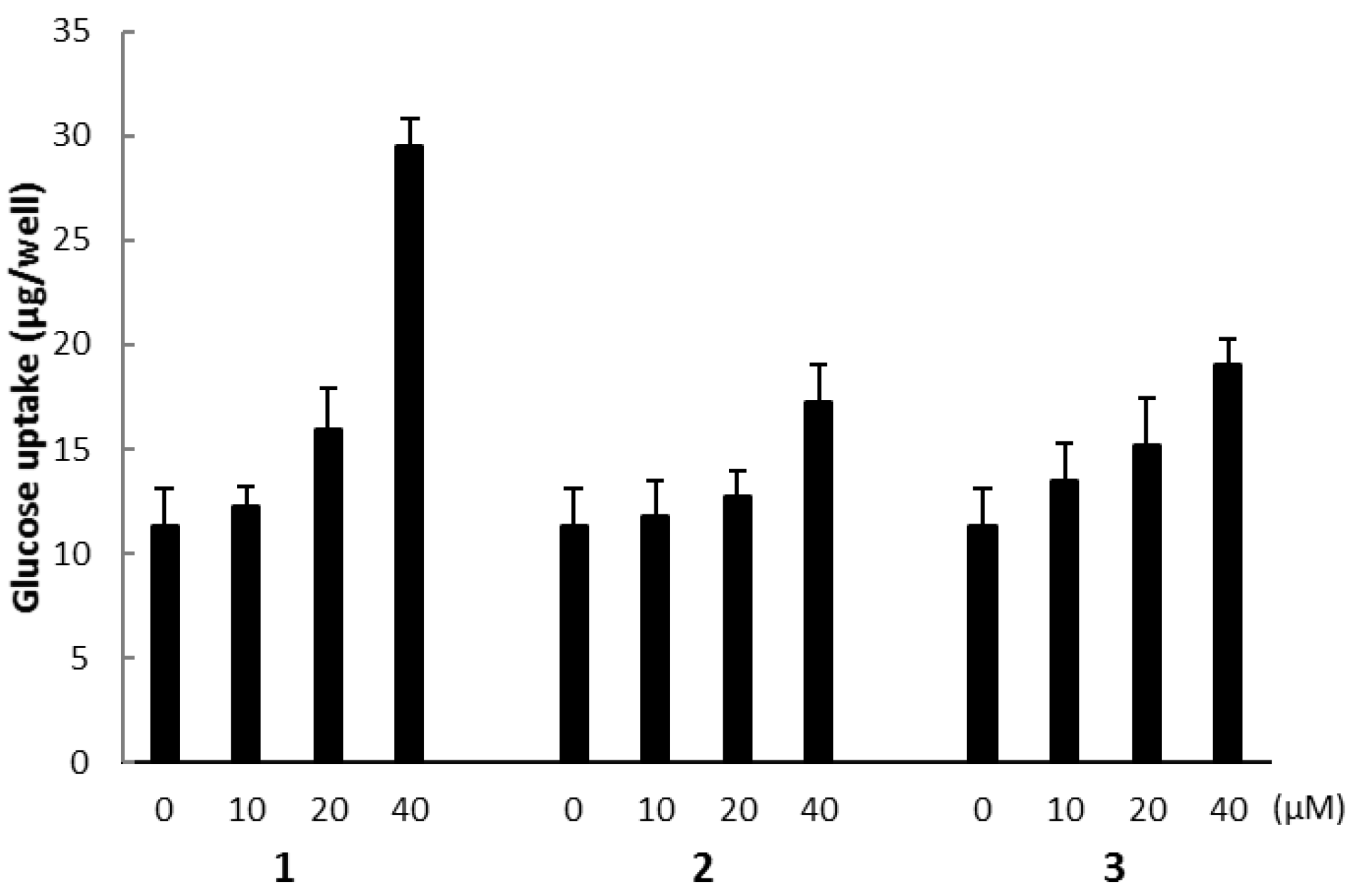
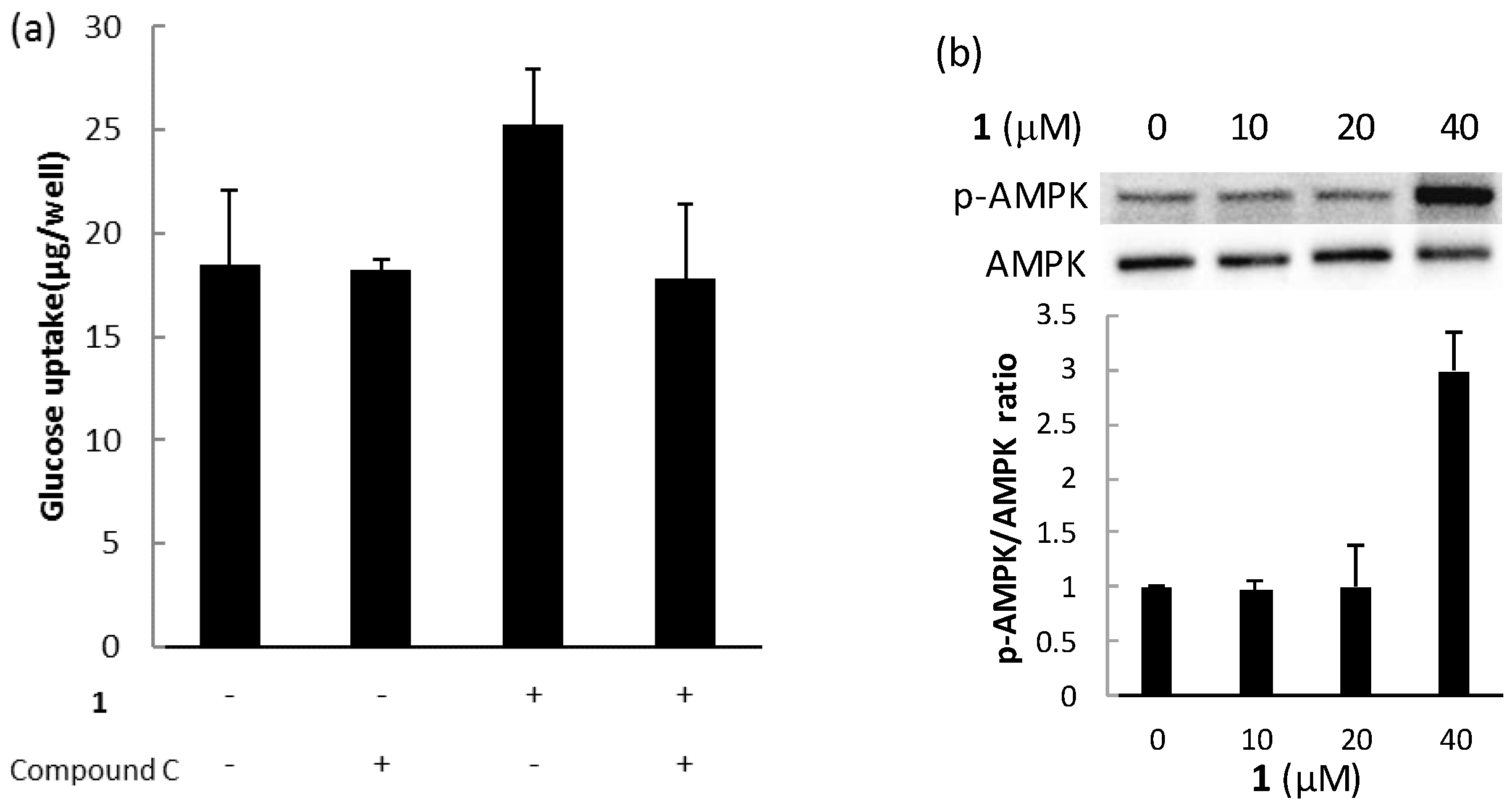
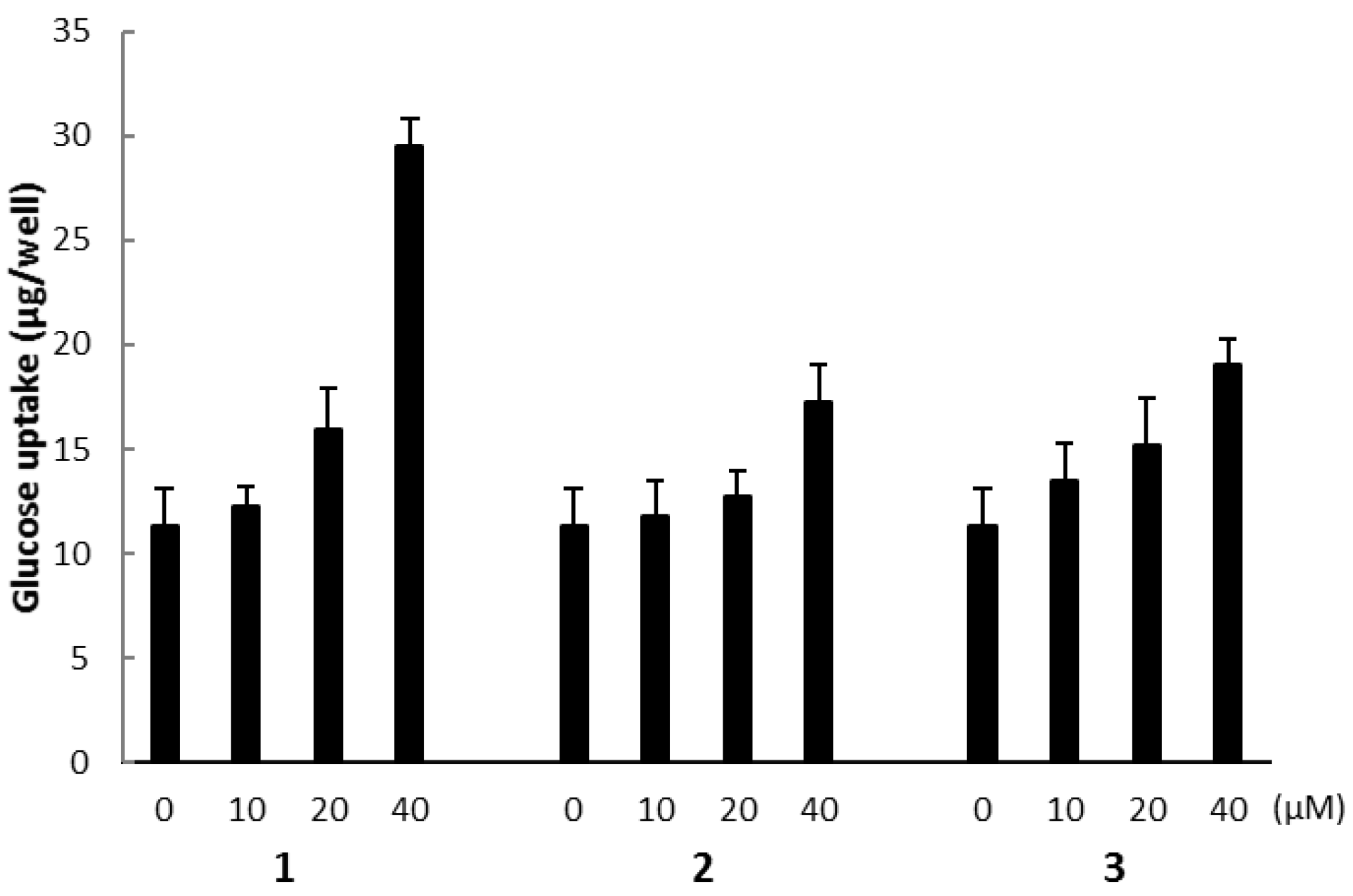
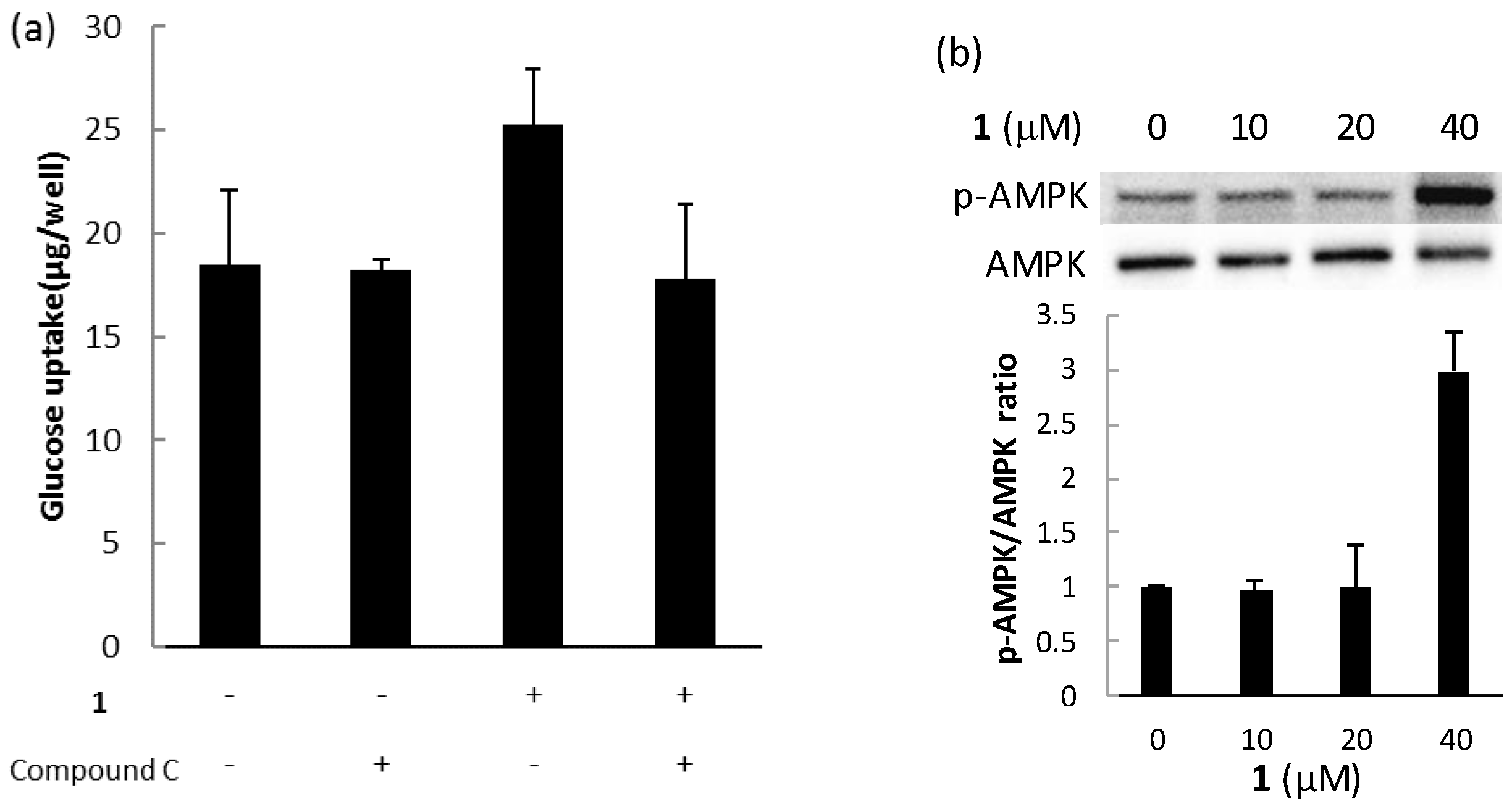
2.3. Biological Activities
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection, Extraction, and Isolation
3.3. Identification of the Marine Cyanobacterium
3.4. Preparation of MTPA Esters of 2
3.5. Preparation of Acetylated Compound 2
3.6. Base Hydrolysis of Compounds 2 and 3
3.7. Preparation of MTPA Ester of Compound 3
3.8. Culture of L6 Myoblasts
3.9. Determination of Glucose Uptake
3.10. Western Blotting
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009, 11, 5062–5065. [Google Scholar] [CrossRef] [PubMed]
- Sumiya, E.; Shimogawa, H.; Sasaki, H.; Tsutsumi, M.; Yoshita, K.; Ojika, M.; Suenaga, K.; Uesugi, M. Cell-morphology profiling of a natural product library identifies bisebromoamide and miuraenamide A as actin filament stabilizers. ACS Chem. Biol. 2011, 6, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Ōmura, S.; Suenaga, K. Isolation and total synthesis of hoshinolactam, an antitrypanosomal lactam from a marine cyanobacterium. Org. Lett. 2017, 19, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Cardellina, J.H., II; Marner, F.J.; Moore, R.E. Malyngamide A, a novel chlorinated metabolite of the marine cyanophyte Lyngbya majuscule. J. Am. Chem. Soc. 1979, 101, 240–242. [Google Scholar] [CrossRef]
- Gerwick, L.; Boudreau, P.; Choi, H.; Mascuch, S.; Villa, F.A.; Balunas, M.J.; Malloy, K.L.; Teasdale, M.E.; Rowley, D.C.; Gerwick, W.H. Interkingdom signaling by structurally related cyanobacterial and algal secondary metabolites. Phytochem. Rev. 2013, 12, 459–465. [Google Scholar] [CrossRef]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Kaneda, M.; Sueyoshi, K.; Teruya, T.; Ohno, H.; Fujii, N.; Oishi, S. Total synthesis of odoamide, a novel cyclic depsipeptide, from an Okinawan marine cyanobacterium. Org. Biomol. Chem. 2016, 14, 9093–9104. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Kudo, T.; Yamano, A.; Sumimoto, S.; Iwasaki, A.; Suenaga, K.; Teruya, T. Odobromoamide, a terminal alkynyl bromide-containing cyclodepsipeptide from the marine cyanobacterium Okeania sp. Bull. Chem. Soc. Jpn. 2017, 90, 436–440. [Google Scholar] [CrossRef]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, R.D.; Barchi, J.J.; Kuniyoshi, M.; Moore, R.E.; Mynderse, J.S. Structure of mlyngamide C. J. Org. Chem. 1985, 50, 2859–2862. [Google Scholar] [CrossRef]
- Milligan, K.E.; Márquez, B.L.; Williamson, R.T.; Davies-Coleman, M.; Gerwick, W.H. Two new malyngamides from a Madagascan Lyngbya majuscula. J. Nat. Prod. 2000, 63, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Shaala, L.A.; Youssef, D.T.A.; McPhail, K.L.; Elbandy, M. Malyngamide 4, a new lipopeptide from the Red Sea marine cyanobacterium Moorea producens (formerly Lyngbya majuscule). Phytochem. Lett. 2013, 6, 183–185. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Cardellina, J.H., II; Dalietos, D.; Marner, F.J.; Mynderse, J.S.; Moore, R.E. (−)-trans-7(S)-Methoxytetradec-4-enoic acid and related amides from the marine cyanophyte Lyngbya majuscule. Phytochemistry 1978, 17, 2091–2095. [Google Scholar] [CrossRef]
- Williamson, R.T.; Márquez, B.L.; Gerwick, W.H.; Kover, K.E. One- and two-dimensional gradient-selected HSQMBC NMR experiments for the efficient analysis of long-range heteronuclear coupling constants. Magn. Reson. Chem. 2000, 38, 265–273. [Google Scholar] [CrossRef]
- Kan, Y.; Fujita, T.; Nagai, H.; Sakamoto, B.; Hokama, Y. Malyngamides M and N from the Hawaiian red alga Gracilaria coronopifolia. J. Nat. Prod. 1998, 61, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.; McPhail, K.L.; Goeger, D.E.; Valeriote, F.A.; Gerwick, W.H. Two cytotoxic stereoisomers of malyngamide C, 8-epi-malyngamide C and 8-O-acetyl-8-epi-malyngamide C, from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2010, 71, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.S.; Gerwick, W.H. Malyngamide I from the tropical marine cyanobacterium Lyngbya majuscula and the probable structure revision of stylocheilamide. Tetrahedron Lett. 1995, 36, 7837–7840. [Google Scholar] [CrossRef]
- Hayashi, T.; Hirshman, M.F.; Kurth, E.J.; Winder, W.W.; Goodyear, L.J. Evidence for 5’AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998, 47, 1369–1373. [Google Scholar] [PubMed]
- Hayashi, T.; Hirshman, M.F.; Fujii, N.; Habinowski, S.N.; Witters, L.A.; Goodyear, L.J. Metabolic stress and altered glucose transport. Diabetes 2000, 49, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Rottacker, E.C.; Kaštovský, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komárek, J.; Gerwick, W.H. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Microbiol. 2012, 62, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Yonezawa, T.; Son, M.J.; Woo, J.T.; Ohba, S.; Chung, U.I.; Yagasaki, K. Antidiabetic effect of nepodin, a component of Rumex roots, and its modes of action in vitro and in vivo. Biofactors 2014, 40, 436–447. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δC a | δH, Mult (J in Hz) b | HMBC | δC a | δH, Mult (J in Hz) b | HMBC | |
| 1a | 41.6 | 3.95, ddd (16.5, 5.8, 1.8) | 2, 3, 4, 1’ | 41.7 | 3.95, ddd (16.3, 5.6, 1.6) | 2, 3, 4, 1’ |
| 1b | 4.23, dd (16.5, 6.7) | 2, 3, 4, 1’ | 4.24, dd (16.3, 6.8) | 2, 3, 4, 1’ | ||
| 2 | 136.3 | - | - | 136.5 | - | - |
| 3 | 121.6 | 6.29, d (1.8) | 1, 2, 4 | 121.4 | 6.27, d (1.6) | 1, 2, 4 |
| 4 | 56.1 | 4.27, s | 1, 2, 3, 5, 9, 10 | 55.1 | 4.36, s | 1, 2, 3, 5, 9, 10 |
| 5 | 201.9 | - | - | 203.3 | - | - |
| 6 | 72.4 | 5.41, dd (12.9, 6.7) | 5, 7, 6-OAc | 72.7 | 5.60, dd (12.9, 6.8) | 5, 7, 6-OAc |
| 7a | 31.3 | 2.32, m | 5, 6, 8, 9 | 34.1 | 2.26, m | 5, 6, 9 |
| 7b | 2.60, ddd (13.3, 12.9, 2.6) | 6 | 2.54, dt (12.9, 2.3) | 5, 6 | ||
| 8 | 74.8 | 5.11, dd (2.6, 2.6) | 4, 6, 9, 10, 8-OAc | 73.8 | 3.90, dd (6.1, 3.0) | 4, 6, 9, 10 |
| 9 | 78.9 | - | - | 80.4 | - | - |
| 10 | 23.5 | 1.26, s | 4, 5, 8, 9 | 23.9 | 1.34, s | 4, 8, 9 |
| NH | - | 6.68, dd (6.7, 5.8) | 1, 1’ | - | 6.84, dd (6.8, 5.6) | 1, 1’ |
| 1’ | 173.8 | - | - | 173.8 | - | - |
| 2’ | 36.2 | 2.27, m | 1’, 3’ | 36.2 | 2.27, m | 1’, 3’, 4’ |
| 3’ | 28.7 | 2.28, m | 4’ | 28.7 | 2.32, m | 1’, 2’, 4’, 5’ |
| 4’ | 130.7 | 5.45, m | 3’, 6’ | 130.7 | 5.45, m | 2’, 3’, 5’ |
| 5’ | 128.3 | 5.47, m | 4’, 7’ | 128.1 | 5.47, m | 3’, 4’, 7’ |
| 6’ | 36.3 | 2.18, m | 4’, 5’, 7’, 8’ | 36.3 | 2.19, m | 4’, 5’, 7’, 8’ |
| 7’ | 80.7 | 3.17, m | 5’, 8’, 9’, 15’ | 80.8 | 3.17, m | 5’, 8’, 9’, 15’ |
| 8’ | 33.3 | 1.42, m | 6’, 7’, 9’, 10’ | 33.4 | 1.43, m | 7’, 9’, 10’ |
| 9’ | 25.5 | 1.27, m e | - | 25.5 | 1.26, m f | - |
| 10’ | 29.9 | 1.27, m e | - | 29.9 | 1.26, m f | - |
| 11’ | 29.4 | 1.27, m e | - | 29.4 | 1.26, m f | - |
| 12’ | 32.0 c | 1.27, m e | - | 31.9 d | 1.26, m f | - |
| 13’ | 22.8 c | 1.27, m e | - | 22.8 d | 1.26, m f | - |
| 14’ | 14.2 | 0.87, t (6.7) | 12’, 13’ | 14.2 | 0.87, t (6.8) | 12’, 13’ |
| 15’ | 56.5 | 3.31, s | 7’ | 56.5 | 3.31, s | 7’ |
| 6-OAc | 170.3 | - | - | 170.4 | - | - |
| 20.8 | 2.16, s | 6-OAc | 20.9 | 2.15, s | 6-OAc | |
| 8-OAc/OH | 169.9 | - | - | - | 3.00, d (3.9) | 7, 8, 9 |
| 21.3 | 2.19, s | 8-OAc | - | - | - | |
| 9-OH | - | 5.69, s | 8, 9, 10 | - | 5.33, s | 8, 9, 10 |
| Position | δC a | δH, mult (J in Hz) b | HMBC |
|---|---|---|---|
| 1 | 37.3 | 4.08, d (6.1) | 2, 3, 4, 1’ |
| 2 | 135.1 | - | - |
| 3 | 120.9 | 6.29, s | 1, 2, 4 |
| 4 | 61.4 | - | - |
| 5 | 204.2 | - | - |
| 6 | 51.0 | 2.41, m | 5, 7, 8, 10 |
| 7 | 68.7 | 3.81, m | 6, 8, 9, 10 |
| 8a | 31.7 | 2.08, ddd (14.9, 8.5, 2.0) | 6, 7, 9 |
| 8b | 2.61, ddd (14.9, 4.7, 2.6) | 4, 6, 7 | |
| 9 | 62.1 | 3.26, dd (2.6, 2.0) | 4, 7, 8 |
| 10 | 12.4 | 1.24, d (7.0) | 5, 6, 7 |
| NH | - | 6.03, t (6.1) | 1, 1’ |
| 1’ | 173.0 | - | - |
| 2’ | 36.4 | 2.21, m | 1’, 3’, 4’ |
| 3’ | 28.6 | 2.30, m | 2’, 4’, 5’ |
| 4’ | 130.7 | 5.45, m | 3’, 5’ 6’ |
| 5’ | 128.0 | 5.47, m | 3’, 4’, 6’, 7’ |
| 6’ | 36.4 | 2.18, m | 7’, 8’ |
| 7’ | 80.8 | 3.16, m | 5’, 8’, 9’, 15’ |
| 8’ | 33.5 | 1.42, m | 7’, 9’, 10’ |
| 9’ | 25.5 | 1.27, m d | - |
| 10’ | 29.9 | 1.27, m d | - |
| 11’ | 29.4 | 1.27, m d | - |
| 12’ | 32.0 c | 1.27, m d | - |
| 13’ | 22.8 c | 1.27, m d | - |
| 14’ | 14.2 | 0.88, t (6.8) | 12’, 13’ |
| 15’ | 56.6 | 3.31, s | 7’ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sueyoshi, K.; Yamano, A.; Ozaki, K.; Sumimoto, S.; Iwasaki, A.; Suenaga, K.; Teruya, T. Three New Malyngamides from the Marine Cyanobacterium Moorea producens. Mar. Drugs 2017, 15, 367. https://doi.org/10.3390/md15120367
Sueyoshi K, Yamano A, Ozaki K, Sumimoto S, Iwasaki A, Suenaga K, Teruya T. Three New Malyngamides from the Marine Cyanobacterium Moorea producens. Marine Drugs. 2017; 15(12):367. https://doi.org/10.3390/md15120367
Chicago/Turabian StyleSueyoshi, Kosuke, Aki Yamano, Kaori Ozaki, Shimpei Sumimoto, Arihiro Iwasaki, Kiyotake Suenaga, and Toshiaki Teruya. 2017. "Three New Malyngamides from the Marine Cyanobacterium Moorea producens" Marine Drugs 15, no. 12: 367. https://doi.org/10.3390/md15120367
APA StyleSueyoshi, K., Yamano, A., Ozaki, K., Sumimoto, S., Iwasaki, A., Suenaga, K., & Teruya, T. (2017). Three New Malyngamides from the Marine Cyanobacterium Moorea producens. Marine Drugs, 15(12), 367. https://doi.org/10.3390/md15120367